

Abstract
Polychlorinated biphenyls (PCBs) comprise a group of persistent organic pollutants that differ significantly in their physicochemical properties, their persistence, and their biological activities. They can be metabolized via hydroxylated and dihydroxylated metabolites to PCB quinone intermediates. We have recently demonstrated that both dihydroxy PCBs and PCB quinones can form semiquinone radicals (SQ•−) in vitro. These semiquinone radicals are reactive intermediates that have been implicated in the toxicity of lower chlorinated PCB congeners. Here we describe the synthesis of selected PCB metabolites with differing degrees of chlorination on the oxygenated phenyl ring, e.g. 4,4′-dichloro-biphenyl-2,5-diol, 3,6,4′-trichloro-biphenyl-2,5-diol, 3,4,6,-trichloro-biphenyl-2,5-diol and their corresponding quinones. In addition two chlorinated ortho-hydroquinones were prepared, 6-chloro-biphenyl-3,4-diol and 6,4′-dichloro-biphenyl-3,4-diol. These PCB (hydro-)quinones readily react with oxygen or via comproportionation to yield the corresponding semiquinone free radicals, as detected by electron paramagnetic resonance spectroscopy (EPR alias ESR). The greater the number of chlorines on the (hydro-)quinone (oxygenated) ring, the higher the steady-state level of the resulting semiquinone radical at near neutral pH.
Keywords: Biaryl, Chlorination, Suzuki-coupling, Semiquinone radical, PCB, EPR
Introduction
Polychlorinated biphenyls (PCBs) are a class of organic compounds containing one to ten chlorine substituents covalently attached to biphenyl.1,2 PCBs have been produced in industrial scale by batch chlorination of biphenyl in the presence of an iron catalyst. They have been (and still are) used as coolants and insulating fluids for transformers and capacitors. PCBs have also been sold commercially for use as stabilizing additives in flexible PVC coatings of electrical wiring and electronic components, cutting oils, flame retardants, hydraulic fluids, sealants, and adhesives. These industrial applications exploit the physicochemical properties of PCBs, such as the high thermal and chemical stability and high dielectric constant. The commercial production and sale of PCBs was banned in the United States in the late 1970s because of their persistence and broad range of adverse biological effects, including (neuro-)developmental toxicity,3,4 and carcinogenesis.5
Mechanisms of PCB toxicity are still incompletely understood. One reason is that commercial PCB products contain a complex mixture of many of the possible 209 PCB derivatives or congeners. Depending on the degree of chlorination and the substitution pattern, PCB congeners have different modes of action. In particular, environmentally persistent PCB congeners interact with a number of cellular receptors and cause adverse effects by interfering with inter- and intracellular signalling processes and gene expression. Other PCB congeners, especially lower chlorinated PCB congeners and congeners with vicinal hydrogen atoms, are more rapidly metabolized2,6 and can form reactive metabolites.7 These metabolites, which include PCB epoxides, dihydroxy PCBs, and PCB quinones, have been shown to react rapidly with cellular nucleophiles, such as glutathione, amino acids, and DNA, in vitro and in vivo.7-10 A quinone metabolite of 4-chlorobiphenyl (PCB 3), 2-(4-chlorophenyl)-[1,4]benzoquinone, has also been implicated as the ultimate carcinogen in the Solt-Faber protocol, an in vivo liver carcinogenesis test.11
A recent electron paramagnetic resonance (EPR) study from our laboratory demonstrated that lower chlorinated dihydroxy PCBs and PCB quinones derived from PCB 3 and other PCB congeners can form PCB reactive semiquinone free radicals by autoxidation under basic conditions (pH > 7) as well as by (enzymatic) one-electron transfer, and comproportionation reactions.12 This study provides direct evidence that PCB metabolites can form reactive PCB semiquinone radicals, which are likely to play an important, but poorly understood, role in the toxicity of PCBs. PCB semiquinone radicals can facilitate the generation of superoxide radicals (O2•−), which in the presence of superoxide dismutase, forms hydrogen peroxide; H2O2 can lead to the production of hydroxyl radicals (HO•). Subsequently, these reactive oxygen species and, possibly, PCB semiquinone radicals themselves can react with cellular nucleophiles, such as protein and non-protein sulfhydryls, resulting in oxidative stress.7
In addition to lower chlorinated PCBs, some higher chlorinated PCBs can be rapidly metabolized to mono- and dihydroxylated PCB metabolites.13 It is currently unclear how the degree of chlorination in the metabolites of these higher chlorinated PCBs alters the formation, persistence* and properties of the corresponding PCB semiquinone radicals. Unfortunately the synthesis of oxygenated PCB-derivatives with differing degrees of chlorination of the oxygenated ring system still represents a synthetic challenge. PCB catechols, hydroquinones and quinones have been traditionally synthesized using a variety of approaches, such as the Cadogan14 and Ullmann coupling reactions.13 These reactions typically have poor yields, require tedious cleanup procedures and/or result in the formation of undesired15 and potentially toxic byproducts.16,17 In addition, chlorinated starting materials with suitable substitution patterns are frequently not available. Novel synthetic strategies to overcome these difficulties are, therefore, needed to make reactive PCB metabolites available for further studies of the corresponding PCB quinones and semiquinone radicals.
Here we report strategies for the synthesis of oxygenated PCB congeners with differing degrees of chlorination (1 to 3 Cl substituents in the oxygenated ring system). In addition, we report an initial investigation of the semiquinone radicals formed by these compounds using electron paramagnetic resonance. We show that the yield of the PCB-derived semiquinone radicals resulting from comproportionation of quinone and hydroquinone depends on the oxygen and chlorine substitution pattern and especially the number of chlorines on the semi-quinone ring.
