

Abstract
Bypass across DNA lesions by specialized polymerases is essential for maintenance of genomic stability. Human DNA polymerase ι (polι) is a bypass polymerase of the Y family. Crystal structures of polι suggest that Hoogsteen base pairing is employed to bypass minor groove DNA lesions, placing them on the spacious major groove side of the enzyme. Primer extension studies have shown that polι is also capable of error-free nucleotide incorporation opposite the bulky major groove adduct N-(deoxyguanosin-8-yl)-2-acetyl-aminofluorene (dG-AAF). We present molecular dynamics simulations and free energy calculations suggesting that Watson-Crick base pairing could be employed in polι for bypass of dG-AAF. In polι with Hoogsteen paired dG-AAF the bulky AAF moiety would reside on the cramped minor groove side of the template. The Hoogsteen-capable conformation distorts the active site, disrupting interactions necessary for error-free incorporation of dC opposite the lesion. Watson-Crick pairing places the AAF rings on the spacious major groove side, similar to the position of minor groove adducts observed with Hoogsteen pairing. Watson-Crick paired structures show a well-ordered active site, with a near reaction-ready ternary complex. Thus our results suggest that polι would utilize the same spacious region for lesion bypass of both major and minor groove adducts. Therefore, purine adducts with bulk on the minor groove side would use Hoogsteen pairing, while adducts with the bulky lesion on the major groove side would utilize Watson-Crick base pairing as indicated by our MD simulations for dG-AAF. This suggests the possibility of an expanded role for polι in lesion bypass.
Introduction
Cancer initiation by bulky chemical carcinogens can stem from mutations induced by DNA adducts in oncogenes or tumor suppressors which govern cell cycle control (1, 2). Such bulky adducts can stall replicative polymerases, leading to a switch to lesion bypass polymerases (3-5). Human DNA polymerase ι (polι) is a lesion bypass polymerase of the Y family (Figure 1A) (6-8). Though evolutionarily related to polη (6, 9), polι exhibits several properties unique among DNA polymerases characterized to date. Polι has both the lowest processivity of any known DNA polymerase as well as the highest rate of misincorporation (8, 10, 11). The properties of polι misincorporation are also unique, in that polι exhibits differential fidelity dependent on the identity of the templating base: templating dA has the highest fidelity, with a misincorporation rate of ∼10−4, while polι actually prefers to insert dG opposite dT instead of the correct partner dA (10, 12). This preferential misinsertion on undamaged template proved difficult to explain in structural or functional terms until the crystal structure of a polι ternary complex became available (13).
Figure 1. Human DNA polymerase ι with incoming dCTP+.
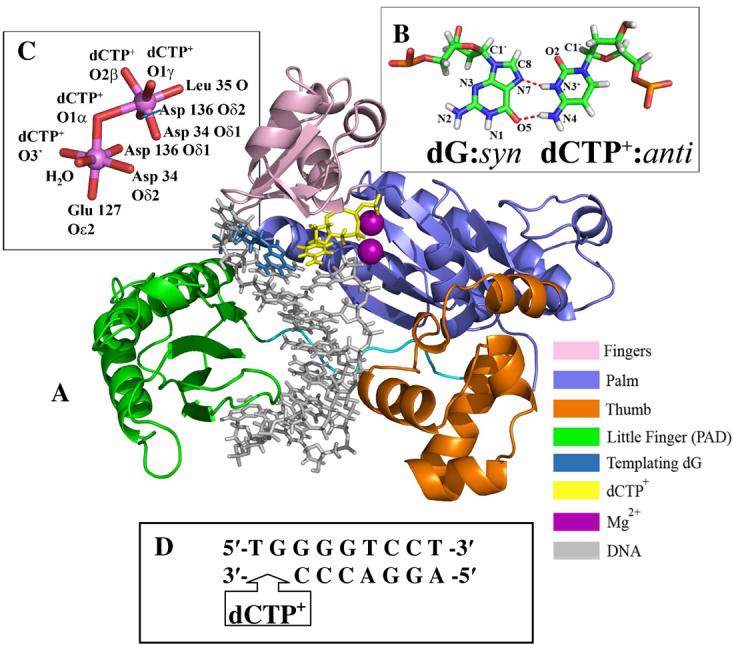
A: Polymerase ι remodeled based on the ternary crystal structure PDB ID: 2ALZ (14) (see Experimental Procedures for details).
B: Catalytic Mg2+ coordination in the initial model. Liganding distances are given in Table S8.
C: The active site Hoogsteen base pair. Oxygen: red; nitrogen: blue; carbon: green; phosphorus: orange.
D: DNA sequence used in all simulations.
