

Abstract
A number of alanine and more conservative mutants of residues in the fourth domain of thrombomodulin (TM) were prepared and assayed for protein C activation and for thrombin binding. Several of the alanine mutations appeared to cause misfolding or structural defects as assessed by poor expression and/or NMR HSQC experiments, while more conservative mutations at the same site appeared to fold correctly and retain activity. Several of the conservative mutants bound more weakly to thrombin despite the fact that the fourth domain does not directly contact thrombin in the crystal structure of the thrombin-TM complex. A few of the mutant TM fragments bound thrombin with similar affinity as wild type, but showed decreases in kcat for protein C activation. These mutants were also less able to cause a change in the steady state fluorescence of fluorescein-EGR-chloromethylketone bound to the active site of thrombin. These results suggest that some residues within the fourth domain of TM may primarily interact with protein C but others are functionally important for altering the way TM interacts with thrombin. Residues in the fourth domain that primarily affect kcat for protein C activation may do this by changing the active site of thrombin.
Keywords: Thrombin, protein C, serine protease, anticoagulant pathway
When thrombin binds thrombomodulin (TM), the substrate specificity of thrombin changes from cleavage of fibrinogen to cleavage of protein C. This also changes the role of thrombin in the blood clotting cascade from procoagulant to anticoagulant (1). TM contains several domains including six EGF-like domains of which the fourth, fifth and sixth are necessary and sufficient for thrombin binding and subsequent cleavage of protein C by the thrombin-TM complex (2, 3). The fourth EGF-like domain is necessary for protein C activation, while the fifth and sixth EGF-like domains are involved in binding thrombin (4, 5).
Alanine scanning mutagenesis of thrombin and TMEGF456 in two separate studies identified residues in both TM and thrombin that are important for protein C activation (6, 7). Alanine scanning of thrombin revealed anion binding exosite 1 (ABE1), the site of fibrinogen binding, as the TM binding site and this was later confirmed by amide H/2H exchange experiments and x-ray crystallography (8, 9). Residues in the active site of thrombin were also identified as important for TM binding (7).
The alanine scan of TMEGF456 identified 22 residues that were critical for protein C activation. These residues were located throughout the fourth, fifth, and sixth EGF-like domains. All of the critical residues were highly conserved in TM from several different species (Figure 1) (6). Several negatively charged residues in TMEGF456 were critical, which was expected because ABE1 of thrombin is positively charged. A crystal structure of thrombin in complex with TMEGF456 clarified the importance of many of these critical residues from TMEGF456 but raised questions about others (9). Figure 2 shows the important residues from the alanine scan of TM mapped onto the thrombin-TMEGF456 crystal structure. Several residues in the fifth and sixth EGF-like domains of TM are in direct contact with thrombin. Mutation of these residues to alanine resulted in weakened TM binding (6).
Figure 1.

Sequence alignment of TMEGF456 from five different species. Several of the critical residues identified by alanine scanning are indicated with arrows above the sequence.
Figure 2.

A) Schematic drawing of TMEGF45 showing the residues identified as important for the cofactor activity of TM. Residues are colored according to location: fourth domain in green, linker region in purple, Met388 in cyan, and fifth domain in red. B) Crystal structure of the thrombin-TMEGF456 complex. The important TM residues are shown as sticks and colored as in A. ABE1 of thrombin is blue and shown in sticks. The active site catalytic triad of thrombin is shown as green sticks.
Surprisingly, the crystal structure showed that critical TM residues in the fourth EGF-like domain were far from the thrombin molecule (9). Despite the lack of direct contact between the fourth EGF-like domain and thrombin in the crystal structure, studies have shown that the fourth EGF-like domain is required to induce changes in the active site of thrombin. Ye et al. performed fluorescence experiments to investigate how two different TM fragments might affect the active site of thrombin. When fluorescein-FPR or ANS-FPR was attached to the active site histidine, the FPR linkage positioned the fluorophore away from the active site serine. Interestingly, this fluorescent label responded specifically to TMEGF1-6 and not to TMEGF56 (10). Amide H/2H exchange experiments further confirmed these findings and showed that the 90s loop in thrombin was altered when TM fragments containing the fourth EGF-like domain were bound (11, 12). The amide H/2H exchange results provide a putative “allosteric” mechanism for the alleviation of substrate repulsion suggested by the elegant studies of Rezaie’s group (13, 14).
Thus, the fourth EGF-like domain of TM is required for alteration of the thrombin active site, but it does not directly contact thrombin according to the crystal structure. There are two possible explanations of this apparent discrepancy. Either the crystal structure has captured an inactive state and under solution conditions the fourth EGF-like domain does contact thrombin, or it may communicate with the thrombin active site through the fifth EGF-like domain. The latter hypothesis would require communication between the fourth and fifth EGF-like domains. Indeed, NMR studies of the minimally active fragment of TM, TMEGF45, showed communication between the fourth and fifth EGF-like domains by way of Met 388 in the linker between the two domains (15, 16). Our hypothesis is that the fourth EGF-like domain of TM is responsible for a number of functions. Besides interacting directly with protein C, it may alter the binding of the fifth EGF-like domain to thrombin so that together these two domains of TM alleviate repulsion of protein C both at Arg 35 (17) and at Asp 102 (14).
