

Abstract
OBJECTIVE— Recent progress suggests that exenatide, a mimetic of glucagon-like peptide-1 (GLP-1), might lower glycemia independent of increased β-cell response or reduced gastrointestinal motility. We aimed to investigate whether exenatide stimulates glucose turnover directly in insulin-responsive tissues dependent or independent of insulinemia.
RESEARCH DESIGN AND METHODS— An intraportal glucose infusion clamp was used in dogs to measure glucose turnover to encompass potent activation of the putative glucose/GLP-1 sensor in the porto-hepatic circulation with exenatide. The modified glucose clamp was performed in the presence of postprandial hyperinsulinemia and hyperglycemia with exenatide (20 μg) or saline injected at 0 min. Furthermore, the role of hyperglycemia versus hyperinsulinemia in exenatide-mediated glucose disposal was studied.
RESULTS— With hyperinsulinemia and hyperglycemia, exenatide produced a significant increase in total glucose turnover by ∼30%, as indicated by portal glucose infusion rate (saline 15.9 ± 1.6 vs. exenatide 20.4 ± 2.1 mg · kg−1 · min−1, P < 0.001), resulting from increased whole-body glucose disposal (Rd, ∼20%) and increased net hepatic uptake of exogenous glucose (∼80%). Reducing systemic hyperglycemia to euglycemia, exenatide still increased total glucose turnover by ∼20% (saline 13.2 ± 1.9 vs. exenatide 15.6 ± 2.1 mg · kg−1 · min−1, P < 0.05) in the presence of hyperinsulinemia, accompanied by smaller increments in Rd (12%) and net hepatic uptake of exogenous glucose (45%). In contrast, reducing hyperinsulinemia to basal levels, exenatide-increased total glucose turnover was completely abolished despite hyperglycemia (saline 2.9 ± 0.6 vs. exenatide 2.3 ± 0.3 mg · kg−1 · min−1, P = 0.29).
CONCLUSIONS— Exenatide directly stimulates glucose turnover by enhancing insulin-mediated whole-body glucose disposal and increasing hepatic uptake of exogenous glucose, contributing to its overall action to lower postprandial glucose excursions.
Exenatide is the synthetic form of exendin-4 and a long-acting mimetic of the incretin hormone glucagon-like peptide-1 (GLP-1) (1). Originally isolated from salivary secretions of the lizard Gila monster, exendin-4 shows a 53% amino acid sequence identity to GLP-1 (2) and shares many actions with GLP-1 via pancreatic GLP-1 receptor (3). Exenatide stimulates glucose-dependent insulin response (4–6), suppresses glucagon secretion (5), and inhibits gastrointestinal motility (7). Exenatide has also been implicated in regulating food intake (8) and β-cell proliferation (9). Exenatide is resistant to digestion by dipeptidyl peptidase-IV and thus has a longer plasma half-life than GLP-1 (10).
Exenatide is currently used in the treatment of type 2 diabetes because of its effect to lower glycemia and improve glycemic control through multiple mechanisms as mentioned above. Exenatide (13 weeks) in diabetic db/db mice led to 50% lower glycosylated hemoglobin (A1C) than in nontreated animals (4). Type 2 diabetic patients who had not attained A1C goals ≤8% with sulfonylureas and/or metformin achieved a ∼0.9% reduction with exenatide (4 weeks) (11).
However, it appears that exenatide-improved glycemic control cannot be completely explained by its currently established actions. In particular, exenatide may have a direct effect on insulin-responsive tissues, although this latter effect is still under debate. In obese Zucker rats, exenatide (6 weeks) dramatically enhanced insulin sensitivity while preventing the progressive increase in A1C and fasting insulin (12). When compared with pair-fed animals with matching A1C, fasting glucose, insulin, and lipids, insulin sensitivity was still 64% higher with exenatide. In another study, exenatide administered with meals to type 2 diabetic patients resulted in reduced glucose excursions facing significantly lower postprandial insulin levels, not higher, as expected based on the insulinotropic effect of exenatide (5). The reduced postprandial glucose excursion has been attributed to exenatide inhibition of gastric emptying and/or glucagon secretion (5,7). However, by simulating intraportal rate of glucose appearance (Ra) in portal vein after meal with or without exenatide (thus bypassing its gastrointestinal effect), we have shown that exenatide lowered glycemia in the presence of similar insulin and glucagon levels (13). Although some studies have failed to show acute effects of exenatide on insulin action (14,15), our previous result suggests a potential exenatide effect to increase insulin sensitivity. The goal of our current study was to investigate whether exenatide directly stimulates glucose turnover dependent or independent of insulinemia in insulin-responsive tissues.
RESEARCH DESIGN AND METHODS
Experiments were conducted in male mongrel dogs (Harlan, Indianapolis, IN), housed under controlled conditions and fed once daily (Labdiet; PMI Nutrition International, Richmond IN). Animals were used for experiments only if they were judged to be in good health as determined by body weight, food intake and stools, temperature, hematocrit, and direct observation. All surgical and experimental procedures were approved by USC Institutional Animal Care and Use Committee.
Surgical procedures.
At least 1 week before the first experiments, chronic catheters (Tygon, inner diameter = 0.05 inch; Norton Plastics, Akron, OH) were implanted under general anesthesia. One catheter was placed in jugular vein (with the tip advanced into right atrium) and the other in portal vein (4-cm upstream from porta hepatis). All catheters, filled with heparinized saline (10 units/ml), were led subcutaneously to the back of the neck, exteriorized, and secured in place.
Experimental design.