Crystal structures of ternary complexes of this polymerase with adenine or guanine as the templating bases reveal syn purines with Hoogsteen (HG) pairing (Figure 1B) in the active site (13-15). These structures suggest that HG pairing with protonated dCTP (dCTP+) opposite templating guanine is favored due to specific features of the polι active site. For example, residues in the active site (Gln59, Lys60, and Leu62) grip the sugar of the templating base, and induce a short C1′-C1′ distance in the nascent base pair that accommodates a narrow (∼8.7Å) HG (dG syn) pair, but not the wider (∼10.6Å) Watson-Crick (WC) (dG anti) pairing (15). Related Y-family polymerases η, κ, and Dpo4, all of which employ WC pairing in the active site, have less bulky residues in the equivalent position (16-18). Crystal structures of polι ternary complexes show a preformed active site that is open and exposed to solvent on the major groove side of the nascent double helix (13-15, 19). The polymerase is close to the nascent base pair on the minor groove side, leaving little space between enzyme and the DNA. Thus, HG pairing is a novel and effective way for the polymerase to move bulky minor groove adducts away from the cramped minor groove side of the nascent base pair through rotation of the glycosidic bond to the syn domain. This allows for nucleotide incorporation opposite damage that compromises WC hydrogen bonding. For clarity we note that a major groove adduct would reside on the major groove side of the adducted base in normal B-DNA with WC base pairing, while a minor groove adduct would reside on the minor groove side. Numerous primer extension experiments with minor groove lesions such as N2-dG adducts derived from benzo[a]pyrene diol epoxide (20), γ-hydroxy-1,N2-propano-dG (21, 22), 3-methyl adenine (23, 24) and N2-methyl, -ethyl, -isobutyl, and –benzyl dG (25) reveal polι's capability for bypass of minor groove lesions. A ternary crystal structure of polι with the Watson-Crick edge obstructing lesion 1,N6-ethenodeoxyadenosine (19) as well as biochemical data with base analogs incapable of WC hydrogen bonding (26, 27) also support this hypothesis.
Polι, like other Y-family polymerases, does not use stringent steric selection or a dNTP-binding induced-fit to select the incoming dNTP (12, 15). Furthermore, a strong case has been made for the primacy of hydrogen bonding in selecting a matching incoming nucleotide in polι (26, 27) and other Y-family polymerases (12, 28, 29). This elegantly explains the template-based differential fidelity of polι. For example, dA is able to form two hydrogen bonds with dT in both syn (HG) and anti (WC) conformations; however, if the incoming dCTP is protonated as in the polι ternary crystal structure (14), forming dCTP+, then two HG hydrogen bonds can be formed with the templating dG syn. Thus polι has higher fidelity with templating dA, and lower with dG, because the AT HG pair has two hydrogen bonds without the energetic cost of protonation that the G-C HG pair requires. When polι encounters a templating pyrimidine the incorporation spectrum resembles that of an abasic site, which may indicate that the pyrimidine is somehow ejected from the active site (15, 30).
2-acetylaminofluorene (AAF) is a mutagenic and carcinogenic aromatic amine first identified as a carcinogen during toxicity trials to determine its suitability as a pesticide (31). After metabolic activation, 2-acetylaminofluorene-derived carcinogens selectively react with dG to produce two major adducts: N-(deoxyguanosin-8-yl)-2-aminofluorene (dG-AF), and N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) (Figure 2), as well as a minor N2-dG-AAF adduct; early work describing this field is reviewed in (32, 33). The present study focuses on dG-AAF. More recently the dG-AAF adduct's mutagenic properties have been characterized in a wide variety of in vitro and in vivo contexts (34-43). Other investigations have focused on studies by NMR (44-48).
Figure 2. dG-AAF structure with torsion angles designated by arrows.
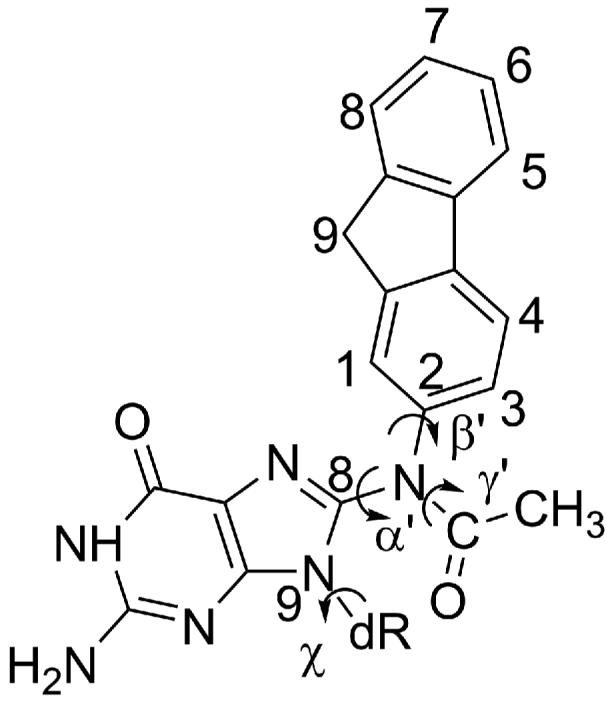
Torsion angles at the linkage site are defined as follows: α′, N9–C8–N–C2; β′, C8–N–C2–C1; γ′, C8–N–C–C(methyl); χ, glycosidic torsion angle O4′–C1′–N9–C4, where O4′, C1′, N9, C4 and C8 are from dG and other atoms are from AAF.