In order to further investigate the role of the critical amino acids in the fourth EGF-like domain, mutations were introduced in TMEGF456 and TMEGF45. Both were assayed for cofactor activity in thrombin-mediated activation of protein C. The TMEGF456 mutants are amenable to direct binding assays using surface plasmon resonance (SPR) whereas the TMEGF45 mutants dissociate too quickly to obtain reliable SPR measurements (18, 19). The TMEGF45 mutants could be prepared in large enough quantity for fluorescence experiments whereas the TMEGF456 mutants could not. Mutations included both alanine substitutions and more conservative changes. Direct thrombin binding assays by SPR showed that some mutations caused decreases in TM binding to thrombin despite the fact that they are not near the binding interface. Some mutants caused a loss of kcat for protein C activation but retained nearly full thrombin-binding affinity. These mutants were further analyzed for their ability to cause changes in the active site of thrombin as assessed by changes in active site fluorescent labels in experiments similar to those previously described by Ye et al. (20). Taken together, the results show definitively that certain residues in TM, which are far from thrombin in the crystal structure, are important for TM-binding and TM-mediated changes in the active site of thrombin.
EXPERIMENTAL PROCEDURES
Site-directed mutants of TMEGF45 and TMEGF456
Mutants of TMEGF45 and TMEGF456 were made using the QuikChange site directed mutagenesis kit from Stratagene (San Diego, CA). Double stranded pPIC9K vector with the synthetic TMEGF45 and TMEGF456 genes with E. coli optimized codons was used as a template (5). The sequence of each mutated gene was determined by DNA sequencing.
Each TMEGF fragment was expressed in Pichia pastoris yeast as described by White et al. (5). The protein was first purified by anion-exchange chromatography (HiLoad 26/10 Q Sepharose) followed by reverse-phase HPLC as described previously (21). Active HPLC fractions were lyophilized, reconstituted in 50 mM Tris, pH 7.4, and 150 mM NaCl and then further purified by HiLoad 16/60 Superdex 75 size-exclusion chromatography (Amersham/ GE Healthcare).
The mutants were prepared in both the context of TMEGF456 and TMEGF45. The TMEGF456 preparations bind to thrombin 10-fold more tightly than TMEGF45 preparations. We used the TMEGF456 mutants for SPR experiments, for which TMEGF45 binds too weakly. On the other hand, the TMEGF45 proteins can be purified in high yields for biophysical studies such as the fluorescence experiments presented here.
Preparation of thrombin
Human thrombin was obtained from purified prothrombin (Haematologic Technologies) as described previously (22). Optimal yields were obtained when the prothrombin concentrate was dissolved in 50 mM Tris pH 7.5, 150 mM NaCl, 10 mM CaCl2, 1mg/mL PEG-8000 so that the final prothrombin concentration was 1.6 mg/mL. The prothrombin was then activated for two hours at 37° C with 5 mg/mL Echis carinatus venom, and purified by cation exchange chromatography on a Mono-S HR 10/10 column (Amersham/GE Healthcare) using a gradient of 100 - 500 mM NaCl in 25 mM KH2PO4, pH 6.5. Alpha-thrombin was identified by fibrinogen clotting assay, and protein concentration was determined by absorbance at 280 nm (ε=1.92 cm mL unit-1 mg-1). Bovine thrombin was prepared from bovine blood as previously described (12).
Protein C activation assay
To measure the thrombin-TM activation of protein C, active thrombin (0.1nM in the assay for active mutants and 1.0 nM in the assay for less active mutants) was mixed with TMEGF45 (10 – 500 nM in the assay) for 10 min at 25°C to allow the thrombin-TM complex to form. Human protein C (0.1 – 0.8 ∝M; Haematologic Technologies, Essex Junction, VT) was added and protein C activation was allowed to occur for 20 min in a microtiter plate, volume 150∝L, in TBS with 0.1% BSA and 5 mM CaCl2 added. The reaction was quenched by addition of 40 ∝L of a solution of heparin (0.075 mg/mL, Calbiochem, San Diego, CA) and antithrombin III (0.070 mg/mL, Haematologic Technologies, Essex Junction, VT) for 10 min. The amount of activated protein C was determined using a chromogenic substrate, S-2366 (Diapharma, West Chester, OH). The amount of activated protein C was determined by comparison to a standard curve prepared with activated protein C under the same conditions (5). A non-linear fit of the Michaelis-Menten plot was used to determine the KM of TMEGF45 when the concentration of thrombin was 0.1 nM. This concentration of TMEGF45 was then used in a protein C activation assay in which varying concentrations of protein C (from 0.1 to 1∝M) were used. The results of the two assays were combined and the values of KM,TM, KM,PC, and kcat were determined.