Results and Discussion
Synthesis of PCB hydroquinones/quinones with one chlorine substituents on the oxygenated ring
Metabolism of PCB congeners by cytochrome P-450 enzymes frequently results in the formation of PCB metabolites with 2,5- and 3,4-dihydroxy substitution patters.13,18 If no chlorine substituents are present in the oxygenated ring system, these catechol and hydroquinone metabolites can be easily synthesized, for example from 3-bromo-1,2-dimethoxy-benzene and a chlorinated benzene boronic acid using the Suzuki coupling reaction.19-21 In contrast, the synthesis of PCB metabolites with a single chlorine substituent in the oxygenated ring system is less straightforward because suitable starting materials are not available. Typically, the approaches used for the synthesis of these metabolites result in product mixtures because of poor selectivity and give the desired product in poor yields.13,14 We investigated the selective chlorination of dimethoxy-benzenes 1 or 2 using H2O2/HCl to overcome these problems and to obtain starting materials that can be used to synthesize putative metabolites of several lower chlorinated biphenyls using the Suzuki coupling reaction.
Chlorination of aromatic compounds with H2O2/HCl system was developed several decades ago and, for example, used to fully chlorinate dimethoxy-benzene 2 for the synthesis of a dihydroxy metabolite of PCB 136 (2,2′,3,3′,6,6′-hexachlorobiphenyl).13 However, controlling the regioselectivity of this chlorination reaction appears to be a challenge,22 possibly because this chlorination reaction is a radical reaction. Here we report that dimethoxy-benzenes 1, 2 and 5 can be chlorinated regioselectively with H2O2/HCl (Scheme 1A). The best yields were obtained at molar ratios of dimethoxy-benzene : H2O2 : HCl of 1 : 4 : 8. However, the monochlorinated products 3, 4 and 6 were even isolated when an excess of chlorination reagent (8 equiv of HCl) was employed. The yields for iodo compound 2 (24%) were significantly lower compared to bromo compound 1 (71%).
Scheme 1. Synthesis of PCB hydroquinones/quinones with one chlorine on the oxygenated ring.

GC-MS analysis showed that mono-chlorination of dimethoxy-benzenes 1 and 2 resulted in the formation of traces of 4,5-dichloro-1,2-dimethoxy-benzene and 4,5-dibromo- (or 4,5-diiodo-) 1,2-dimethoxy-benzene. Similar side products were observed with dimethoxy-benzene 5. These side reactions, which are consistent with a radical mechanism, explain the lower yield with the iodo compound 2. The yield of debrominated side products (i.e., the respective chlorinated dimethoxy benzenes) increased when an excess of H2O2/HCl was employed. Overall, chlorination with H2O2/HCl is not a straightforward approach for the preparation of bromo (or iodo) dimethoxy-benzenes with two or three chlorine substituents because the impurities are difficult to remove.
The brominated dimethoxy-benzenes 3 and 6 were subsequently converted into dihydroxy compounds 9-10 and 13-14. These compounds are putative metabolites of 2-chlorobiphenyl (PCB 1), 2,4′-dichlorobiphenyl (PCB 8), 4-chlorobiphenyl (PCB 3) and 4,4′-dichlorobiphenyl (PCB 15), respectively (Scheme 1B). In the first step of the synthesis, the corresponding dimethoxy PCBs were synthesized with the Suzuki coupling of 3 and 6 with benzene or 4-chlorobenzene boronic acid.19-21 This cross-coupling reaction was highly efficient in the case of 4-chlorophenylboronic acid (98% yield for compounds 8 and 12), whereas lower yields were obtained for Suzuki coupling reactions involving benzene boronic acid (66% and 71% for compounds 7 and 11, respectively). These comparatively low yields are probably due to the more difficult purification of lower chlorinated biphenyls with ortho substituents, consistent with earlier observations from our laboratory.19-21 Subsequently, the dimethoxy PCBs were demethylated with BBr3 in dichloromethane.20,21 Recrystallization using CHCl3 : n-hexanes = 1: 3 (v/v) gave the desired PCB catechol and hydroquinone metabolites in moderate-to-good yields. The only exception was compound 9, which was relatively unstable at ambient temperature.
The structure of PCB hydroquinone 13 was further characterized by single-crystal X-ray diffraction analysis (Figure 1a and 1b). The solid state twist angle of compound 13, an important structural determinant of the toxicity of PCBs and their metabolites, is 43.3°.
Figure 1. Molecular structures derived from the crystal structure of 13 and 20.
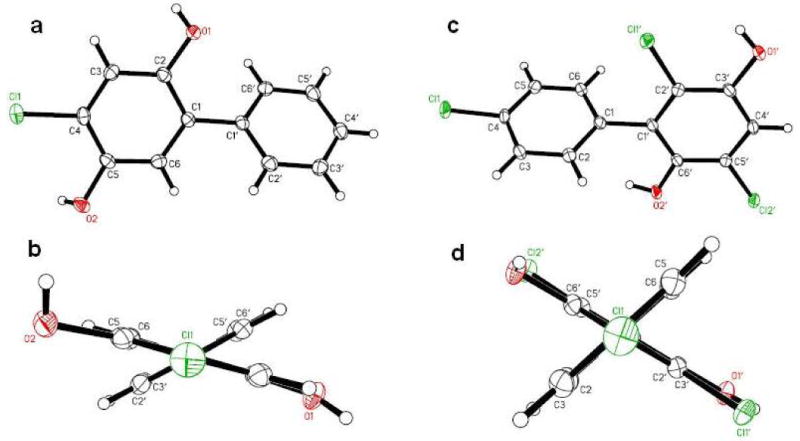
(a) Molecular structure of 4-chloro-biphenyl-2,5-diol (13) showing the atom-labeling scheme and (b) view of 4-chloro-biphenyl-2,5-diol (13) along the C1–C1′ axis illustrating the non-planar conformation of the molecule; (c) molecular structure of 3,6,4′-trichloro-biphenyl-2,5-diol (20) showing the atom-labeling scheme and (d) view of 3,6,4′-trichloro-biphenyl-2,5-diol (20) along the C1–C1′ axis illustrating the non-planar conformation of the molecule. Displacement ellipsoids are drawn at the 50% probability level.