Primer extension studies with dG-AAF have revealed that polι predominantly incorporates dC opposite the lesion (49) in an error-free manner. In this experiment no extension beyond the lesion was observed, as is frequently the case in polι lesion bypass (21, 30). Early work at the monomer level suggested that dG-AAF favors the syn domain due to difficulty in avoiding steric clashes between the acetyl group on the AAF moiety and the damaged nucleotide, adopting a conformation with carcinogen-base stacking (50-52). NMR solution structures later showed that, as had been predicted, in double-stranded DNA dG-AAF predominantly adopts a base-displaced intercalated conformation in solution with syn glycosidic torsion angle (44, 46), with some NMR evidence for an external conformation available as well (53). A base-displaced intercalated conformation displaces the templating dG, inconsistent with the error-free selection of dCTP/dCTP+ opposite dG-AAF by polι. Therefore the AAF fluorenyl rings must be placed on the major or minor groove side of the nascent base pair, with anti or syn glycosidic bond conformations, respectively. While a syn conformation for dG-AAF is consistent with HG pairing in polι, if the C8 adduct is syn (HG) the bulky AAF fluorenyl rings would be placed in a small, crowded region of the enzyme on the minor groove side of the template. With WC pairing, on the other hand, the AAF ring system would be well accommodated in the large space on the major groove side. Recently it has been shown through MD simulations that dG-AAF can adopt the anti (WC) conformation with WC pairing through a modest alteration in sugar pucker in the modified nucleoside. This alleviates the steric hindrance between the acetyl and the adjacent sugar (54).
In the present study we investigated both WC and HG paired dG-AAF in polι using a molecular modeling and molecular dynamics (MD) approach supplemented by potential of mean force (PMF) analysis. Our results show that with WC pairing the AAF adduct is well accommodated on the major groove side of the growing double helix, with frequent sampling of a near reaction-ready state. HG paired models led to severe distortion of the active site, and in no case did they sample a near reaction-ready state. In addition, free energy calculations indicate a relatively small (∼5kcal/mol) energy difference between the WC and HG paired dG-dCTP/dCTP+ in the active site of polι. These results suggest that the dG-AAF adduct can be bypassed by polι utilizing a WC pairing scheme which places the bulky adduct on the spacious major groove side of the nascent base pair, similar to the accommodation of bulky minor groove lesions via HG base pairing in the same space. Thus polι could play a role in bypassing adducts on both the major and minor groove sides of a damaged base, adding to its versatility in human lesion bypass.
Experimental Procedures
Molecular Modeling of Initial Unmodified Control Structures for MD
We used the polι ternary complex crystal structure containing a dG-dCTP+ HG base pair (PDB (55) ID: 2ALZ (15)) as the basis for our initial models. Missing loops containing residues 371 to 378 and residues 395 to 403 were modeled with the program MODELLER (56) on the Modloop web server (57). The DNA sequence (Figure 1D) was taken from the crystal structure (14). While the crystal contained three residues in the single-stranded 5′ overhang, the last two were not resolved and are not included in our simulations. This structure was used as our HG unmodified control simulation (Figure 1A). dCTP+ was modeled from dCTP by protonation of N3. For the first Watson-Crick unmodified control simulation the glycosidic torsion angle χ (Figure 2) on the templating dG was altered from the crystal value of 64.9° (syn) to 254.1 (anti) which permitted WC pairing. A second unmodified WC structure was derived from the WC-AAF2 trajectory's most representative structure (see below). AAF atoms were deleted and replaced by a hydrogen atom, and the resulting WC unmodified structure was subjected to our molecular dynamics protocol (see below).