Biacore surface plasmon resonance
Surface plasmon resonance experiments were performed using a BIACORE 3000 surface plasmon resonance instrument (Biacore, Inc., Piscataway, NJ) as described in detail elsewhere (18). Biotin-labeled TMEGF456 (570 response units) was coupled to a SA (streptavidin) sensor chip on a BIACORE 3000 instrument. Sensorgrams were collected for PPACK-thrombin as the flowing analyte (0.78 nM, 1.56 nM, 3.125 nM, 6.25nM, 12.5 nM, 25 nM) in 10 mM Hepes buffer, 150 mM NaCl, 2.5 mM CaCl2 (pH 7.4) at a flow rate of 80 μl/min and at a sampling rate of 5 Hz. No surface regeneration was required for the thrombin-TMEGF456 interaction. Rate constants for association (ka) and dissociation (kd) and the dissociation constant (KD) were obtained by globally fitting the data from five injections of thrombin using the BIAevaluation software version 3.0 using the simple 1:1 Langmuir binding model. Statistical analysis of the curve fits for both dissociation and association phases of the sensorgrams show low χ2 values. We have previously shown that the TMEGF456 binding kinetics are identical for human and bovine thrombin (18). In addition, the direct binding kinetics are the same for active thrombin and for PPACK-thrombin (18).
NMR spectroscopy
TMEGF45 Y358A was uniformly labeled with 15N during fermentation as described (21). Samples were 0.25 mM with a final volume of 0.45 mL in 90% H2O/10% 2H2O with 2 mM NaN3, pH 6.5. The pH was adjusted by adding aliquots of 100 mM NaOH. 1H-15N HSQC were collected at 310 K on a Bruker DRX600 or a Bruker DMX500 as previously described (15, 23).
Fluorescence Spectroscopy
Bovine thrombin was fluorescently labeled with either fluorescein-D-Glu-Gly-Arg chloromethylketone or fluorescein-D-Phe-Pro-Arg chloromethylketone (FEGRCK, FFPRCK; Haematalogic Technologies, Essex Junction, VT) using the method developed by Bock (24). Briefly, 27 μM thrombin was reacted with a 10-fold molar excess of FEGRCK or FFPRCK in 25 mM sodium phosphate, 150 mM NaCl, pH 6.5 at 25°C for 3 hr. Excess inhibitor removal and buffer exchange were accomplished by size exclusion chromatography (Sephadex 75, GE Healthcare) in 50 mM Tris, 150 mM NaCl, 2.5 mM CaCl2, pH 7.4. The stock thrombin samples were 2 ∝M and were stored in small aliquots at -80 °C until use but not more than 2 weeks.
Samples of TMEGF45 mutants were prepared from lyophilized protein which was reconstituted in Tris buffer (50 mM Tris, 150 mM NaCl, 2.5 mM CaCl2, pH 7.4) to make stock solutions of 200 ∝M concentration. These samples were stored at -80 °C until use.
All fluorescence spectra were acquired on a Fluoromax-2 spectrofluorometer (Jobin Yvon Inc., Edison, NJ) using an excitation wavelength of 490 nm and an emission range of 500 to 600 nm. Typically, spectra were measured with excitation and emission slit widths set at 2.0 nm and 2.5 nm respectively. The titrations of TMEGF variants (22 ∝M in 50 mM Tris, 150 mM NaCl, 2.5 mM CaCl2, pH 7.4) into fluorescein-labeled thrombin derivatives were performed by the addition of TMs into 25 nM thrombin at 25 °C to a 20-fold molar excess, mixing thoroughly after each addition. Wavelengths from 500 nM to 600 nM were scanned in 1 nM increments. The emission intensity was measured after each addition of titrant and corrected for dilution. All data were collected at 25 °C and are reported as F/Fo values. Experiments were repeated at least once, and for the FEGRCK, variation was less than 10% between experiments. Dissociation constants were determined from Scatchard analysis using non-linear regression in a manner similar to that described previously (10).
RESULTS
Direct binding of TMEGF456 variants to thrombin
Direct binding of some of the TMEGF456 variants to thrombin was measured by SPR as described previously (18). In this assay, the TMEGF456 variant was specifically biotinylated at the N-terminus and attached to a streptavidin surface. Thrombin was flowed over the surface and binding kinetics were measured. As reported previously, the binding kinetics of bovine and human thrombin are identical in this assay (18). The measured Kd value was under 10 nM for the TMEGF456M388L control (5, 6). The striking result from these direct binding assays is that many of the alanine mutants show binding defects, including many such as D349A, D357A, Y358A and F376A that are not directly contacting thrombin in the crystal structure (first column in Table 1).