Experiments were performed on animals in a conscious relaxed state. Animals were fasted for 14–16 h with free access to water. For all experiments, animals were brought to the laboratory at ∼6:00 a.m. and placed into a Pavlov sling. Three types of experiments were performed in paired experiments with subcutaneous injection of either exenatide or saline to examine the effect of exenatide on glucose turnover. In all studies, we used a novel glucose clamp technique, the intraportal glucose infusion–glucose clamp. Unlike the classical clamp where exogenous glucose is given systemically, in these experiments, we infused exogenous glucose directly into portal vein to maintain the desired systemic glycemia. Previous evidence has suggested the presence of glucose sensors in the porto-hepatic circulation that may interact with GLP-1 to affect glucose clearance (16–22). Intraportal glucose infusion was used to stimulate any extant portal/hepatic sensors, as would happen during postprandial nutritional entry of carbohydrate into the systemic circulation. The intraportal glucose infusion–glucose clamp was performed under the following conditions: study 1, in the presence of hyperinsulinemia and hyperglycemia; study 2, in the presence of hyperinsulinemia but systemic euglycemia; and study 3, in the presence of hyperglycemia but basal insulin levels.
Study 1: Effect of exenatide on glucose turnover in the presence of postprandial hyperinsulinemia and hyperglycemia.
Paired experiments with or without a single subcutaneous injection of exenatide were performed in nine animals (eight with tracer infusions), 28.8 ± 0.5 kg body wt. The modified glucose clamp was composed of two periods: a 2-h equilibrium period (−120 to 0 min), followed by injection of saline or exenatide, and a 4-h experimental period (0–240 min; Fig. 1A). At −120 min, immediately after a systemic injection of 25-μCi bolus of [3-3H]d-glucose (“tracer”; DuPont-NEN, Boston, MA), a continuous infusion of tracer at 0.25 μCi/min was initiated via a peripheral venous catheter. Ninety minutes later, blood samples for the basal period were taken every 10 min from −30 to 0 min. At 0 min, peripheral infusions of somatostatin (Bachem, Torrance, CA), at 1 μg · kg−1 · min−1, and porcine insulin (Eli Lilly, Indianapolis, IN), at 0.75 mU · kg−1 · min−1, were initiated to obtain systemic hyperinsulinemia of ∼250 pmol/l. An intraportal infusion of porcine glucagon (Sigma, St. Louis, MO), at 0.65 ng · kg−1 · min−1, was simultaneously started to maintain plasma glucagon at basal levels. At 0 min, a single subcutaneous injection of either exenatide (20 μg Byetta; Amylin, San Diego, CA) or saline of equal volume was administered in paired experiments. During the experimental phase (0–240 min), blood samples were taken from another peripheral venous catheter every 10 min; plasma glucose were measured immediately. Exogenous glucose (50% dextrose, 454.5 mg/ml; B Braun, Irvine, CA) was given via the portal vein catheter at variable rates to maintain systemic glucose at 150 mg/dl. An additional tracer infusion was given at variable rates via the jugular vein catheter to minimize fluctuations in systemic plasma specific activity; the ratio between the rate of the additional systemic tracer infusion and that of intraportal glucose infusion was kept at 1.8 μCi/g. The average of the four samples taken from 150–180 min was defined as the clamp steady state when plasma glucose, insulin, and exogenous glucose infusion rate were least variable.
FIG. 1.

A: Intraportal glucose infusion clamp. Basal and clamp SS indicate the steady state during the basal and clamp period, respectively. Porcine insulin was infused at either 0.75 (studies 1 and 2) or 0.15 mU · kg−1 · min−1 (study 3) to achieve postprandial hyperinsulinemia or maintain basal insulin levels. Exogenous glucose was given into portal vein to achieve systemic hyperglycemia (studies 1 and 3) or maintain systemic euglycemia (study 2). B: Calculation of NHGA. Glucose label indicates the constant and the variable tracer infusions.
Study 2: Effect of exenatide on glucose turnover in the presence of hyperinsulinemia but systemic euglycemia.
Paired experiments with or without a single subcutaneous injection of exenatide were conducted in eight animals (six with tracer infusions), 28.2 ± 0.7 kg body wt. The modified glucose clamp was used to measure glucose turnover as discussed above, except that intraportal glucose infusion was given to maintain systemic glucose at basal levels (Fig. 1A).
Study 3: Effect of exenatide on glucose turnover in the presence of hyperglycemia but basal insulin levels.
Paired experiments with or without a single subcutaneous injection of exenatide were carried out in six animals (six with tracer infusions), 29.5 ± 0.8 kg body wt. The modified glucose clamp was used to measure glucose turnover as discussed in study 1, except that plasma insulin was maintained at basal levels by a low infusion rate of 0.15 mU · kg−1 · min−1 while systemic glucose was raised to 150 mg/dl (Fig. 1A).
Blood sampling.
Samples for the determination of glucose, free fatty acids (FFAs), insulin, C-peptide, glucagon, and tracer were collected as previously described (16,23). All samples were immediately centrifuged, and plasma was separated and stored at −80°C until analysis. To prevent triglyceride breakdown, FFAs samples were kept on ice and either immediately assayed or kept at −80°C for a limited time before assay, as previously described (16).
Assays.
Glucose was measured with a YSI 2700 autoanalyzer (Yellow Springs Instruments, Yellow Springs, OH). Insulin was measured using a human insulin ELISA kit (Linco/Millipore, Billerica, MA) adapted for dog plasma (24). C-peptide and glucagon were measured using radioimmunoassay kits (Linco/Millipore). FFAs were determined using an enzymatic colorimetric assay (NEFA C; Wako Pure Chemical Industries, Richmond, VA). Samples for [3-3H]-d-glucose were deproteinized, dried, resuspended in scintillation fluid (Readysafe; Beckman, Fullerton, CA), and then read in a β-scintillation counter.