Molecular Modeling of Initial dG-AAF Structures for MD
Structures from the last frame (10 nanoseconds (ns)) of the unmodified control production dynamics simulations were used as the initial structures for the dG-AAF simulations (HG control for HG-AAF, WC control for WC-AAF). A high resolution AAF structure (58) was obtained from the Cambridge Structural Database (59) (refcode ACAFLR) and bonded to C8 of the templating dG. Then the α′ and β′ linkage torsion angles (Figure 2) of the resultant dG-AAF were surveyed over their 360° range at 10° intervals starting at 5°, with glycosidic torsion χ in the syn (HG) conformation (64.9°), as observed in the crystal structure (14) for a total of 1296 structures created. These structures were evaluated for steric clashes with the bumpcheck utility of InsightII (Accelrys Inc.). For these searches γ′ (Figure 2) was retained in the conformation of the AAF crystal structure (58), i.e. 340.7°. For the anti WC-AAF simulations the last frame (10 ns) of the WC control structure (χ = 192.7°) was used for a similar search of an additional 1296 structures. After these initial searches were completed α′, β′ combinations that exhibited minimal close contacts were selected for further investigation. For the HG-AAF structures we identified two narrow regions that showed the least close contacts, one placing the fluorenyl rings 3′ of the templating dG (HG-AAF1 and HG-AAF2), and one 5′ (HG-AAF3 and HG-AAF4). For the WC-AAF structures, the AAF could be accommodated either in the broad range of α′ 260° to 284° on the exposed major groove side of the nascent base pair (WC-AAF1 and WC-AAF3), or in a narrow pocket in the little finger domain (WC-AAF2 and WC-AAF4). For both WC-AAF and HG-AAF structures each region could accommodate a pair of structures with the same α′ torsion angles, but with the methylene bridge (C9 in Figure 2) rotated ∼180° about the long axis of the fluorenyl rings, which is governed by β′. Finally, the α′, β′, and γ′ torsions of all structures were further adjusted to minimize close contacts (Figure S1, Table S1).
Force Field
The Cornell et al. force field (60) with modifications (61, 62) and the PARM99 parameter set (63) were employed for all simulations. For the partial charge calculations a syn dG-AAF model with no steric clashes was modeled using a generic B-DNA dG from the Biopolymer module of InsightII (Accelrys Inc.) linked to a high resolution AAF structure (58) from the Cambridge Structural Database (59) (refcode ACAFLR). The structure had C2′ endo envelope sugar pucker and χ = 34.2°, α′ = 135°, β′ = 83°, and γ′ = 165°. Partial charges for the dG-AAF and for dCTP+ (Table S2, S3) were calculated with the HF method and the 6-31G* basis set (64) using Gaussian 03 (65), and the restrained electrostatic potential fitting algorithm RESP (66, 67) was employed to fit the charge to each atom center. dCTP charges were taken from previous work (68). Parameters for atom types in dG-AAF and dCTP+ not found in the PARM99 parameter set were taken from the GAFF (69) parameter set or developed by analogy to chemically similar atom types in the PARM99 and GAFF parameter sets. Parameters are provided in Table S4.
Molecular Dynamics Protocol
All minimizations and MD simulations used the SANDER module of the AMBER 8.0 software suite (70). The LEaP module of AMBER 8.0 (70) was used to add hydrogen atoms and neutralize the system with sufficient Na+ atoms to bring the net charge to zero. Hydrogen atoms of the solute (DNA, polymerase and incoming dCTP/dCTP+) were minimized with implicit solvent using a distance dependent dielectric function of ε = 4.0r (where r is the distance between an atom pair) for 600 steps of steepest descent, followed by 600 steps of conjugate gradient. The resulting ternary complex was reoriented with SIMULAID (71) to minimize the number of water molecules needed to solvate the system. A periodic TIP3P (72) rectangular water box with a buffer distance of 10 Å between each wall and the closest solute atom in each direction was added with the LEaP module of AMBER. Box dimensions were approximately 80 ×75 × 76 Å, with a total of ∼14,500 water molecules.
All systems employed the following equilibration and MD protocols:
(i) minimization of the counterions and solvent molecules for 2500 steps of steepest descent and 2500 steps of conjugate gradient, with 50 kcal/mol restraints on the solute atoms; (ii) 30 picoseconds (ps) initial MD at 10 K with 25.0 kcal/mol restraints on the solute to allow the solvent to relax; (iii) heat-up from 10 to 310°K (37°C) at constant volume over 80 ps with 10 kcal/mol restraint on the solute; (iv) 20 ps MD at constant volume and 310°K, with 10.0 kcal/mol restraints on the solute; (v) 30, 40 and 50 ps MD at 1 atm and 310° K with decreasing restraints of 10, 1, and 0.1 kcal/mol, respectively, on solute atoms, and an increasing time constant of 1.0, 2.0, and 4.0 ps, respectively, for heat bath coupling of the system; (vi) production MD was conducted at 1 atm, 310° K for 10 ns, with 1 ps coupling constants for both pressure and temperature.
In all MD simulations, long-range electrostatic interactions were treated with the particle mesh Ewald method (73, 74). A 9 Å cutoff was applied to the non-bonded Lennard–Jones interactions. The SHAKE algorithm (75) was applied to constrain all bonds involving hydrogen atoms with relative geometrical tolerance of 10−5Å. The Berendsen coupling algorithm (76) was used for temperature scaling. A 2 femtosecond (fs) time step was used, and the translational/rotational center-of-mass motion was removed every 0.5 ps (77).