Table 1.
Steady state kinetic constants measured for TMEGF456 mutants
| Protein | KD(nM)1 TM456 | KM,TM(nM) TMEGF456 | KM,PC(∝M) TMEGF456 | kcat(s-1) TMEGF456 |
|---|---|---|---|---|
| wild type | ND | 12±0.6 | 0.6±0.2 | 4.6±0.1 |
| D349A | > 100 | 700±60 | 0.6 | 1.8±0.4 |
| D349N | 4.2 | 21±2 | 1.7±0.2 | 4.2±0.8 |
| E357A | > 50 | 212 | 0.5 | 0.17±0.04 |
| E357Q | 5.2 | 30±12 | 0.7±0.5 | 0.33±0.03 |
| E357D | ND | ND | ND | ND |
| Y358A | > 100 | > 100 | > 3 | < 0.1 |
| Y358F | 2.6 | 14.0±0.3 | 0.90±0.06 | 7.5±0.2 |
| M388L | 4.4 | 12±3 | 0.6±0.2 | 6.0±0.7 |
| F376A | 20 | ND | ND | <0.1 |
| F376W | ND | 127±1 | 1.9±0.2 | 3±0.2 |
Kd was determined by SPR.
Protein C activation by TMEGF mutants
To correlate the direct binding results with TM activity, a steady state kinetic assay was used to measure the Michaelis constants for the production of activated protein C from thrombin (the enzyme) and TM (an essential activator) (5).
As shown in Scheme 1, the KM for binding of each TM variant to thrombin (KM,TM) was measured by varying the concentration of each TM variant keeping the thrombin and protein C concentrations constant. The KM for binding of protein C to the thrombin-TM variant complex (KM,PC) was determined by varying the protein C concentration in a second experiment carried out at the measured KM for the particular TM variant. The kcat was determined from the apparent Vmax measured in both experiments. This assay was used previously to demonstrate that TMEGF45 has the same kcat and KM,PC for activation of protein C as full-length TM, and has a 10-fold weaker KM,TM (5). Table 1 summarizes the steady state kinetic data we collected for the TMEGF456 mutants. We surveyed both alanine mutants and conservative mutations at the same sites in order to differentiate between structural defects and the role of specific side chains for TM function. The KM,TM was higher for all mutants that showed a binding defect by SPR (Table 1). For the TMEGF45 variants, we could not perform direct binding assays by SPR, but in all cases where binding was defective in the TMEGF456 mutants, KM,TM was also higher for the TMEGF45 mutants (Tables 1 and 2). Again, alanine mutants seemed to be more deleterious than conservative mutations at the same position. Mutations that decreased kcat but had little effect on KMTM were taken on for further study to see if they were able to cause TM-mediated changes in the thrombin active site as assessed by fluorescence experiments that probe changes in the thrombin active site.
Scheme 1.

Table 2.
Steady state kinetic constants measured for TMEGF45 mutants
| Protein | KD, (nM)1 TMEGF45 | KM,TM(nM) TMEGF45 | KM,PC(∝M) TMEGF45 | kcat(s-1) TMEGF45 |
|---|---|---|---|---|
| wild type | 68 | 140±12 | 0.32±0.1 | 5.0±0.1 |
| D349N | 43 | 440±38 | 0.77±0.2 | 0.69±0.01 |
| E357Q | 55 | 149±29 | 1.0±0.2 | 0.36±0.06 |
| E357D | ND | 542±41 | 0.62±0.26 | 0.22±0.02 |
| Y358A | ND | 623±100 | 1.3±0.3 | 0.08±0.003 |
| Y358H | ND | 163±16 | 1.6±0.16 | 0.2±0.02 |
| Y358W | ND | 61±9 | 0.8±0.5 | 5.6±1.6 |
| F376W | ND | 208±19 | 0.9±0.14 | 15±4 |
| M388L | 57 | 140±40 | 0.38±0.1 | 7.8±0.5 |
| D400N | ND | 616±131 | 0.7±0.04 | 0.04±0.001 |
| D400E | 49 | 237±16 | 1.36±0.1 | 0.17±0.01 |
Kd was determined by Scatchard analysis of the fluorescein-EGR-chloromethylketone-labeled thrombin binding data.
Alanine substitutions affected folding of the TM fragments
Alanine substitution of Asp 349, Tyr 358 and Phe 376 drastically affected thrombin binding despite the fact that these mutations are far from the thrombin binding site in TMEGF456 (9). Direct thrombin binding as assessed by SPR was barely detectable for all of these mutants (Table 1) and the protein C activation activity was low for these mutations in both TMEGF456 and TMEGF45 contexts. To ascertain whether the alanine substitutions were affecting the structure of TMEGFs, heteronuclear single quantum coherence (HSQC) NMR spectra were collected on the Y358A and F376A mutants. The HSQC spectrum of the Y358A mutant of TMEGF45 is shown in Figure 3. Many fourth domain cross peaks are missing (E346, C351, F352, A354, C356, E357, Q359, C360, L363, Q365, T366, L369, C370, V371, C372, A373, G375, I379, E382, H384), but peaks from the fifth domain are still present (N402, T403, S406, G418) consistent with the idea that this mutant has a folding defect in the fourth domain.