PCB ortho-quinones are typically synthesized in situ by oxidation of the corresponding catechols with Ag2O.11 This approach has the drawback that PCB catechols themselves are unstable and can not be stored for extended periods of time without considerable degradation. Therefore, we explored the direct oxidation of dimethoxy PCBs 8 or 12 to the corresponding PCB quinones using CAN, a strong one-electron oxidizing reagent that can be used as a reagent for a wide variety of organic transformations, including the oxidation of 1,4-hydroquinones and methoxy compounds to the corresponding quinones.23 Unfortunately, oxidation of compound 8 with CAN failed to yield the desired PCB ortho-quinone as the major product and in a purity sufficient for biological studies. However, the oxidation of dimethoxy PCB 12 with CAN gave the PCB para-quinone 15 in good yield (Scheme 1C).
Synthesis of PCB hydroquinones/quinones with two chlorine substituents in the oxygenated ring
Reaction of 1, 2 and 5 with excess of H2O2/HCl resulted in partial substitution of bromine (or iodine) with chlorine. This approach was abandoned because the resulting byproducts were tedious to separate from the desired products. Chlorination of 1 and 5 with other chlorination reagents, such as NCS, was also not successful. Therefore, we explored a different synthetic strategy and investigated the introduction of a substituent suitable for the Suzuki coupling reaction (e.g., bromine) into a dichlorinated dimethoxy-benzene, such as 16. Although the bromination of bulky and unreactive aromatic compounds is well documented in the literature, the bromination of compound 16 proved to be a challenge. Bromination of 16 with a series of bromination reagents (such as Br2/AcOH,24 pyridinium bromide perbromide,25 NBS26 and NBS/HBF4·Et2O27) did not yield the desired product. Only Br2 in the presence of iron powder28 yielded a brominated product in 43% yield (Scheme 2). Surprising, the product was not the desired dimethoxy-benzene 18, but the demethylated product 17. The dimethoxy-benzene 18 was obtained by methylation with Me2SO4. Subsequently, the dimethoxy PCB 19 was obtained in moderate yield by the Suzuki coupling of 18 with 4-chlorobenzene boronic acid.
Scheme 2. Synthesis of PCB hydroquinones/quinones with two chlorines on the oxygenated ring.

Demethylation with BBr3 gave the desired hydroquinone 20. The structure of compound 20 was confirmed by single-crystal X-ray diffraction analysis (Figure 1c and 1d). The solid state twist angle of compound 20 is 73.2° and, because of the ortho chlorine substituent, larger compared to the twist angle of compound 13. Oxidation of 19 with CAN yielded para-quinone PCB 21 (Scheme 2). The oxidation of compound 19 to 21 was slower than the oxidation of the monochlorinated compound 12 to 15 (1 hour vs. 3 hours), which suggests that multiple chlorine substituents reduce the reaction rate of CAN oxidations.
Synthesis of PCB hydroquinones/quinones with three chlorines on the oxygenated ring
We explored the synthesis of PCB hydroquinones and quinones with three chlorines on the oxygenated ring via the Suzuki coupling reaction with the goal to exploit the selectivity and versatility of this approach. The Suzuki coupling of a chlorinated (instead of a brominated) benzene derivative with a boronic acid is generally not suitable for the synthesis of PCB derivatives because of the unselective coupling of all chlorine substituents. However, the Suzuki coupling of symmetrical chlorinated dimethoxy-benzenes, such as 2,3,5,6-tetrachloro-1,4-dimethoxy-benzene 23, with benzene boronic acids can be used for the synthesis of the desired dimethoxy biphenyls, such as 24 (Scheme 3A). The Suzuki-coupling reaction of 23 with benzene boronic acids using Pd(PPh3)4 with Na2CO3-toluene19-21 or Pd(OAc)2 with di-tert-butylphosphino-biphenyl as ligand and potassium phosphate as base did not yield the desired product 24. However, crude compound 24 (containing 12.5 % of 2,3,5-trichloro-1,4-dimethoxy-benzene) was obtained with Pd(OAc)2 in 32% yield using 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (DPDB) as the ligand and potassium phosphate as the base.29-31 Subsequently, demethylation of crude 24 with BBr320,21 yielded 3,4,6-trichloro-biphenyl-2,5-diol 25 in good yield (Scheme 3A).
Scheme 3. Synthesis of PCB hydroquinones/quinones with three chlorines on the oxygenated ring.

Several approaches were investigated for the synthesis of 2,3,5-trichloro-6-phenyl-[1,4]benzoquinone 26, including CAN oxidation of 24 (as described above), coupling of chloranil and benzene boronic acid with Pd(OAc)2/2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl/potassium phosphate, and chlorination of biphenyl-2,5-diol 27 with H2O2/HCl/FeCl3.32 However, the purification of 26 proved to be difficult because the compound rapidly degraded during handling, even at sub-ambient temperatures.
EPR studies of PCB semiquinones
Dihydroxylated aromatic compounds, for example dopamine,33 catechol estrogens,34,35 or stilbene catechols,36 form reactive semiquinone radicals that play an important role in their toxicity. Similarly, PCB hydroquinones, catechols and quinones can form semiquinone radicals12 that have been implicated as reactive intermediates in PCB toxicity.9,37,38 Unfortunately, little is known about the effects of chemical structure (e.g., substitution pattern and degree of chlorination) on the properties of PCB semiquinone radicals (SQ•−) and, ultimately, their toxicity. Here we investigate the SQ•− formed from PCB hydroquinones and quinones with varying numbers of chlorines on the oxygenated ring.
Autoxidation of the 2,5-dihydroxylated PCB metabolite 28 (4′-chloro-biphenyl-2,5-diol) resulted in a weak but distinct approximate 1:3:3:1 four-line spectrum (aH3 = aH4 = 2.1 G and aH6 = 2.5 G at pH 7.412,39), Figure 2a. This spectrum is typical for oxygenated PCB metabolites with no chlorine substituent on the oxygenated phenyl ring.12,40 We have shown previously, that neither chlorine nor hydrogen substituents on the non-oxygenated phenyl ring tend to exhibit hyperfine splittings in the EPR spectra, but they can contribute to the line width.12 Under identical experimental conditions (100 μM, pH 7.4), the autoxidation of 4-chloro-biphenyl-2,5-diol (13) gave an approximate 1:2:1 three-line spectrum with aH3 = 1.7 G and aH6 = 2.3 G (Figure 2b). Interestingly, the yield of SQ•− of 13 was 15-fold higher compared to SQ•− from 28, suggesting that the electronegative chlorine substituent contributes to this increase in the yield of SQ•−. As expected, compound 14, with one chlorine substituent on each phenyl ring gives an approximate 1:2:1 three-line spectrum for its SQ•− (Figure 2c). Interestingly, the intensity of this spectrum is about 30% greater than the EPR spectrum for SQ•− from 13, again consistent with the electronegative chlorine stabilizing SQ•−.