Potential of Mean Force Calculations
Thirty six initial structures were generated by taking the most representative structure from the stable region of the HG control dynamics simulations (6-10 ns ; see Figure S2) and resetting χ at 10° intervals from 5° to 355°. The most representative structure was identified using the trajectory clustering function in the MOIL-VIEW (78) program with all residues within 8.0Å of any atom in the nascent base pair selected for analysis. Initial structures were minimized and equilibrated as described below. MD simulations were then run for 200 ps, with a 30 kcal/mol harmonic restraint centered on the initial value of χ, and χ angle values in each 200 ps ensemble were collected at 20 fs intervals. We performed three separate sets of thirty six simulations with identical initial conditions and different random initial velocity distributions. All χ angle values from all three trials were used as input to a program devised by A. Grossfield to implement the weighted histogram analysis method (WHAM) (available at http://membrane.urmc.rochester.edu/) for calculating potential of mean force (relative free energy).
Stability of MD Simulations
All simulations showed reasonable stability in the active site after 4 ns, and stability in the whole enzyme after ∼6 ns. This was evaluated by inspecting the RMSD of the enzyme as compared to the initial state after equilibration over time (Figure S2). Therefore all analyses presented here were performed on the final 4 ns of the trajectories.
Results
In vitro primer extension data for polι containing a dG-AAF lesion as the templating base revealed that polι very strongly selects the correct partner, dC, opposite the lesion (49). Thus we investigated structures with incoming dCTP/dCTP+ in order to determine the features that led to incorporation of the correct nucleotide opposite the adduct. We were specifically interested in whether the damaged base assumes a syn (HG) or anti (WC) conformation in the active site of the polymerase. A syn (HG) conformation places the bulky adduct on the cramped minor groove side of the growing double helix, while an anti (WC) conformation places the AAF lesion on the spacious major groove side. We performed eleven molecular dynamics simulations: three unmodified controls, one with undamaged syn (HG) and two with undamaged anti (WC) dG as the template, and eight dG-AAF damaged template simulations: four syn (HG) and four anti (WC). Initial α′ and β′ torsion angles (Figure 2, Table S1, Figure S1) for the adduct were selected through an extensive conformational search of 2592 structures as detailed in the Experimental Procedures. We performed 10 ns of production MD for each structure, with the final 4 ns used for analysis.
Criteria for a near reaction-ready state
Using the well-organized active site of a high resolution polβ crystal structure containing two Mg2+ ions and a primer terminal 3′ OH (79) and the results of a quantum mechanical molecular modeling study (80) as our guides we utilize the following structural criteria for a near reaction-ready state: (1) a Pα-O3′ distance of less than 3.5Å; (2) an O3′-Pα-O3α angle of attack of 170±10°; and (3) reasonable coordination of the two catalytic Mg2+ ions (Figure 1B) (discussed below). Furthermore, as a reasonably stringent criterion for a near reaction-ready active site we evaluated the percentage of structures in our trajectories that satisfy all three of the above criteria simultaneously and term that the near reaction-ready occupancy (Table 1).
Table 1.
Near reaction-ready occupancy and number of hydrogen bonds formed between the incoming dCTP(WC)/dCTP+(HG) and the templating dG(control)/dG-AAF(damaged).
| Structures | HG Control | WC Control 1 | WC Control 2 | HG-AAF1 | HG-AAF2 | HG-AAF3 | HG-AAF4 | WC-AAF1 | WC-AAF2 | WC-AAF3 | WC-AAF4 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Near reaction- ready occupancy |
51% | 82% | 83% | 0% | 0% | 0% | 0% | 76% | 75% | 0.4% | 70% |
| Number of hydrogen bonds |
2 | 3 | 3 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 3 |
We also considered the extent of hydrogen bonding between the incoming dCTP/dCTP+ and the templating base in order to determine if proper HG or WC hydrogen bonding was taking place (Table S5). In addition we analyzed hydrogen bonding between the nascent base pair and the enzyme, using the unmodified control HG simulation as our benchmark (Table S6, S7). Our criteria for hydrogen bonding are a donor and acceptor heavy atom distance of <3.4Å, and a donor-hydrogen-acceptor angle of 180°±55°. Finally we have used WHAM (81) to derive the free energy profile around the glycosidic bond for the HG unmodified control simulation to gain energetic insight into the overall cost of forming an anti structure in the active site.
WC and HG unmodified control simulations share some common features in the active site
Both HG and WC unmodified control simulations were performed and analyzed as described above. The most representative active site structure was identified using the trajectory clustering function in the MOIL-VIEW (78) program with all residues within 8.0Å of any atom of the nascent base pair selected for analysis. The fingers domain of the enzyme shows the highest degree of stability, with a fingers domain backbone RMSD of <1Å between the most representative active site structure of any of the unmodified control simulations and the crystal structure. All of the active site hydrogen bonds between the incoming nucleotide and the enzyme observed in the crystal are maintained in the HG and both WC unmodified control simulations, with the exception of the hydrogen bond between Tyr68 and the incoming dCTP/dCTP+, which is replaced by two hydrogen bonds with water in all three simulations (Table S6). In other respects the unmodified control simulations differ, as described below.