Figure 3.

Overlay of the NMR HSQC spectra of TMEGF45 wild type with the mutant Y358A. The wild type spectrum is in blue, and the mutant spectrum is in red. Samples were 0.25 mM with a final volume of 0.45 mL in 90% H2O/10% 2H2O with 2 mM NaN3, pH 6.5. 1H-15N HSQC were collected at 310 K on a Bruker DRX600 (wild type) or a Bruker DMX500 (Y358A). Many of the cross peaks that are missing from the spectrum of the Y358A mutant can be assigned to the fourth EGF-like domain suggesting that mutation of Tyr 358 to Ala results in a defect in folding of this domain. Some of the fourth domain peaks are labeled, and some fifth domain peaks that are observed in both spectra are labeled with boxes.
Using the protein C activation assay as a measure of function, we screened a number of other mutations at these sites. Mutation of Tyr358 to Leu in TMEGF45 was also highly deleterious, but mutation to His resulted in protein with measurable activity, and mutation to Phe or Trp improved activity. In contrast, mutation of Phe 376 to Leu or His yielded proteins devoid of any measurable activity. Both Tyr 358 and Phe 376 tolerated mutation to Trp. Whereas mutation of Asp 349 to Ala was deleterious, mutation to Asn resulted in a protein with near wild type activity. These results suggested that more conservative changes at the critical residues might be required to avoid structural defects. It is possible that because TM is a non-globular protein, it may be more sensitive to folding defects upon single site mutation than a globular protein (25).
Mutations that improved the activity of TM fragments
As reported previously, mutation of Met388 to Leu results in hyperactivity (26). This linker substitution improved kcat for protein C activation by approx. 1.5 fold but did not affect TM binding thrombin (Table 1). Several other substitutions also resulted in hyperactivity. Y358F in TMEGF456 had a 1.5 fold increase in kcat for protein C activation. Y358W in TMEGF45 bound to thrombin with a 2-fold improved affinity. F376W in TMEGF45 also resulted in hyperactivity with a 3-fold increase in kcat. This mutation was actually deleterious in TMEGF456, which also contained the M388L mutation. When the F376W mutation was made in TMEGF45 also containing the M388L mutation, it was also deleterious. Thus, the hyperactivity associated with the F376W mutation requires Met at 388. These results show that there are many ways to improve the activity of TM. Interestingly, none of these residues are anywhere near thrombin in the crystal structure (Figure 2).
Ability of TMEGF mutants to alter the thrombin active site
To monitor changes in the active site of thrombin upon TM binding, we employed the elegant experimental approach of Esmon, Johnson, and coworkers (10, 20). These researchers used fluorescein-FPR-chloromethylketone to place a fluorescent label at the active site of thrombin that would sense binding of TM, an allosteric effector of thrombin, at anion binding exosite 1. By comparing TMEGF1-6 with TMEGF56, they were able to show that the fluorescence decreased upon binding of TMEGF1-6 but not upon binding of the cofactor-inactive TMEGF56 (20). We first showed that we could recapitulate these results using TMEGF45, the smallest cofactor-active fragment of TM (Figure 4A). We also labeled thrombin with fluorescein-EGR-chloromethylketone since this substrate is thought to more closely mimic protein C. This label showed a more dramatic decrease in fluorescence upon TMEGF45 binding whereas TMEGF56 binding still caused very little change (Figure 4B). Several mutants that contained conservative changes at key fourth domain residues were found to primarily affect kcat for protein C activation while retaining nearly wild type binding by the measures employed in Tables 1 and 2. In addition, non-linear Scatchard analysis of the change in fluorescence vs. TMEGF concentration gave KDs in the range of 40 – 70 nM, which agreed with estimates from the SPR experiments that could not be quantitated (Baerga-Ortiz unpublished data). These mutants were: D349N, E357Q, and D400E (bold in Table 2). Binding of these mutants to fluorescein-EGR-chloromethylketone-labeled thrombin was analyzed to see if any of these mutations affected the way in which TM alters the active site of thrombin. Figure 5 shows that M388L causes nearly the same fluorescence decrease as the wild type TMEGF45, however the E357Q, D349N and D400E mutants all were significantly less able to decrease the fluorescence at the active site of thrombin upon TM binding.
Figure 4.