Calculations.
Two metabolic parameters can be calculated from the intraportal glucose infusion clamp: whole-body glucose disposal and a parameter we named “net hepatic glucose addition” (NHGA). The time course of whole-body glucose disposal [rate of glucose disappearance (Rd)] was calculated using Steele's equation with a labeled glucose infusion (25) after data smoothing using OOPSEG (26). Because exogenous glucose was infused directly into portal vein, a fraction of intraportally given glucose would be taken up by the liver without entering the systemic circulation; referred to as 1st-pass hepatic glucose uptake (1st-pass HGU). Therefore, the Ra equals portal glucose infusion (PoGinf) plus endogenous glucose production (EGP) minus 1st-pass HGU (Eq. 1). The difference between EGP and 1st-pass HGU, which is defined as NHGA, is thus derived from the difference between Ra and PoGinf (Eq. 2; Fig. 1B). Ra itself was calculated using Steele's equation, similar to Rd.
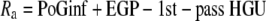 |
(1) |
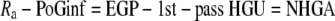 |
(2) |
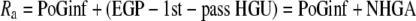 |
(3) |
In an intraportal glucose infusion–glucose clamp, NHGA, as the name implies, reflects the net addition of glucose by the liver to portally infused exogenous glucose (Eq. 3). During the basal period, PoGinf and 1st-pass HGU both equal 0; thus, Ra = EGP = NHGA. During the experimental period, under the influence of elevated insulin and/or glucose levels, EGP decreases while 1st-pass HGU increases. A positive NHGA indicates EGP>1st-pass HGU, i.e., there is a net addition of glucose by the liver to portally infused glucose and thus systemic Ra>PoGinf. A negative NHGA indicates EGP<1st-pass HGU, i.e., there is a net uptake of glucose by the liver from portally infused glucose and thus systemic Ra<PoGinf. Physiologically, the parameter NHGA (EGP − 1st-pass HGU) represents, in postprandial situations, the net effect of the liver on Ra in the systemic circulation by changing EGP and 1st-pass HGU (Ra = PoGinf + NHGA). It is noteworthy that NHGA is different from net hepatic glucose balance (NHGB). The latter is calculated as the arteriovenous difference of hepatic glucose input and output; NHGB = EGP − total HGU (not just 1st-pass HGU).
Statistics.
All experimental data are expressed as means ± SE. Two-way ANOVA was used to compare time course data with or without exenatide. Paired Student's t test was used to compare the basal or clamp steady-state parameters between saline and exenatide treatments. Differences were considered statistically significant at P < 0.05.
RESULTS
Study 1: The effect of exenatide on glucose turnover in the presence of postprandial hyperinsulinemia and hyperglycemia.
We first studied the effect of exenatide on glucose turnover in the presence of postprandial insulin and glucose levels. The time courses of plasma insulin and glucose were matched between the saline and exenatide groups, with insulin raised to ∼225 pmol/l and glucose raised to ∼150 mg/dl at the clamp steady state (Table 1).
TABLE 1.
Intraportal glucose infusion clamp in the presence of postprandial hyperinsulinemia and hyperglycemia: basal and clamp steady-state parameters in the saline- and exenatide-treated groups
| Basal
|
Clamp steady state
|
|||
|---|---|---|---|---|
| Saline | Exenatide | Saline | Exenatide | |
| Glucose (mg/dl) | 92.6 ± 1.6 | 89.4 ± 2.1* | 150.2 ± 2.8† | 157.4 ± 4.3† |
| Insulin (pmol/l) | 41.3 ± 4.5 | 45.8 ± 4.0 | 231.5 ± 10.5† | 217.9 ± 11.5*† |
| C-peptide (ng/ml) | 0.24 ± 0.06 | 0.25 ± 0.06 | 0.06 ± 0.06† | 0.05 ± 0.04† |
| Glucagon (ng/l) | 42.5 ± 4.2 | 50.8 ± 6.4 | 33.3 ± 3.2 | 33.5 ± 3.9† |
| FFAs (mmol/l) | 0.58 ± 0.06 | 0.55 ± 0.11 | 0.02 ± 0.01† | 0.03 ± 0.03† |
Data are means ± SE.
P < 0.05 significantly different from the saline group.
P < 0.05 significantly different from basal in each group.
The total glucose turnover, as indicated by portal glucose infusion rate, was elevated with exenatide, increasing ∼30% at steady state (15.9 ± 1.6 saline vs. 20.4 ± 2.1 exenatide mg · kg−1 · min−1, P < 0.001; Fig. 2A). Increased glucose turnover was a result of both increased whole-body glucose disposal (Rd) and increased net hepatic uptake of portal exogenous glucose. Rd was increased by ∼20% (15.5 ± 1.5 saline vs. 18.3 ± 2.1 exenatide mg · kg−1 · min−1, P < 0.05; Fig. 2B). NHGA was quickly switched from net addition to net uptake (1st-pass), which was increased by ∼80% with exenatide (−1.2 ± 0.2 saline vs. −2.2 ± 0.3 exenatide mg · kg−1 · min−1, P < 0.05; Fig. 2C).
FIG. 2.

Intraportal glucose infusion clamp in the presence of postprandial hyperinsulinemia and hyperglycemia. Time course (left) and clamp steady-state (right) data of intraportal glucose infusion rate (PoGinf) (A), Rd (B), and NHGA (C) in the saline (SAL, ○) and exenatide (EX, •) groups. *P < 0.05, significantly different from saline.