Hoogsteen unmodified control is faithful to the crystal structure
All ternary complex crystal structures of polι to date show a HG pair formed between the incoming dNTP and the templating base (13-15, 19). In our unmodified control simulation the syn (HG) conformation of the templating base is maintained throughout the HG unmodified control simulation (Figure S3). The structural features of the active site are similar to the crystal, with an HG compatible C1′-C1′ distance of 8.8±0.2Å (Figure S4). Good base stacking is observed between the incoming dCTP+ and the primer terminus (Figure S5). The Mg2+ ions are well coordinated (discussed below) (Figure S6, S7, Table S8), though the inter-Mg2+ distance does increase somewhat, from 3.4Å to 4.1±0.1Å. Overall near reaction-ready occupancy for the HG unmodified control is 51% (Table 1), a figure that is not surprising given polι's extremely slow rate of incorporation (8, 10). Hydrogen bonding in the nascent base pair is good, with both HG hydrogen bonds present at >99% occupancy (Table 1, Table S5). Other features of the active site resemble the ternary crystal structure (14), with the close C1′-C1′ distance enforced by the tight coordination of the incoming dCTP+ via numerous hydrogen bonds (Table S6) and other polar interactions coupled with the stabilization of the templating base's sugar by Gln59, Lys60, and Leu62 (Figure 3A). Lys60 forms a hydrogen bond with the free phosphates in the templating dG backbone, contributing to the HG compatible C1′-C1′ distance (Figure 3A, Table S7). Ser307, on the other hand, forms a hydrogen bond with the dT 5′ overhang phosphate group, as opposed to hydrogen bonding with the dG backbone phosphate as in the crystal structure, due to a shift in the position of the mobile little finger domain (12). This change in hydrogen bonding does not appear to impact the organization of the nascent base pair or the active site as a whole..
Figure 3.

Active site organization and hydrogen bonds formed by the templating dG/dG-AAF. Polι is green, duplex DNA is grey, and key active site features are colored by atom. Oxygen: red; nitrogen: blue; carbon: green; phosphorus: purple. Stereo views are available in Supplementary Material, Figure S11.
A: Unmodified HG control simulation. Two HG hydrogen bonds are formed between the incoming dCTP+ and the template. Hydrogen bonds between Ser307 and the terminal dT O5′ and between Lys60 and a backbone oxygen from the previously incorporated dG (labeled as OP) stabilize the base position. Note the stacking of the dT single stranded overhang with Tyr61.
B: HG-AAF4. This structure was selected as the least distorted HG-AAF structure (Table 1). Note the absence of hydrogen bonding between the templating dG-AAF and the enzyme, and the single hydrogen bond between dG-AAF and dCTP+.
C: Unmodified WC control simulation. Three WC hydrogen bonds are formed between dG and dCTP. The hydrogen bond between Ser307 and the terminal O5′ is replaced by a hydrogen bond between Arg347 and a backbone oxygen from the templating dG, and the 5′ dT overhang stacks with His354 instead of Tyr61. Lys60 forms a hydrogen bond with a backbone oxygen from the previously incorporated dG (labeled as OP) as in the HG control. The dG sugar ring is pulled away from residues Gln59, Lys60, and Leu62. A novel hydrogen bond not possible in the HG conformation is formed between Gln59 and N2 of the templating dG.
D: WC-AAF2. The active site is stabilized by hydrogen bonds from a templating dG backbone oxygen to Lys60 and from a backbone oxygen on the previously incorporated dG (labeled OP) to Ser307, as well as the novel hydrogen bond between Gln59 and N2 of the templating dG observed in the WC control. Three WC hydrogen bonds are formed in the nascent base pair, and the 5′ dT overhang stacks with Tyr61 as in the HG control.
Watson-Crick unmodified control loses important contacts with the polymerase
In order to evaluate the feasibility of accommodating a WC pair in the active site of polι we performed two simulations:
(1)WC unmodified simulation 1, where the initial structure was identical to the HG unmodified control, except that the templating base was initiated with an anti (WC) conformation (Table S1), and
(2) WC unmodified simulation 2, a WC unmodified simulation using the most representative structure from the WC-AAF2 simulation, with the AAF moiety replaced by a hydrogen atom, as the initial state; WC-AAF2 showed the highest reaction ready occupancy of the WC-AAF simulations. This simulation was suggested by a reviewer.