Figure 2. EPR spectra of semiquinones generated from PCB hydroquinones with differing numbers and positions of chlorine on the phenyl rings.

(Final concentrations of the original hydroquinones were 100 μM in phosphate buffer, pH = 7.4). (a) SQ•− generated from 4′-chloro-biphenyl-2,5-diol (28); (b) SQ•− generated from 4-chloro-biphenyl-2,5-diol (13); (c) SQ•− generated from 4,4′-dichloro-biphenyl-2,5-diol (14); (d) SQ•− generated from 4′-chloro-biphenyl-3,4-diol (30); (e) SQ•− generated from 6-chloro-biphenyl-3,4-diol (9); (f) SQ•− generated from 6,4′-dichloro-biphenyl-3,4-diol (10).
In contrast to SQ•− derived from PCB para-hydroquinones, the EPR spectra of SQ•− formed by oxidation PCB ortho-hydroquinones 30 (4′-chloro-biphenyl-3,4-diol) and 9 are quite weak, yielding an irregular spectrum12 and a 1:2:1 three-line spectrum (aH3 = 2.1 G and aH6 = 2.1 G), respectively (Figure 2d and 2e). Introducing a second chlorine such that both the oxygenated and non-oxygenated rings have a chlorine (10) results in a somewhat stronger EPR spectrum for the corresponding SQ•− (1:2:1 three-line, aH3 = 2.1 G and aH6 = 2.1 G) compare Figure 2f to 2d and 2e). However, the apparent stabilizing-effect of these chlorines on the corresponding SQ•− is much less for PCB ortho-SQ•− than PCB para-SQ•− The greater steady-state concentration for the PCB para-SQ•− compared to PCB ortho-SQ•− is most likely due a slower rate of disproportionation for the para-SQ•−.
Because of the dramatic influence of chlorine on the persistence of SQ•− derived from PCB para-hydroquinone (Figure 2b and 2c), we investigated the influence of additional chlorines on the persistence of the corresponding SQ•−. Figure 3 shows the EPR spectra of SQ•− formed by autoxidation of a series of PCB para-hydroquinones and quinones with 0 to 3 chlorines on the oxygenated phenyl ring; as the number of chlorines increases, the persistence of the corresponding SQ•− increases. As we have reported, the SQ•− formed from 4′-chloro-biphenyl-2,5-diol (28) and 2-(4-chloro-phenyl)-[1,4]benzoquinone (29) are essentially identical (Figure 3a and 3b).12 Similarly, the SQ•− formed by PCB hydroquinones 14, 20 and 25 are identical to the SQ•− formed by the corresponding PCB quinones 15, 21 and 26, respectively. As anticipated, the spectra change with increasing chlorine (and decreasing hydrogen) substitution from 1:3:3:1 four-line (28 and 29) to 1:2:1 three-line (14 and 15, aH3 = 1.7 G and aH6 = 2.3 G) to 1:1 two-line (20 and 21, aH4 = 1.9 G) to one-line spectra (25 and 26, no hyperfine splitting). Another interesting phenomenon is that the g-factor of each of the SQ•− changed, with the increasing of numbers of chlorine: 2.0047 (28 and 29); 2.0050 (14 and 15); 2.0051 (20 and 21); and 2.0054 (25 and 26). This systematic increase in g-value is consistent with the increased spin-orbit coupling that is expected with increasing number of chlorines.
Figure 3. EPR spectra of semiquinones generated from PCB hydroquinones and quinones with differing numbers of chlorine on the oxygenated phenyl rings.

(a) SQ•− generated from 1 mM of 4′-chloro-biphenyl-2,5-diol (28) (yield: 0.000 092); (b) SQ•− generated from 1 mM of 2-(4-chloro-phenyl)-[1,4]benzoquinone (29) (yield: 0.000 12); (c) computer simulation of SQ•− with three hydrogens; (d) SQ•− generated from 100 μM of 4,4′-dichloro-biphenyl-2,5-diol (14) (yield: 0.004 5); (e) SQ•− generated from 100 μM of 2-chloro-5-(4-chloro-phenyl)-[1,4]benzoquinone (15) (yield: 0.003 5); (f) computer simulation of SQ•− with two hydrogens; (g) SQ•− generated from 10 μM of 3,6,4′-trichloro-biphenyl-2,5-diol (20) (yield: 0.026); (h) SQ•− generated from 10 μM of 2,5-dichloro-3-(4-chloro-phenyl)-[1,4]benzoquinone (21) (yield: 0.025); (i) computer simulation of SQ•− with one hydrogen; (j) SQ•− generated from 1 μM of 3,4,6-trichloro-biphenyl-2,5-diol (25) (yield: 0.23); (k) SQ•− generated from 1 μM of 2,3,5-trichloro-6-phenyl-[1,4]benzoquinone (26) (yield: 0.23); (l) computer simulation of SQ•− with no hydrogen. These spectra were collected using a modulation amplitude of 1.0 Gauss. Lowering the modulation amplitude to 0.1 Gauss (for spectra d and e) or 0.01 Gauss (for spectra g, h, j, and k) did not reveal any possible hyperfine splittings from the chlorines on the semiquinone moiety.
The most intriguing observation is the apparent exponential increase in the initial yield of SQ•− with increasing degree of chlorination in the oxygenated ring system, Figure 3. The apparent yield of SQ•− increases from 0.000 092/0.000 12 for 28/29 to 0.23/0.23 for 25/26. Thus, as the degree of chlorination increases, the persistence of the radical also increases. The three-dimensional structure did not influence SQ•− persistence (e.g., the dihedral angle; Figure 1b and 1d). To examine this systematically, we generated each of the radicals shown in Figure 3 via the comproportionation reaction of quinone and hydroquinone.