Endogenous insulin secretion was suppressed with somatostatin, as reflected by plasma C-peptide levels, which were significantly reduced at steady state in both groups. Plasma glucagon was replaced near basal levels and matched between groups. Plasma FFAs were quickly and completely suppressed in both groups (Table 1).
Study 2: The effect of exenatide on glucose turnover in the presence of hyperinsulinemia but systemic euglycemia.
To dissect the role of hyperglycemia in exenatide-mediated glucose turnover, the effect of exenatide on glucose turnover was examined in the presence of hyperinsulinemia but systemic euglycemia (reduced glycemia intraportally). The time courses of plasma insulin and glucose were superimposable between the saline and exenatide groups, with insulin raised to ∼245 pmol/l and glucose clamped at basal (∼94 mg/dl) at the clamp steady state (Table 2). At systemic euglycemia, total glucose turnover was still increased with exenatide but to a lesser degree, increasing ∼20% at steady state (13.2 ± 1.9 saline vs. 15.6 ± 2.1 exenatide mg · kg−1 · min−1, P < 0.05; Fig. 3A). Accordingly, Rd was only increased by ∼12%, but the increment was still significant (13.0 ± 2.4 saline vs. 14.5 ± 2.4 exenatide mg · kg−1 · min−1, P < 0.05; Fig. 3B). The increment in net hepatic uptake of portal exogenous glucose was 45%, which was significant over the entire time course (−1.1 ± 0.1 saline vs. −1.6 ± 0.5 exenatide mg · kg−1 · min−1, P < 0.005 time course, whereas P = 0.33 steady state; Fig. 3C). Again, no difference was found in plasma C-peptide, glucagon, and FFAs between groups (Table 2).
TABLE 2.
Intraportal glucose infusion clamp in the presence of hyperinsulinemia but systemic euglycemia: basal and clamp steady-state parameters in the saline- and exenatide-treated groups
| Basal
|
Clamp steady state
|
|||
|---|---|---|---|---|
| Saline | Exenatide | Saline | Exenatide | |
| Glucose (mg/dl) | 93.1 ± 1.9 | 93.0 ± 1.5 | 91.2 ± 2.9 | 96.2 ± 2.5 |
| Insulin (pmol/l) | 39.5 ± 5.5 | 47.7 ± 4.0 | 246.3 ± 11.7* | 242.8 ± 10.9* |
| C-peptide (ng/ml) | 0.21 ± 0.05 | 0.23 ± 0.05 | 0.04 ± 0.03* | 0.06 ± 0.03* |
| Glucagon (ng/l) | 47.3 ± 4.6 | 47.8 ± 5.8 | 31.1 ± 4.0* | 30.1 ± 5.2* |
| FFAs (mmol/l) | 0.64 ± 0.06 | 0.57 ± 0.07 | 0.04 ± 0.01* | 0.03 ± 0.01* |
Data are means ± SE.
P < 0.05 significantly different from basal in each group.
FIG. 3.

Intraportal glucose infusion clamp in the presence of hyperinsulinemia but systemic euglycemia. Time course (left) and clamp steady-state (right) data of intraportal glucose infusion rate (PoGinf) (A), Rd (B), and NHGA (C) in the saline (SAL, ○) and exenatide (EX, •) groups. *P < 0.05, significantly different from saline.
Study 3: The effect of exenatide on glucose turnover in the presence of hyperglycemia but basal insulin levels.
To investigate whether hyperinsulinemia is required for the effect of exenatide on glucose disposal, the effect of the compound was examined in the presence of hyperglycemia but basal insulin levels. The time courses of plasma insulin and glucose were matched between the saline and exenatide groups, with insulin maintained at basal levels (∼45 pmol/l) and systemic glucose elevated to ∼150 mg/dl (Table 3). In sharp contrast to studies 1 and 2, in the absence of hyperinsulinemia, exenatide did not cause an increase in total glucose turnover despite elevated glycemia (2.9 ± 0.6 saline vs. 2.3 ± 0.3 exenatide mg · kg−1 · min−1 at steady state, P = 0.29; Fig. 4A). Hence, without hyperinsulinemia, whole-body glucose disposal did not differ between groups; a slight increment from basal levels resulting from elevated glycemia occurred similarly in both groups (4.1 ± 0.5 saline vs. 4.0 ± 0.3 exenatide mg · kg−1 · min−1, P = 0.77; Fig. 4B). Without hyperinsulinemia, exenatide did not induce net hepatic uptake of portally infused glucose either; net addition was simply suppressed to ∼50% of basal levels (1.3 ± 0.1 saline vs. 1.4 ± 0.2 exenatide mg · kg−1 · min−1, P = 0.77; Fig. 4C). Again, no difference was found in plasma C-peptide, glucagon, and FFAs profiles between groups (Table 3).
TABLE 3.
Intraportal glucose infusion clamp in the presence of hyperglycemia but basal insulin levels: basal and clamp steady-state parameters in the saline- and exenatide-treated groups
| Basal
|
Clamp steady state
|
|||
|---|---|---|---|---|
| Saline | Exenatide | Saline | Exenatide | |
| Glucose (mg/dl) | 89.5 ± 0.8 | 90.0 ± 1.7 | 151.0 ± 2.8* | 153.4 ± 4.3* |
| Insulin (pmol/l) | 41.1 ± 4.5 | 54.3 ± 3.3† | 44.4 ± 1.8 | 42.2 ± 2.7* |
| C-peptide (ng/ml) | 0.20 ± 0.03 | 0.19 ± 0.05 | 0.01 ± 0.01* | 0.00 ± 0.00* |
| Glucagon (ng/l) | 45.3 ± 7.3 | 50.8 ± 8.9 | 38.1 ± 6.3* | 39.2 ± 7.2* |
| FFAs (mmol/l) | 0.44 ± 0.05 | 0.43 ± 0.07 | 0.23 ± 0.04* | 0.22 ± 0.03* |
Data are means ± SE.