A) Change in fluorescence emission at 520 nm for fluorescein-FPRchloromethylketone-labeled thrombin (25 nM) upon addition of either TMEGF45 (■) or TMEGF56 (●) (up to 20-fold molar excess). B) Change in fluorescence emission at 520 nm for fluorescein-EGRchloromethylketone-labeled thrombin (25 nM) upon addition of either TMEGF45 (■) or TMEGF56 (●) (up to 20-fold molar excess).
Figure 5.
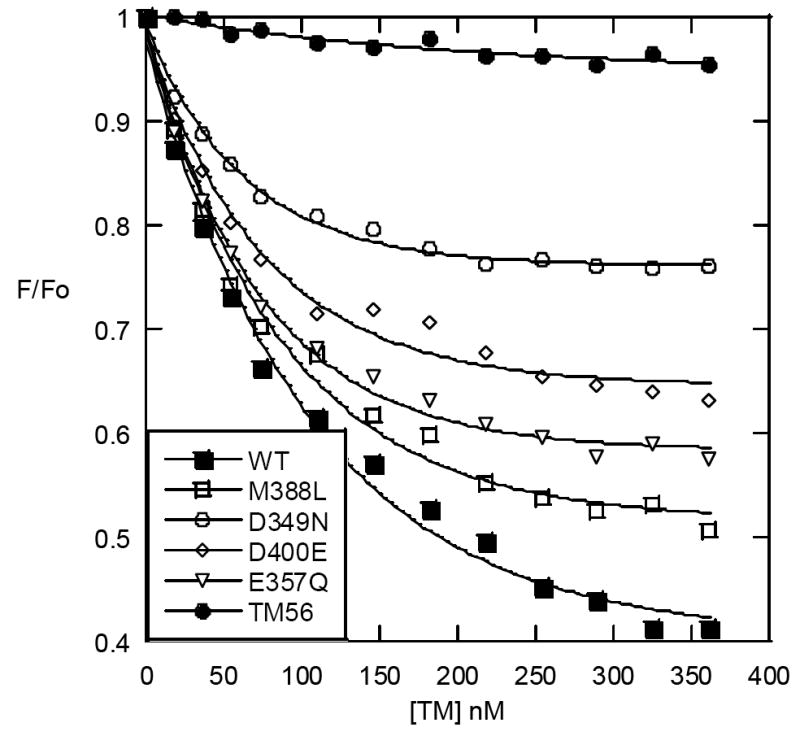
Change in fluorescence emission at 520 nm for fluorescein-EGRchloromethylketone-labeled thrombin (25 nM) upon addition of TMEGF45 wt (■), M388L (□), D349N (○), D400E (◊), E357Q (v), or TMEGF56 (●) (up to 20-fold molar excess).
DISCUSSION
Discovery of other hyperactive TM mutants
The results presented here expand the survey of TM mutations beyond those previously reported. One surprise was the number of ways activity could be increased by mutation. Substitution of Tyr358 with either Phe or Trp resulted in activity increases similar to those seen for the M388L mutant. We also observed improved kcat for F376Y mutation, but this was only observed in the context of the smallest (TMEGF45) active fragment of TM. We have previously shown using NMR that the residue at position 388 is critical for communication between the fourth and fifth EGF-like domains of TM (15, 16, 23). It is interesting that Tyr358 is “connected” to the residue at position 388 through long-range Nuclear Overhauser effects (NOEs). More specifically, long range NOEs were observed between the Leu388 and Tyr358 even though these had not been observed for Met 388. Tyr358 also showed significant changes in backbone dynamics when the linker Met 388 was oxidized (16). We hypothesized at the time that the hyperactivity of the M388L may be linked to stronger connectivity through the fourth domain. It will be interesting to investigate whether the substitution of Tyr358 with Phe and/or Trp increases the NOE connectivities in the context of Met 388, providing a mechanism for the hyperactivity observed for these mutants as well.
Structural defects of alanine mutants
Alanine scanning mutagenesis experiments often rely on the implicit assumption that replacement of one residue, particularly a surface one, with alanine will not cause structural defects. Indeed, in globular proteins this is most often the case (25). TM, however, is not a globular protein and to our surprise many of the alanine mutants either could not be expressed or showed large structural defects. Thus, for TM, many of the residues thought to be important for protein C activation are actually important for the structural integrity of TM itself. In order to discover functional rather than structural defects, we made conservative changes to TM at the same sites shown to be important by alanine scanning (6). These experiments revealed that Tyr358 could be substituted with Phe retaining wild type activity and with His retaining low activity, whereas Phe376 substitutions to anything but Trp were deleterious. Thus, the requirement for an aromatic residue at these positions is likely structural, and may not be involved in direct interactions with thrombin consistent with their positions in the thrombin-TM crystal structure (9).