The expression for the equilibrium constant for this reaction has the form K = [SQ•−]2/[hydroquinone][quinone], assuming constant pH. EPR spectra were collected and quantitated from solutions with equal concentrations of PCB hydroquinone and PCB quinone and the resulting concentrations used to estimate the equilibrium constant for the comproportionation reaction of each set of PCB hydroquinones and quinones of Figure 3. As seen in Figure 4, the estimated equilibrium constant increases by approximately 102.3 = 200 with each addition of chlorine to the oxygenated ring. It is quite amazing that nearly 64% of the trichloro hydroquinone/quinone equilibrium system (25/26) exists as the semiquinone radical. These observations demonstrate the additive nature of the ring chlorines and the size of the MO coefficients to the persistence of the corresponding radical.
Figure 4. The equilibrium constant for the comproportionation reaction to form SQ•− increases with the number of chlorines on the oxygenated ring.

Assay conditions: quinone and hydroquinone mixture in 100 mM PBS buffer, pH 7.4. (a) Zero chlorine, 50 μM of 28 and 50 μM of 29 yields [SQ•−]ss = 63 nM; (b) one chlorine, 5 μM of 14 and 5 μM of 15 yields [SQ•−]ss = 70 nM; (c) two chlorines, 500 nM of 20 and 500 nM of 21 yields [SQ•−]ss = 87 nM; (d) three chlorines, 50 nM of 25 and 50 nM of 26 yields [SQ•−]ss = 65 nM. Where K = [SQ•−]2 / [hydroquinone][quinone]. This equilibrium constant will actually be dependent on pH because the pKas for the hydroquinones will be above pH or near 7, while the pKa of the semiquinone radicals will be somewhat acidic; for example, the pKa of the semiquinone radical of unsubstituted benzoquinone/hydroquinone is 4.25.41
In summary, in this work we have presented efficient and high-yielding synthetic routes for the preparation of a series of oxygenated PCB metabolites with differing degrees of chlorination. The synthesis of these compounds is straightforward. These oxygenated PCBs readily react with oxygen or via comproportionation to yield corresponding semiquinone free radicals. The greater the number of chlorines on the oxygenated ring, the more stable the semiquinone radical.
Experimental
General procedure for the preparation of chlorinated bromo (or iodo-)dimethoxy-benzenes 3, 4 and 6: 1-bromo-4-chloro-2,5-dimethoxy-benzene (6)13
Hydrogen peroxide (30%, 15 mL, 145 mmol) was added over 1 h to a rapidly stirred solution of 1-bromo-2,5-dimethoxy-benzene 5 (7.6 g, 35 mmol) in CHCl3 (30 mL) and concentrated HCl (24 mL, 280 mmol). After 16 h at room temperature, the mixture was extracted with CHCl3 (3 × 30 mL). The organic extracts were combined, washed with 5% NaHSO3 (25 mL), dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The product was purified by column chromatography on silica gel with n-hexanes : CHCl3 = 1 : 1 (v/v), yielding 5.7 g (69%) of 6 as white crystals. mp 134-135°C; 1H NMR (400 MHz, CDCl3): δ 7.12(s, 1H), 6.94(s, 1H), 3.85(s, 3H), 3.84(s, 3H); 13 NMR (100 MHz, CDCl3): δ 150.3, 149.6, 121.8, 117.4, 114.3, 109.6, 57.1, 57.0; MS m/z(relative intensity): 250(61, M•+), 235(7); Anal. Calcd for C8H8BrClO2: C, 38.18; H, 3.21. Found: C, 38.33; H, 3.03.
General Suzuki-coupling procedure A for the synthesis of 7, 8, 11, 12 and 19: 4,4′-dichloro-2,5-dimethoxy-biphenyl (12)19-21
Sodium carbonate (20 mL, 2 M aq.) was added to a solution of 1-bromo-4-chloro-2,5-dimethoxy-benzene (6) (5.2 g, 20.0 mmol) and Pd(PPh3)4 (0.72 g, 3% molar ratio) in toluene(80 mL). A solution of a 4-chloro-phenylboronic acid (4.7 g, 30.0 mmol) in ethanol : toluene : H2O = 8 : 1 : 1 (40 mL total) was added slowly to the mixture under a nitrogen atmosphere. The reaction mixture was maintained at 80 °C for 12 h. Hydrogen peroxide (30%, 2.0 mL) was added slowly to the warm reaction mixture to destroy unreacted boronic acid. The mixture was stirred at room temperature for an additional 4 h and diluted with diethyl ether (120 mL). The reaction mixture was extracted once with NaOH (40 mL, 2 M aq.) and three times with water (40 mL). The organic phase was dried over MgSO4 and the solvents were removed under reduced pressure. Column chromatography over silica gel with n-hexanes : CHCl3 = 3 : 1 (v/v) as eluent followed by recrystallization with n-hexanes : CHCl3 = 3 : 1 (v/v) yielded 5.8 g (98%) of 12 as white solid. mp 123-124 °C; 1H NMR (400 MHz, CDCl3): δ 7.46-7.37(m, 4H), 7.02(s, 1H), 6.89(s, 1H), 3.89(s, 3H), 3.77(s, 3H); 13 NMR (100 MHz, CDCl3): δ 150.7, 149.6, 136.3, 133.6, 131.0, 128.9, 128.5, 122.3, 115.2, 114.4, 57.2, 56.6; MS m/z(relative intensity): 282(88, M•+), 267(43), 232(100), 217(30), 189(17), 161(11) 126(16); Anal. Calcd for C14H12Cl2O2: C, 59.36; H, 4.27. Found: C, 59.24; H, 4.19.