P < 0.05 significantly different from basal in each group.
P < 0.05 significantly different from the saline group.
FIG. 4.

Intraportal glucose infusion clamp in the presence of hyperglycemia but basal insulin levels. Time course (left) and clamp steady-state (right) data of intraportal glucose infusion rate (PoGinf) (A), Rd (B), and NHGA (C) in the saline (SAL, ○) and exenatide (EX, •) groups.
In Fig. 5, we summarized clamp steady-state values of Rd and NHGA versus their respective plasma insulin or glucose levels from the three studies. In the absence of elevated insulin levels (study 3), no difference was induced by exenatide either in Rd or in NHGA despite the presence of hyperglycemia (Rd, Fig. 5A; and NHGA, Fig. 5C). In contrast, raising insulin to postprandial levels, a significant enhancement in whole-body glucose disposal with exenatide occurred at similar systemic hyperglycemia (study 3 vs. study 1; Fig. 5A). At hyperinsulinemia, a greater net hepatic uptake of portal exogenous glucose was also induced with exenatide facing similar hepatic glucose load (estimated by plasma glucose × hepatic flow + portal glucose infusion; study 3 vs. study 2; Fig. 5C). Hyperinsulinemia is a prerequisite for exenatide-mediated glucose disposal, pointing to an insulin-sensitizing effect of exenatide. In addition, at similar hyperinsulinemia, raising systemic glycemia from basal levels to ∼150 mg/dl (study 2 vs. study 1), the increments in Rd (Fig. 5B) and in net hepatic uptake of exogenous glucose (Fig. 5D) were further increased. Hyperglycemia further enhanced exenatide-mediated glucose disposal only in the presence of elevated insulinemia, again indicating increased insulin sensitivity with exenatide.
FIG. 5.

The clamp steady-state data of Rd and NHGA expressed against their respective plasma insulin (A and C) or glucose (B and D) levels. ○, saline; •, exenatide. Study 1, postprandial hyperinsulinemia plus hyperglycemia; study 2, hyperinsulinemia plus systemic euglycemia; study 3, hyperglycemia plus basal insulin levels. *P < 0.05, significantly different from saline.
DISCUSSION
It has been shown that exenatide improves glycemic control primarily by reducing postprandial hyperglycemia (27). Multiple mechanisms have been implicated in this regard, including the effect of exenatides on the pancreas, the gastrointestinal tract, and the brain (3). The central finding of the present study is that exenatide also directly stimulates glucose turnover by enhancing insulin-mediated whole-body glucose disposal and increasing uptake of exogenous glucose by the liver, contributing to its overall action to lower postprandial glucose excursion.
Currently, available information regarding the effect of exenatide or GLP-1 on glucose turnover is not consistent (14,28–30). Although the discrepancies may be explained by different study designs or subjects used, one explanation could be a GLP-1/glucose sensor in the porto-hepatic region to mediate the glucose-lowering effect of exenatide and GLP-1. The existence of such a GLP-1 sensing mechanism has been suggested by anatomical and functional studies, showing GLP-1 receptors expressed on nerve terminals in portal vein (21) and increased vagal discharge rate by intraportal GLP-1 infusion (20). Moreover, concurrent portal hyperglycemia appears to be required for the activation of GLP-1 sensor (16–19,22). We have shown that intraportal GLP-1 does not lower systemic glucose unless it is paired with intraportal (but not systemic) glucose infusion (16). Therefore, we used the intraportal glucose infusion clamp to measure exenatide-mediated glucose turnover, which mimics the postprandial nutritional entry into the systemic circulation, potentially stimulating the putative portal sensor with exenatide. Although administered subcutaneously, portal exenatide concentration should increase effectively because of its longer plasma half-life than GLP-1 (3). Exenatide was given at 20 μg, a recommended clinical daily dose. We have previously confirmed that this dose induces a significant improvement in glucose tolerance and is well tolerated in dogs (13), so we expected it to provide important information regarding the mechanism of action of exenatide. In the future, dose-response studies can be done to determine the relative importance of the different effects of this agent to enhance glucose tolerance (delayed gastric emptying, glucagon suppression, enhancement of insulin response, and now, improved insulin action).
Using such a modified glucose clamp, we first measured a significant increase in total glucose turnover with exenatide in the presence of postprandial insulin and glucose levels, as a result of increased whole-body glucose disposal and increased net hepatic uptake of portal exogenous glucose (study 1). This finding supports the concept that exenatide directly stimulates glucose turnover. This finding also raised the question of whether exenatide might increase glucose disposal via potentiating insulin action or enhancing glucose-mediated glucose disposal, or independent of elevated insulinemia and glycemia. When lowering systemic hyperglycemia to basal levels (study 2), the increased glucose disposal was maintained, although reduced, both in glucose utilization and in NHGA (net uptake) in the presence of elevated insulinemia. In sharp contrast, reducing insulinemia from postprandial to basal levels (study 3), the increased glucose disposal was completely abolished both for glucose utilization and NHGA despite elevated glycemia. Therefore, it appears that hyperinsulinemia is a prerequisite for exenatide-mediated glucose disposal, suggesting an insulin-sensitizing effect of exenatide. Hyperglycemia further enhances exenatide-mediated glucose disposal only in the presence of elevated insulinemia, again supporting increased insulin action with exenatide that has been amplified by the elevation of glycemia.