In both simulations the incoming dCTP was unprotonated so as to allow three WC hydrogen bonds to form. These simulations performed surprisingly well, both attaining a standard WC C1′-C1′ distance of 10.6±0.2Å (82) which was then maintained throughout the stable region of the trajectory (Figure S4). For both simulations base stacking between the nascent pair and the previously incorporated pair is not disrupted (Figure 3C, Figure S5). Mg2+ coordination does not differ significantly from the HG unmodified control in either case (Figure S6, S7, Table S8), and the inter-Mg2+ distance is 3.9±0.1Å in both simulations. Both simulations have two Watson-Crick hydrogen bonds that are present at >99% occupancy, with the third at ∼86% occupancy in WC unmodified 1 and ∼98% in WC unmodified 2 (Table S5). In addition a hydrogen bond between Gln59 and N2 of the templating dG is formed in both simulations, with ∼98% occupancy in WC unmodified 1 and ∼77% occupancy in WC unmodified 2 (Table S7). This hydrogen bond is not possible with the syn template in the HG pair. Moreover, the near reaction-ready occupancy is 82% for WC unmodified 1, and 83% for WC unmodified 2 (Table 1).
We sought to define the structural rearrangements associated with forming such a near reaction-ready Watson-Crick pair in the unmodified control simulations. In both cases the hydrogen bonds between the incoming dCTP and the polymerase are similar to the HG unmodified control (Table S6). However, in order to stabilize a wider C1′-C1′ distance the dT 5′ to the templating dG moves towards the major groove side of the nascent base pair, pulling the DNA backbone away from the polymerase. In WC unmodified control 1 the single stranded overhang stacks with a histidine (His354) in the little finger domain, pulling the C1′ carbon of the templating dG ∼2Å away from the active site (Figure 3C). In WC unmodified control 2 the 5′ OH of the overhang hydrogen bonds with N7 of the templating dG. Since the incoming dCTP remains stably positioned relative to the fingers domain of the protein in both cases, the movement of the overhang and backbone away from the confines of the polymerase allows the C1′-C1′ distance to increase to ∼10.6Å, appropriate for a WC pair (82) (Figure S4), and the sugar pucker is a B-DNA standard C2′ endo (82) (Table S9). In both cases the DNA template backbone also loses the hydrogen bond with Ser307 found in the HG unmodified control simulation due to the rearrangement of the backbone. This hydrogen bond is replaced with a hydrogen bond to Arg347 in the little finger domain in WC control 1, but is lost entirely in WC control 2 (Figure 3C, Table S7). This major groove position of the single stranded base 5′ to the template is not seen in any of the other simulations, and neither of these structures would be feasible if the 5′ overhang were longer (Figure 3C) due to a dearth of room on the major groove side of the duplex. This implies that such a WC permissive conformation in the enzyme with unmodified DNA is likely to be disfavored during replication, consistent with the crystallographic observations to date.
Hoogsteen AAF simulations show a variety of distortions that prevent achievement of a near reaction-ready state
Of the 1296 initial models of dG-AAF with HG pairing that we screened (see Experimental Procedures and Supplementary Data) we selected four with minimal close contacts between the adduct and the enzyme for further study. HG-AAF MD simulations differed in the combined initial values of the torsion angles α′, β′, and γ′, and therefore the placement of the lesion relative to the DNA and polymerase (Figure S1, Table S1). Two initial structures placed the AAF rings 3′ to the templating dG (HG-AAF1 and HG-AAF2), and two placed the AAF rings 5′ to the templating dG (HG-AAF3 and HG-AAF4) (Figure S1). Both members of each pair of structures place the fluorenyl rings in the same region relative to dG, but the methylene bridge (C9 in Figure 2) is rotated ∼180° around the long axis of the fluorenyl rings in the pair members (Figure S1). After 10 ns of simulation the active site is severely distorted in all four cases, with no common motifs for placement of the AAF (Figure 3B, S8, S9). Hydrogen bonding between the incoming dCTP+ and templating dG-AAF is poor or absent (Table 1, Table S5), Pα-O3′ distances are long (Figure S4), and base stacking between the incoming dCTP+ and the primer terminus is often disrupted (Figure S5). While hydrogen bonds between the incoming dCTP+ and the enzyme are mostly maintained (Table S6), the interactions between the templating base and the enzyme are lost or altered in the majority of the HG dG-AAF simulations (Figure3B, Table S7). Most importantly the HG dG-AAF structures, without exception, have a near reaction-ready occupancy of 0% (Table 1), strongly suggesting that incorporation of the incoming dCTP+ would be disfavored.
The HG dG-AAF structures attempt to accommodate the lesion in different ways. The bulky hydrophobic fluorenyl moiety stacks with the cytosine ring of the incoming dCTP+ in the HG-AAF1, HG-AAF2, and HG-AAF4 simulations (Figure S8, S9). In the HG-AAF2 and HG-AAF4 cases this causes the cytosine ring to rotate ∼90° around the glycosidic bond, disrupting hydrogen bonding with the dG-AAF (Figure 3B). Accommodation of the lesion has the additional effect of displacing the modified guanine from the double helix in all trials save HG-AAF3 (Figure S8, S9), resulting in increased C1′-C1′ distances (Figure S4). The primer terminus disengages from the active site, in the case of HG-AAF2 moving as far as 11Å away from the incoming dCTP+ Pα (Figure S4). The acetyl moiety of dG-AAF is placed 3′ to the dG-AAF when there is significant displacement of the base, and otherwise is placed 5′ of the guanine rings (Figure S8, S9).