General demethylation procedure for the synthesis of 9, 10, 13, 14, 20 and 25: 4,4′-Dichloro-biphenyl-2,5-diol (14)20,21
Boron tribromide (18.0 mL, 1M in n-hexanes, 2.2 equiv) was added to a solution of the 4,4′-dichloro-2,5-dimethoxy-biphenyl (12) (2.36 g, 8.3 mmol) in anhydrous dichloromethane (50 mL) under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 14 hours, cooled with ice/salt and hydrolyzed with an equal volume of ice-cold water. The aqueous phase was extracted with dichloromethane (2 × 50 mL). The combined organic layers were washed with water (3 × 25 mL), dried over MgSO4 and the solvent was removed under reduced pressure. The product was purified by column chromatography on silica gel with n-hexanes : ethyl acetate = 1 : 1 (v/v) as eluent followed by recrystallization using n-hexanes : CHCl3 = 3 : 1 (v/v) yielded 1.18 g (55%) of 14 as white solid. mp 118-120 °C; 1H NMR (400 MHz, CDCl3): δ 7.47-7.38(m, 4H), 6.98(s, 1H), 6.91(s, 1H), 5.19(s, 1H), 4.75(s, 1H); 13 NMR (100 MHz, CDCl3): δ 146.4, 145.7, 134.7, 134.5, 130.5, 129.6, 127.6, 119.8, 117.2, 116.6; MS m/z(relative intensity): 398(47, [M+TMS2]•+), 348(19), 239(13), 93(27), 73(100, [Si(CH3)3]+); Anal. Calcd for C12H8Cl2O2: C, 56.48; H, 3.16. Found: C, 56.31; H, 3.12.
General procedure for the synthesis of PCB quinones 15 and 21 from methoxylated PCB derivatives: 2-Chloro-5-(4-chloro-phenyl)-[1,4]benzoquinone (15)42
4,4′-Dichloro-2,5-dimethoxy-biphenyl 12 (0.54 g, 1.9 mmol) was dissolved in acetonitrile (10 mL) at 50 °C, a solution of CAN (5.8 g, 10.0 mmol) in water (15 mL) was added, and the reaction mixture was stirred at room temperature for 1 h. The solution was extracted with chloroform (2 × 20 mL) and washed with water (2 × 50 mL). The combined organic phase was and dried over MgSO4 and the product was purified by column chromatography on silica gel with CHCl3 : n-hexanes = 2 : 1 (v/v) as eluent, yielding 0.38 g (80%) of 15 as a yellow solid. mp 137-139 °C (Lit.: 147-148 °C43); 1H NMR (400 MHz, CDCl3): δ 7.44(s, 4H), 7.10(s, 1H), 7.00(s, 1H); 13 NMR (100 MHz, CDCl3): δ 184.3, 179.7, 145.4, 144.2, 137.3, 134.2, 132.3, 130.9, 130.5, 129.2; MS m/z(relative intensity): 254(30, [M+2H]•+), 217(100); Anal. Calcd for C12H6Cl2O2: C, 56.93; H, 2.39. Found: C, 56.84; H, 2.35.
2-Bromo-3,6-dichloro-4-methoxy-phenol (17)28
A solution of bromine (7.0 g, 44 mmol) in dichloromethane (10 mL) was added slowly over 15 min to a solution of 1,4-dichloro-2,5-methoxy-benzene 16 (8.5 g, 41 mmol) and iron powder (1.24 g, 22 mmol) in dichloromethane (50 mL) heated under reflux. After 4 h, another portion of iron powder (1.24 g, 22 mmol) was added and the reaction heated for an additional 4 h. The reaction mixture was allowed to cool to ambient temperature, the iron powder was filtered off, and the product was purified by column chromatography on silica gel with CHCl3 : n-hexanes = 2 : 1 (v/v) as eluent to yield 4.8 g (43%) of 17 as white crystals. mp 126-127 °C; 1H NMR (400 MHz, CDCl3): δ 6.96(s, 1H), 5.60(s, 1H), 3.87(s, 3H); 13 NMR (100 MHz, CDCl3): δ 150.2, 144.2, 123.4, 118.9, 113.1, 112.9, 57.4; MS m/z(relative intensity): 270(80, M+), 255(100), 227(20); Anal. Calcd for C7H5BrCl2O2: C, 30.90; H, 1.85. Found: C, 30.84; H, 1.73.
Suzuki-coupling procedure B for the synthesis of 3,4,6-trichloro-2,5-dimethoxy-biphenyl (24)29-31
K3PO4 (4.25 g, 20 mmol), Pd(OAc)2 (44 mg, 2% molar ratio), and 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (164 mg, 4% molar ratio) were added to a solution of 1,2,4,5-tetrachloro-3,6-dimethoxy-benzene (23)(2.62 g, 10.0 mmol) and phenylboronic acid (1.83 g, 15.0 mmol) in toluene (50 mL) under nitrogen atmosphere. The reaction mixture was then stirred at 110 °C for 48 hours. Hydrogen peroxide (2.0 mL, 30%) was added slowly to the warm reaction mixture to destroy unreacted boronic acid. The mixture was stirred at room temperature for an additional 4 h and diluted with diethyl ether (60 mL). After filtration, the solvent was extracted once with NaOH (20 mL, 2 M aq.) and three times with water (20 mL). The organic phase was dried over MgSO4 and the solvents were removed under reduced pressure. Column chromatography over silica gel with n-hexanes: CHCl3 = 3:1 (v/v) as eluent followed by recrystallization with n-hexanes: CHCl3 = 3:1 (v/v) yielded 1.03 g (32%) of crude 24 (contained 12.5 % of 2,3,5-trichloro-1,4-dimethoxy-benzene) as white solid. mp 49-51 °C; 1H NMR (400 MHz, CDCl3): δ7.50-7.41(m, AA′XX′ system, 3H), 7.33-7.27(m, AA′XX′ system, 2H), 3.93(s, 3H), 3.44(s, 3H); 13 NMR (100 MHz, CDCl3): δ 151.8, 150.2, 135.6, 134.3, 130.1, 128.45, 128.41, 127.9, 127.2, 111.6, 61.0, 60.9; MS m/z(relative intensity): 316(100, M•+), 301(40, M-CH3), 266(80, M-CH3Cl); Anal. Calcd for (C14H11Cl3O2 + 1/8 C14H11Cl3O2): C, 51.80; H, 3.44. Found: C, 51.79; H, 3.38.