An enhancement of insulin sensitivity with exenatide using the hyperinsulinemic-euglycemic clamp has been reported in chronic studies performed in diabetic or insulin-resistant animal models (12,28,31). But, concurrent changes in food intake, body weight, and metabolic parameters can affect insulin sensitivity and make it difficult to differentiate the direct versus indirect effects of exenatide on insulin action. In one study, food intake, body weight, plasma glucose, insulin, and lipid levels were matched in pair-fed animals (12). A ∼50% increase in insulin sensitivity with exenatide was maintained, supporting the direct role of the peptide in insulin action. Enhancing insulin sensitivity by exenatide has also been suggested by other studies in which the effect of exenatide on postprandial glycemia was investigated (5,7,27). When exenatide is administered with a meal, significantly lower plasma glucose occurs without a corresponding increase in insulin secretion. Decreased gastric emptying and lower plasma glucagon level partially explain the lowered glucose excursion, although enhanced insulin sensitivity cannot be ruled out as a contributing factor. By simulating portal Ra in portal vein after meal with or without exenatide (thus bypassing gastrointestinal tract), we have shown that exenatide lowers systemic glucose levels in the absence of corresponding changes in plasma insulin and glucagon (13). Similar results have been found in studies using GLP-1, which leads to lowered postprandial plasma glucose accompanied by lowered insulin levels (32,33). In one study, erythromycin was used to antagonize the effect of GLP-1 on gastric emptying (32). With matching gastric emptying between the GLP-1 plus erythromycin and the control groups, plasma insulin rose similarly in both groups after a meal. Yet, plasma glucose was still partially lower than the control. These results suggest a possible role of exenatide to enhance postprandial insulin sensitivity.
In contrast to positive results, some studies failed to reveal an acute effect of exenatide on insulin sensitivity using the hyperinsulinemic-euglycemic clamp (14,15). Several factors might explain the lack of an insulin-sensitizing effect in these negative studies. First, in these latter studies, intraportal glucose was not raised significantly, and thus, the putative portal GLP-1/glucose sensor may have been left inactive. As we mentioned, intraportal GLP-1 lowers systemic glucose only when paired with intraportal but not systemic glucose infusion (16,19). In addition, exendin 9–39 (GLP-1 receptor antagonist) infused portally at a low rate attenuates the glucose-lowering effect exenatide (17). It appears that the putative GLP-1/glucose sensor might at least partially mediate the glucose-lowering effect of GLP-1 and exenatide. In chronic studies, exenatide was given twice a day with meals for 6 weeks (12,28,31), continuously producing the putative portal signals. Significant changes in insulin signaling pathways and/or glucose transporting systems might occur (34), and thus, enhanced insulin sensitivity with exenatide could be more readily detected. Second, cortisol, known to induce insulin resistance, increased with the treatment of exendin-4 in these studies, which might offset possible exenadin-4–increased insulin action (14). Instead, in the present study, plasma cortisol levels were matched between the saline control and exenatide groups (data not shown).
An interesting aspect of the present study is the effect of exenatide on hepatic glucose turnover. In the presence of hyperinsulinemia and hyperglycemia, the liver quickly switched from net production to net uptake (1st-pass), as indicated by negative NHGA. The addition of exenatide further increased net hepatic uptake of portally infused glucose, reducing the amount of portal exogenous glucose entering the systemic circulation. A higher portal glucose infusion associated with exenatide treatment led to a slight increase in hepatic glucose load by ∼12% compared with the saline control, but it cannot completely account for the 83 and 45% increase in NHGA (net uptake) in studies 1 and 2. Importantly, it is known that impaired suppression of endogenous glucose production and reduced splanchnic glucose uptake contribute to postprandial hyperglycemia in type 2 diabetic patients (35,36). The present work suggests that exenatide can increase uptake of exogenous glucose by the liver, limiting the appearance of exogenous glucose in the system similarly to its known action to reduce gastric emptying. The observation that exenatide significantly increased net hepatic glucose uptake (1st-pass) in the liver facing elevated insulin and glucose levels suggests that it might promote the conversion of glucose into glycogen at the cellular level. A few in vitro studies indicate that GLP-1 and exendin-4 exert a stimulatory effect on glycogen synthase a but an inhibitory effect on glycogen phosphorylase a in the liver and muscle (37,38). Glucokinase appears to be another good candidate because it is pivotal for hepatic glycogen synthesis and because its cytoplasmic activity has been shown to be regulated by insulin and glucose (39–42).
In summary, the present study was designed to understand the mechanisms of action of the glucose-lowering effect of exenatide in postprandial conditions. Our results reveal a novel mechanism: Exenatide potentiates insulin-mediated glucose disposal, acting as an insulin sensitizer. Specifically, exenatide enhances whole-body glucose disposal, increasing the Rd while it also increases the uptake of portally delivered exogenous glucose by the liver, decreasing the Ra. Although a direct enhancement of insulin sensitivity by exenatide is confined to healthy animals in the present study, it appears that such a mechanism might as well exist in insulin-resistant conditions. There is evidence that GLP-1 and exendin-4 enhance insulin action, acutely or chronically, in obese, insulin-resistant, or type 2 diabetic humans and animals (28,29,31,43–46). Nonetheless, whether exenatide is capable of reversing or improving insulin sensitivity in insulin-resistant conditions, e.g., a diet-induced obese dog model manifesting central obesity and deterioration of insulin action in the liver and periphery (36,47,48), should be further studied.
Acknowledgments
R.N.B. has received National Institutes of Health Grants DK-27619 and DK-29867. This work was supported by an investigator-initiated research grant from Amylin.
No other potential conflicts of interest relevant to this article were reported.