Although this study avoided mutation of any TM residue in the thrombin binding site, a surprising number of the mutants, including some substitutions at Asp349, Glu357, Tyr358, and Asp400 showed deficiencies in thrombin binding. Asp400 is pointing away from thrombin in the crystal structure, but if the TM molecule retains some flexibility in the complex, it is possible that this residue could contact thrombin directly (9). The rest of the TM sites are so far removed from thrombin in the crystal structure that another explanation must be invoked. For at least some of these residues, NOE connections were observed by NMR leading to the idea that they may play a role in dynamically connecting the fourth and fifth EGF-like domains for optimal TM activity (15, 16, 23). Our hypothesis is that mutations at these residues have large effects on the thrombin binding ability of TM because they transmit changes in the conformation of the fifth domain.
Mechanistic analysis of kcat defective mutants
Many of the mutations had detrimental effects on the kcat for activation of protein C by the thrombin-TM complex, yet retained near-wild type binding affinity towards thrombin and protein C. These included D349N, E357Q, and D400E. The results presented here suggest that TM alters the catalytic activity of thrombin by changing the conformation of the active site, which is sensed by the fluorescence of fluorescein-EGR-chloromethylketone. If the crystal structure is correct, we must conclude that the fourth domain alters the way the fifth domain interacts with thrombin. All of the mutants with reduced kcat for protein C activation were less able to alter the active site of thrombin.
The results solidify the idea that dynamics and communication between the fourth and fifth EGF-like domains is important for function, and that somehow the fourth domain communicates via the fifth domain to increase the catalytic activity of thrombin towards protein C. These results strongly support a dual role for the fourth EGF-like domain. Besides acting as a docking site for protein C, it changes the way the fifth EGF-like domain interacts with thrombin, affecting both TM binding and its affects on the active site. Some residues in TM that are far from the thrombin binding site affect TM binding to thrombin, strongly suggesting that they alter the way the fifth domain interacts with anion binding exosite 1. Although it is unusual that non-contacting residues would cause such long-range (perhaps indirect) effects, the results can be explained by the fact that TM is a non-globular protein and NMR experiments have shown that the conformation of TM is highly plastic and sensitive to mutation (15, 16, 23). At least three different mutations in the fourth EGF-like domain of TM bound to thrombin with similar affinity as wild type, but specifically decreased kcat for protein C activation. All of these mutants concomitantly lost some of their ability to cause conformational changes at the active site of thrombin. These results lend further support for the assertion that TM allosterically regulates thrombin by affecting changes at the thrombin active site that promote protein C activation (12, 13).
Acknowledgments
Financial support for this work was provided by NIH grant RO1-HL070999 to EAK. JK acknowledges support from the Cell and Molecular Genetics Training Program.
ABBREVIATIONS
- TFA
trifluoroacetic acid
- MALDI-TOF
matrix assisted laser desorption time-of-flight
- ABE 1
anion-binding exosite 1
- TM
thrombomodulin
References
- 1.Esmon CT. Regulation of blood coagulation. Biochim Biophys Acta. 2000;1477:349–360. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 2.Stearns DJ, Kurosawa S, Esmon CT. Microthrombomodulin. Residues 310-486 from the epidermal growth factor precursor homology domain of thrombomodulin will accelerate protein C activation. J Biol Chem. 1989;264:3352–3356. [PubMed] [Google Scholar]
- 3.Hayashi T, Zushi M, Yamamoto S, Suzuki K. Further localization of binding sites for thrombin and protein C in human thrombomodulin. J Biol Chem. 1990;265:20156–20159. [PubMed] [Google Scholar]
- 4.Kurosawa S, Stearns DJ, Jackson KW, Esmon CT. A 10-kDa cyanogen bromide fragment from the epidermal growth factor homology domain of rabbit thrombomodulin contains the primary thrombin binding site. J Biol Chem. 1988;263:5993–5996. [PubMed] [Google Scholar]
- 5.White CE, Hunter MJ, Meininger DP, White LR, Komives EA. Large-scale expression, purification and characterization of small fragments of thrombomodulin: the roles of the sixth domain and of methionine 388. Protein Engineering. 1995;8:1177–1187. doi: 10.1093/protein/8.11.1177. [DOI] [PubMed] [Google Scholar]