2,3,5-Trichloro-6-phenyl-[1,4]benzoquinone (26)32
Biphenyl-2,5-diol (27) (2.0 g, 10.7 mmol) was suspended in glacial acetic acid (30 mL) and concentrated HCl (20 mL), followed by anhydrous FeCl3 (3.4 g), was added to the solution. Hydrogen peroxide (20 mL, 30%) was dropped slowly into the reaction mixture (20 min) and the mixture was heated under reflux for an additional 1 h. The mixture was filtered and extracted with CHCl3 (3 × 20 mL). The combined organic phases were washed with saturated NaHCO3 (20 mL) and water (3 × 20 mL), dried over MgSO4, and the solvents were removed under reduced pressure. Recrystallization with n-hexanes : ethyl acetate = 4 : 1 (v/v) yielded 2.8 g (91%) of 26 as a yellow solid. mp 94-95 °C (Lit.: 112-114 °C32); 1H NMR (400 MHz, CDCl3): δ 7.58-7.46(m, 3H), 7.40-7.29(m, 2H); 13 NMR (100 MHz, CDCl3): δ 175.6, 171.9, 143.9, 141.6, 140.6, 140.5, 130.5, 130.3, 129.8, 128.5; MS m/z(relative intensity): 288(100, [M+2H]•+), 251(90, M-Cl), 233(52); HRMS(EI) Calcd for C12H5Cl3O2 m/z([M]•+) 285.9355. Found 285.9350.
EPR Spectroscopy
Electron paramagnetic spectroscopy was performed using a Bruker EMX spectrometer equipped with a High Sensitivity cavity and an Aqua-X sample holder. Spectra were obtained at room temperature. Typical EPR-parameters were: 3510.3 G center field; 15 G scan width; 9.854 GHz microwave frequency; 20 mW power; 2×105 receiver gain; modulation frequency of 100 kHz; modulation amplitude of 1.0 G; with the conversion time and time constant both being 40.96 ms with 5 scans for each 1024-point spectrum. Stock solutions (200 mM each) of PCB hydroquinones and quinones were prepared in DMSO. All experiments were carried out in 100 mM phosphate buffer containing 250 μM DETAPAC (diethylenetriaminepentaacetic acid) as described previously.12
Quantitation was accomplished by double integration of the recorded EPR signals using the WinEPR program with 3-carboxyproxyl as the standard, ε234 = 2370 ± 50 M-1 cm-1.44 To obtain the best quantitative information on all samples and standards, they were dissolved in identical aqueous buffer; after setup, the 4-bore Aqua-X holder was not removed from the cavity for all samples.
Spectral simulations of EPR spectra were performed using the WinSim program developed at the NIEHS by Duling.45 Correlation coefficients of simulated spectra were typically > 0.99.
Supplementary Material
Experimental procedures, and characterisation for compounds 3, 4, 7, 8, 9, 10, 11, 13, 18, 19, 20, 21, 23 and 25; 1H NMR and 13 NMR spectra for compounds 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20, 21, 23, 24, 25 and 26; GC-MS chromatograms and MS spectra for compounds 9, 24 and 26; crystallographic data for 13, 17 and 20 (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by grants ES05605, ES012475 and ES013661 from the National Institute of Environmental Health Sciences, NIH, (HJL and LWR) and R01GM073929 from the National Institute of General Medical Sciences (GRB) and MRI grant #0319176 (SP) from the National Science Foundation. Contents of this manuscript are solely the reponsibility of the authors and do not necessarily represent the official views of the NIEHS/NIH or NSF.
Footnotes
The apparent persistence of semiquinone radicals in the setting of our experiments is a complicated result of the thermodynamic or kinetic properties, or a combination, of all three redox forms, i.e. Q, SQ•−, and H2Q. The work presented here does not address this directly. However, it is clear that the yield of semiquinone radical varies greatly among the (hydro-)quinones studied; both thermodynamic and kinetics will play a role in these apparent yields.
References
- 1.Robertson LW, Hansen LG. Recent advances in the environmental toxicology and health effects of PCBs. University Press of Kentucky; Lexington: 2001. [Google Scholar]
- 2.Hansen LG. The ortho side of PCBs: Occurrence and disposition. Kluwer Academic Publishers; Boston: 1999. [Google Scholar]
- 3.Kodavanti PRS. In: Molecular Neurotoxicology: environmental agents and transcription-transduction coupling. Zawia NH, editor. CRC press; Boca Roton, FL: 2004. pp. 151–182. [Google Scholar]
- 4.Schantz SL, Widholm JJ, Rice DC. Environ Health Perspect. 2003;111:357–376. doi: 10.1289/ehp.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silberhorn E, Glauert HP, Robertson LW. Crit Rev Toxicol. 1990;20:439–496. doi: 10.3109/10408449009029331. [DOI] [PubMed] [Google Scholar]
- 6.Letcher RJ, Klasson-Wehler E, Bergman A. In: The handbook of environmental chemistry Vol 3 Part K New types of persistent halogenated compounds. Paasivirta J, editor. Springer Verlag; Berlin Heidelberg: 2000. pp. 315–359. [Google Scholar]
- 7.Robertson LW, Gupta RC. Molecular Drug Metabolism and Toxicology. 2000:16–32. [Google Scholar]
- 8.Lin PH, Sangaiah R, Ranasinghe A, Upton PB, La DK, Gold A, Swenberg JA. Chem Res Toxicol. 2000;13:710–8. doi: 10.1021/tx000030f. [DOI] [PubMed] [Google Scholar]