We thank Dr. Cathryn Kolka for comments on manuscript; Elza Demirchyan and Rita Thomas for technical assistance; and Dr. Erlinda Kirkman, Edward Zuniga, and Edgardo Paredes for animal care and experiment assistance.
Published ahead of print at http://diabetes.diabetesjournals.org on 14 November 2008.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Holst JJ: Glucagon-like peptide-1: from extract to agent: The Claude Bernard Lecture, 2005. Diabetologia 49: 253–260, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Eng J, Kleinman WA, Singh L, Singh G, Raufman JP: Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom: further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem 267: 7402–7405, 1992 [PubMed] [Google Scholar]
- 3.Nielsen LL, Young AA, Parkes DG: Pharmacology of exenatide (synthetic exendin-4): a potential therapeutic for improved glycemic control of type 2 diabetes. Regul Pept 117: 77–88, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Greig NH, Holloway HW, De Ore KA, Jani D, Wang Y, Zhou J, Garant MJ, Egan JM: Once daily injection of exendin-4 to diabetic mice achieves long-term beneficial effects on blood glucose concentrations. Diabetologia 42: 45–50, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, Taylor K, Kim D, Aisporna M, Wang Y, Baron AD: Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 88: 3082–3089, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Parkes DG, Pittner R, Jodka C, Smith P, Young A: Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 50: 583–589, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Dupre J, Behme MT, McDonald TJ: Exendin-4 normalized postcibal glycemic excursions in type 1 diabetes. J Clin Endocrinol Metab 89: 3469–3473, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Szayna M, Doyle ME, Betkey JA, Holloway HW, Spencer RG, Greig NH, Egan JM: Exendin-4 decelerates food intake, weight gain, and fat deposition in Zucker rats. Endocrinology 141: 1936–1941, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Xu G, Stoffers DA, Habener JF, Bonner-Weir S: Exendin-4 stimulates both β-cell replication and neogenesis, resulting in increased β-cell mass and improved glucose tolerance in diabetic rats. Diabetes 48: 2270–2276, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ: Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 44: 1126–1131, 1995 [DOI] [PubMed] [Google Scholar]
- 11.Fineman MS, Bicsak TA, Shen LZ, Taylor K, Gaines E, Varns A, Kim D, Baron AD: Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care 26: 2370–2377, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Gedulin BR, Nikoulina SE, Smith PA, Gedulin G, Nielsen LL, Baron AD, Parkes DG, Young AA: Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology 146: 2069–2076, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Ionut V, Zheng D, Stefanovski D, Bergman RN: Exenatide can reduce glucose independent of islet hormones or gastric emptying. Am J Physiol Endocrinol Metab 295: E269–E277, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vella A, Shah P, Reed AS, Adkins AS, Basu R, Rizza RA: Lack of effect of exendin-4 and glucagon-like peptide-1-(7,36)-amide on insulin action in non-diabetic humans. Diabetologia 45: 1410–1415, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Degn KB, Brock B, Juhl CB, Djurhuus CB, Grubert J, Kim D, Han J, Taylor K, Fineman M, Schmitz O: Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes 53: 2397–2403, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Ionut V, Hucking K, Liberty IF, Bergman RN: Synergistic effect of portal glucose and glucagon-like peptide-1 to lower systemic glucose and stimulate counter-regulatory hormones. Diabetologia 48: 967–975, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Ionut V, Liberty IF, Hucking K, Lottati M, Stefanovski D, Zheng D, Bergman RN: Exogenously imposed postprandial-like rises in systemic glucose and GLP-1 do not produce an incretin effect, suggesting an indirect mechanism of GLP-1 action. Am J Physiol Endocrinol Metab 291: E779–E785, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Johnson KM, Edgerton DS, Rodewald T, Scott M, Farmer B, Neal D, Cherrington AD: Intraportal GLP-1 infusion increases nonhepatic glucose utilization without changing pancreatic hormone levels. Am J Physiol Endocrinol Metab 293: E1085–E1091, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Johnson KM, Edgerton DS, Rodewald T, Scott M, Farmer B, Neal D, Cherrington AD: Intraportally delivered GLP-1, in the presence of hyperglycemia induced via peripheral glucose infusion, does not change whole body glucose utilization. Am J Physiol Endocrinol Metab 294: E380–E384, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Nakabayashi H, Nishizawa M, Nakagawa A, Takeda R, Niijima A: Vagal hepatopancreatic reflex effect evoked by intraportal appearance of tGLP-1. Am J Physiol 271: E808–E813, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Vahl TP, Tauchi M, Durler TS, Elfers EE, Fernandes TM, Bitner RD, Ellis KS, Woods SC, Seeley RJ, Herman JP, D'Alessio DA: Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 148: 4965–4973, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Burcelin R, Da Costa A, Drucker D, Thorens B: Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50: 1720–1728, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Kim SP, Catalano KJ, Hsu IR, Chiu JD, Richey JM, Bergman RN: Nocturnal free fatty acids are uniquely elevated in the longitudinal development of diet-induced insulin resistance and hyperinsulinemia. Am J Physiol Endocrinol. Metab 292: E1590–E1598, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Steil GM, Ader M, Moore DM, Rebrin K, Bergman RN: Transendothelial insulin transport is not saturable in vivo: no evidence for a receptor-mediated process. J Clin Invest 97: 1497–1503, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finegood DT, Bergman RN, Vranic M: Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps: comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 36: 914–924, 1987 [DOI] [PubMed] [Google Scholar]