- 6.Nagashima M, Lundh E, Leonard JC, Morser J, Parkinson JF. Alanine-scanning mutagenesis of the epidermal growth factor-like domains of human thrombomodulin identifies critical residues for its cofactor activity. J Biol Chem. 1993;268:2888–2892. [PubMed] [Google Scholar]
- 7.Hall SW, Nagashima M, Zhao L, Morser J, Leung LLK. Thrombin interacts with thrombomodulin, protein C, and thrombin-activatable fibrinolysis inhibitor via specific and distinct domains. J Biol Chem. 1999;274:25510–25516. doi: 10.1074/jbc.274.36.25510. [DOI] [PubMed] [Google Scholar]
- 8.Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Nat Acad Sci U S A. 1998;95:14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 10.Ye J, Liu LW, Esmon CT, Johnson AE. The 5th and 6th Growth Factor-Like Domains of Thrombomodulin Bind to the Anion-Binding Exosite of Thrombin and Alter Its Specificity. J Biol Chem. 1992;267:11023–11028. [PubMed] [Google Scholar]
- 11.Mandell JG, Baerga-Ortiz A, Akashi S, Takio K, Komives EA. Solvent accessibility of the thrombin-thrombomodulin interface. J Mol Biol. 2001;306:575–589. doi: 10.1006/jmbi.2000.4416. [DOI] [PubMed] [Google Scholar]
- 12.Koeppe JR, Seitova A, Mather T, Komives EA. Thrombomodulin tightens the thrombin active site loops to promote protein C activation. Biochemistry. 2005;44:14784–14791. doi: 10.1021/bi0510577. [DOI] [PubMed] [Google Scholar]
- 13.Rezaie AR, Yang L. Thrombomodulin allosterically modulates the activity of the anticoagulant thrombin. Proc Nat Acad Sci U S A. 2003;100:12051–12056. doi: 10.1073/pnas.2135346100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rezaie AR, Yang L. Mutagenesis studies toward understanding the mechanism of the cofactor function of thrombomodulin. Biophys Chem. 2005;117:255–261. doi: 10.1016/j.bpc.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Wood MJ, Sampoli Benitez BA, Komives EA. Solution structure of the smallest cofactor-active fragment of thrombomodulin. Nat Struct Biol. 2000;7:200–204. doi: 10.1038/73302. [DOI] [PubMed] [Google Scholar]
- 16.Prieto JH, Sampoli Benitez BA, Melacini G, Johnson DA, Wood MJ, Komives EA. Dynamics of the fragment of thrombomodulin containing the fourth and fifth epidermal growth factor-like domains correlate with function. Biochemistry. 2005;44:1225–1233. doi: 10.1021/bi0478852. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Manithody C, Rezaie AR. Activation of protein C by the thrombin-thrombomodulin complex: Cooperative roles of Arg-35 of thrombin and Arg-67 of protein C. Proc Nat Acad Sci U S A. 2006;103:879–884. doi: 10.1073/pnas.0507700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baerga-Ortiz A, Rezaie AR, Komives EA. Electrostatic dependence of the thrombin-thrombomodulin interaction. J Mol Biol. 2000;296:651–658. doi: 10.1006/jmbi.1999.3447. [DOI] [PubMed] [Google Scholar]
- 19.Baerga-Ortiz A, Bergqvist SP, Mandell JG, Komives EA. Two different proteins that compete for binding to thrombin have opposite kinetic and thermodynamic profiles. Protein Sci. 2004;13:166–176. doi: 10.1110/ps.03120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye J, Esmon NL, Esmon CT, Johnson AE. The active site of thrombin is altered upon binding to thrombomodulin. Two distinct structural changes are detected by fluorescence, but only one correlates with protein C activation. J Biol Chem. 1991;266:23016–23021. [PubMed] [Google Scholar]
- 21.Wood MJ, Komives EA. Production of large quantities of isotopically labeled protein in Pichia pastoris by fermentation. J Biomol NMR. 1999;13:149–159. doi: 10.1023/a:1008398313350. [DOI] [PubMed] [Google Scholar]
- 22.Croy CH, Koeppe JR, Bergqvist S, Komives EA. Allosteric Changes in Solvent Accessibility Observed in Thrombin upon Active Site Occupation. Biochemistry. 2004;43:5246–5255. doi: 10.1021/bi0499718. [DOI] [PubMed] [Google Scholar]
- 23.Wood MJ, Becvar LA, Prieto JH, Melacini G, Komives EA. NMR structures reveal how oxidation inactivates thrombomodulin. Biochemistry. 2003;42:11932–11942. doi: 10.1021/bi034646q. [DOI] [PubMed] [Google Scholar]
- 24.Bock PE. Active site selective labeling of serine proteases with spectroscopic probes using thioester peptide chloromethyl ketones: demonstration of thrombin labeling using N alpha-[(acetylthio)acetyl]-D-Phe-Pro-Arg-CH2Cl. Biochemistry. 1988;27:6633–6639. doi: 10.1021/bi00417a063. [DOI] [PubMed] [Google Scholar]
- 25.Blaber M, Baase WA, Gassner N, Matthews BW. Alanine scanning mutagenesis of the alpha-helix 115-123 of phage T4 lysozyme: effects on structure, stability and the binding of solvent. J Mol Biol. 1995;246:317–330. doi: 10.1006/jmbi.1994.0087. [DOI] [PubMed] [Google Scholar]
- 26.Clarke JH, Light DR, Blasko E, Parkinson JF, Nagashima M, McLean K, Vilander L, Andrews WH, Morser J, Glaser CB. The short loop between epidermal growth factor-like domains 4 and 5 is critical for human thrombomodulin function. J Biol Chem. 1993;268:6309–6315. [PubMed] [Google Scholar]