- 9.Arif JM, Lehmler HJ, Robertson LW, Gupta RC. Chem-Biol Interact. 2003;142:307–316. doi: 10.1016/s0009-2797(02)00141-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhao S, Narang A, Ding X, Eadon G. Chem Res Toxicol. 2004;17:502–511. doi: 10.1021/tx034245b. [DOI] [PubMed] [Google Scholar]
- 11.Espandiari P, Glauert HP, Lehmler HJ, Lee EY, Srinivasan C, Robertson LW. Toxicol Sci. 2004;79:41–46. doi: 10.1093/toxsci/kfh097. [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Wagner BA, Lehmler HJ, Buettner GR. Chem Res Toxicol. 2008;21:1359–1367. doi: 10.1021/tx8000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waller SC, He YA, Harlow GR, He YQ, Mash EA, Halpert JR. Chem Res Toxicol. 1999;12:690–699. doi: 10.1021/tx990030j. [DOI] [PubMed] [Google Scholar]
- 14.Lin PH, Sangaiah R, Ranasinghe A, Ball LM, Swenberg JA, Gold A. Organic & Biomolecular Chemistry. 2004;2:2624–2629. doi: 10.1039/B409373A. [DOI] [PubMed] [Google Scholar]
- 15.Shaikh NS, Parkin S, Lehmler HJ. Organometallics. 2006;25:4207–4214. [Google Scholar]
- 16.Goldstein JA, Hass JR, Linko P, Harvan DJ. Drug Metab Dispos. 1978;6:258–264. [PubMed] [Google Scholar]
- 17.Moron M, Sundström G, Wachtmeister CA. Acta Chem Scand. 1973;27:3121–3122. doi: 10.3891/acta.chem.scand.27-3121. [DOI] [PubMed] [Google Scholar]
- 18.McLean MR, Bauer U, Amaro AR, Robertson LW. Chem Res Toxicol. 1996;9:158–164. doi: 10.1021/tx950083a. [DOI] [PubMed] [Google Scholar]
- 19.Lehmler HJ, Robertson LW. Chemosphere. 2001;45:137–143. doi: 10.1016/s0045-6535(00)00546-4. [DOI] [PubMed] [Google Scholar]
- 20.Lehmler HJ, Robertson LW. Chemosphere. 2001;45:1119–1127. doi: 10.1016/s0045-6535(01)00052-2. [DOI] [PubMed] [Google Scholar]
- 21.Kania-Korwel I, Parkin S, Robertson LW, Lehmler HJ. Chemosphere. 2004;56:735–744. doi: 10.1016/j.chemosphere.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 22.Samant BS, Saraf YP, Bhagwat SS. J Colloid Interface Sci. 2006;302:207–213. doi: 10.1016/j.jcis.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Nair V, Deepthi A. Chem Rev. 2007;107:1862–1891. doi: 10.1021/cr068408n. [DOI] [PubMed] [Google Scholar]
- 24.Clive DLJ, Khodabocus A, Vernon PG, Angoh AG, Bordeleau L, Middleton DS, Lowe C, Kellner D. J Chem Soc, Perkin Trans. 1991;1:1433–1444. [Google Scholar]
- 25.Shimbashi A, Tsuchiya A, Imoto M, Nishiyama S. Bull Chem Soc Japan. 2004;77:1925–1930. [Google Scholar]
- 26.Goldberg Y, Bensimon C, Alper H. J Org Chem. 1992;57:6374–6376. [Google Scholar]
- 27.Oberhauser T. J Org Chem. 1997;62:4504–4506. doi: 10.1021/jo9622993. [DOI] [PubMed] [Google Scholar]
- 28.Liu H, Bernhardsen M, Fiksdahl A. Tetrahedron. 2006;62:3564–3572. [Google Scholar]
- 29.Zapf A, Ehrentraut A, Beller M. Angew Chem Int Edit. 2000;39:4153–4155. doi: 10.1002/1521-3773(20001117)39:22<4153::aid-anie4153>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 30.Wolfe JP, Buchwald SL. Angew Chem Int Ed. 1999;38:2413–2416. doi: 10.1002/(sici)1521-3773(19990816)38:16<2413::aid-anie2413>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 31.Wolfe JP, Singer RA, Yang BH, Buchwald SL. J Am Chem Soc. 1999;121:9550–9561. [Google Scholar]
- 32.Wilson JG. Aust J Chem. 1972;25:2383–2391. [Google Scholar]
- 33.Kalyanaraman B, Premovic PI, Sealy RC. J Biol Chem. 1987;262:11080–7. [PubMed] [Google Scholar]
- 34.Seacat AM, Kuppusamy P, Zweier JL, Yager JD. Arch Biochem Biophys. 1997;347:45–52. doi: 10.1006/abbi.1997.0323. [DOI] [PubMed] [Google Scholar]
- 35.Kalyanaraman B, Sealy RC, Sivarajah K. J Biol Chem. 1984;259:14018–14022. [PubMed] [Google Scholar]
- 36.Kalyanaraman B, Sealy RC, Liehr JG. J Biol Chem. 1989;264:11014–11019. [PubMed] [Google Scholar]
- 37.Sadeghi-Aliabadi H, Chan K, Lehmler HJ, Robertson LW, O'Brien PJ. Chem-Biol Interact. 2007;167:184–192. doi: 10.1016/j.cbi.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 38.McLean MR, Twaroski TP, Robertson LW. Arch Biochem Biophys. 2000;376:449–455. doi: 10.1006/abbi.2000.1754. [DOI] [PubMed] [Google Scholar]
- 39.Ashworth P, Dixon WT. J Chem Soc Perkin Trans. 1972;2:2264–2267. [Google Scholar]
- 40.Pedersen JA. Handbook of EPR Spectra from Quinones and Quinols. CRC Press, Inc.; Boca Raton, FL: 1985. [Google Scholar]
- 41.Yamazaki I, Piette LH. J Am Chem Soc. 1965;87:986–989. doi: 10.1021/ja01083a009. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Alvarado P, Avendano C, Menendez JC. Synthetic Communications. 2002;32:3233–3239. [Google Scholar]
- 43.Bagli JF, L'Ecuyer P. Can J Chem. 1961;39:1037–1048. [Google Scholar]
- 44.Venkataraman S, Martin SM, Schafer FQ, Buettner GR. Free Radical Biol Med. 2000;29:580–585. doi: 10.1016/s0891-5849(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 45.Duling DR. J Magn Res Series B. 1994;104:105–110. doi: 10.1006/jmrb.1994.1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, and characterisation for compounds 3, 4, 7, 8, 9, 10, 11, 13, 18, 19, 20, 21, 23 and 25; 1H NMR and 13 NMR spectra for compounds 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 17, 18, 19, 20, 21, 23, 24, 25 and 26; GC-MS chromatograms and MS spectra for compounds 9, 24 and 26; crystallographic data for 13, 17 and 20 (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.