- 26.Bradley DC, Steil GM, Bergman RN: OOPSEG: a data smoothing program for quantitation and isolation of random measurement error. Comput Methods Programs Biomed 46: 67–77, 1995 [DOI] [PubMed] [Google Scholar]
- 27.Edwards CM, Stanley SA, Davis R, Brynes AE, Frost GS, Seal LJ, Ghatei MA, Bloom SR: Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am J Physiol Endocrinol Metab 281: E155–E161, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Young AA, Gedulin BR, Bhavsar S, Bodkin N, Jodka C, Hansen B, Denaro M: Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48: 1026–1034, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Egan JM, Meneilly GS, Habener JF, Elahi D: Glucagon-like peptide-1 augments insulin-mediated glucose uptake in the obese state. J Clin Endocrinol Metab 87: 3768–3773, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Ryan AS, Egan JM, Habener JF, Elahi D: Insulinotropic hormone glucagon-like peptide-1-(7–37) appears not to augment insulin-mediated glucose uptake in young men during euglycemia. J Clin Endocrinol Metab 83: 2399–2404, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Gedulin BR, Smith P, Prickett KS, Tryon M, Barnhill S, Reynolds J, Nielsen LL, Parkes DG, Young AA: Dose-response for glycaemic and metabolic changes 28 days after single injection of long-acting release exenatide in diabetic fatty Zucker rats. Diabetologia 48: 1380–1385, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Meier JJ, Kemmeries G, Holst JJ, Nauck MA: Erythromycin antagonizes the deceleration of gastric emptying by glucagon-like peptide 1 and unmasks its insulinotropic effect in healthy subjects. Diabetes 54: 2212–2218, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Naslund E, Bogefors J, Skogar S, Gryback P, Jacobsson H, Holst JJ, Hellstrom PM: GLP-1 slows solid gastric emptying and inhibits insulin, glucagon, and PYY release in humans. Am J Physiol 277: R910–R916, 1999 [DOI] [PubMed] [Google Scholar]
- 34.Li L, Yang G, Li Q, Tan X, Liu H, Tang Y, Boden G: Exenatide prevents fat-induced insulin resistance and raises adiponectin expression and plasma levels. Diabetes Obes Metab 10: 921–930, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Ludvik B, Nolan JJ, Roberts A, Baloga J, Joyce M, Bell JM, Olefsky JM: Evidence for decreased splanchnic glucose uptake after oral glucose administration in non-insulin-dependent diabetes mellitus. J Clin Invest 100: 2354–2361, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SP, Ellmerer M, Van Citters GW, Bergman RN: Primacy of hepatic insulin resistance in the development of the metabolic syndrome induced by an isocaloric moderate-fat diet in the dog. Diabetes 52: 2453–2460, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Alcantara AI, Morales M, Delgado E, Lopez-Delgado MI, Clemente F, Luque MA, Malaisse WJ, Valverde I, Villanueva-Penacarrillo ML: Exendin-4 agonist and exendin(9–39)amide antagonist of the GLP-1(7–36)amide effects in liver and muscle. Arch Biochem Biophys 341: 1–7, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Luque MA, Gonzalez N, Marquez L, Acitores A, Redondo A, Morales M, Valverde I, Villanueva-Penacarrillo ML: Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human myocytes. J Endocrinol 173: 465–473, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Chu CA, Fujimoto Y, Igawa K, Grimsby J, Grippo JF, Magnuson MA, Cherrington AD, Shiota M: Rapid translocation of hepatic glucokinase in response to intraduodenal glucose infusion and changes in plasma glucose and insulin in conscious rats. Am J Physiol Gastrointest Liver Physiol 286: G627–G634, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Fajans SS, Bell GI, Polonsky KS: Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 345: 971–980, 2001 [DOI] [PubMed] [Google Scholar]
- 41.Shiota M, Postic C, Fujimoto Y, Jetton TL, Dixon K, Pan D, Grimsby J, Grippo JF, Magnuson MA, Cherrington AD: Glucokinase gene locus transgenic mice are resistant to the development of obesity-induced type 2 diabetes. Diabetes 50: 622–629, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Toyoda Y, Tsuchida A, Iwami E, Shironoguchi H, Miwa I: Regulation of hepatic glucose metabolism by translocation of glucokinase between the nucleus and the cytoplasm in hepatocytes. Horm Metab Res 33: 329–336, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Gutniak M, Orskov C, Holst JJ, Ahren B, Efendic S: Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med 326: 1316–1322, 1992 [DOI] [PubMed] [Google Scholar]
- 44.Mizuno A, Kuwajima M, Ishida K, Noma Y, Murakami T, Tateishi K, Sato I, Shima K: Extrapancreatic action of truncated glucagon-like peptide-I in Otsuka Long-Evans Tokushima Fatty rats, an animal model for non-insulin-dependent diabetes mellitus. Metabolism 46: 745–749, 1997 [DOI] [PubMed] [Google Scholar]
- 45.Sandhu H, Wiesenthal SR, MacDonald PE, McCall RH, Tchipashvili V, Rashid S, Satkunarajah M, Irwin DM, Shi ZQ, Brubaker PL, Wheeler MB, Vranic M, Efendic S, Giacca A: Glucagon-like peptide 1 increases insulin sensitivity in depancreatized dogs. Diabetes 48: 1045–1053, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Zander M, Madsbad S, Madsen JL, Holst JJ: Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 359: 824–830, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Mittelman SD, Van Citters GW, Kim SP, Davis DA, Dea MK, Hamilton-Wessler M, Bergman RN: Longitudinal compensation for fat-induced insulin resistance includes reduced insulin clearance and enhanced β-cell response. Diabetes 49: 2116–2125, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Lottati M, Kolka CM, Stefanovski D, Kirkman EL, Bergman RN: Seven percent visceral fat loss by greater omentectomy markedly improves insulin sensitivity in non-obese dogs (Abstract). Diabetes 57 (Suppl.): A395, 2008 [Google Scholar]