Watson-Crick AAF simulations accommodate the lesion well with little distortion of the active site
When dG-AAF was modeled with an anti (WC) glycosidic torsion angle four structures were again selected as initial models for MD simulation from a scan of 1296 possible conformations (see Experimental Procedures and Supplementary Data) (Table S1, Figure S1). In all four cases the active site is stable and well ordered after MD simulation (Figure 3D, Figure S8, S9). There are three hydrogen bonds between the templating base and dCTP present in all simulations, ranging from 59% occupancy to 100% occupancy (Table 1, Table S5). Base stacking between the incoming dCTP and the primer terminus is well maintained (Figure S5), and the C1′-C1′ distance is near the typical WC value of 10.6Å (Figure S4). The WC-AAF1, WC-AAF2, and WC-AAF3 simulations produce near reaction-ready occupancies (involving good Pα-O3′ distance (Figure S4), Mg2+ coordination (Figure S6, S7, Table S8), and in-line attack angle (Figure S3, Table S9)) of >70%. However, the WC-AAF3 simulation showed only 0.4% occupancy (Table 1) due to an overlong Pα-O3′ distance (Figure S4) resulting from a hydrogen bond between Tyr39 and O3′ of dCTP (Figure S10, Table S6). This hydrogen bond pulls the dCTP Pα away from O3′ of the primer terminus, and it is not observed in other WC-AAF simulations (Table S7).
The dG-AAF does not significantly disrupt the active site in these simulations. Analysis of the four trials reveals two motifs for placement of the fluorenyl rings and acetyl group of the dG-AAF. WC-AAF1 and WC-AAF3 simulations had the fluorenyl rings placed 5′ to the damaged base, thus stacking with the 5′ overhang dT (Figure S8). The acetyl moiety is 3′ to the guanine rings, causing a change in the sugar pucker to the C1′ exo domain in order to avoid close contacts with the acetyl (Figure S3, S8, Table S9). The remaining two simulations have the fluorenyl rings 3′ to the dG, allowing the 5′ overhang to stack with Tyr61. The acetyl is 5′ to the base, and the sugar is C1′ exo (WCAAF2) or O4′ endo (WCAAF4) (Figure S3, S8, Table S9). The alteration of the sugar pucker is consistent with previous results in Dpo4, which also showed that a C1′ exo pucker permitted anti dG-AAF (54). Hydrogen bonding between the incoming dCTP and polι is robust, with no significant differences from the HG unmodified control (Table S6).
In these simulations the increased C1′-C1′ distance necessary for WC pairing was made possible by an adjustment of the sugar on the templating base that moves the C1′ carbon away from the incoming dCTP. Unlike the WC unmodified control there is no stacking between the template 5′ dT overhang and the little finger domain. The templating dGAAF forms strong hydrogen bonds with Gln59 and Lys60 (Figure 3D, Table S7), helping to maintain contact with the hydrophobic residues that hold the dG-AAF sugar. The fingers domain adjusts slightly to accommodate the new placement of the sugar on the templating base, with the residues in the vicinity of the sugar (i.e., Gln 59 to Leu62) moving ∼1Å relative to the rest of the fingers domain. The remainder of the active site protein backbone is relatively static, with an average RMSD of 0.50Å between the WC dG-AAF structures and the crystal structure.
Mg2+ coordination in our simulations is less than ideal
The two catalytic Mg2+ ions in the polι active site are each coordinated by six electronegative oxygen atoms (Figure 1B). We consider good Mg2+ coordination in polι as one Mg2+ ion with all six oxygens within 2.4Å and the other Mg2+ with five of these oxygens within 2.4Å and one oxygen within 3.2Å (see Discussion). In the simulations which exhibit good Mg2+ coordination, all but one of the coordinating oxygens are within 2.4Å. The exception is the only O that is involved in coordinating both Mg2+ ions, dCTP/dCTP+ O1α. The O1α is within 2.4Å of either Mg2+A or Mg2+B, but never both at the same time. Time course analyses and Mg2+ coordination mean distance and standard deviation values for the stable region of the dynamics are shown in Figure S6, S7, and Table S8.
Free energy analysis with incoming dCTP+ shows both syn and anti conformations occupy energy wells, with syn lower in energy
In light of our findings above indicating that a WC pair can be stably accommodated in the active site of polι we sought to estimate the overall energetic cost of maintaining an anti conformation of the templating base relative to a syn conformation. We used the weighted histogram analysis method (WHAM) (81) on a series of umbrella samples (see Experimental Procedures) which scanned over the entire range of the glycosidic torsion angle χ. Analysis of the relative free energy around χ for the most representative HG unmodified control structure with incoming dCTP+ showed that the syn structure is ∼5 kcal/mol lower in energy than the anti structure (Figure 4A). Both the syn and anti structures lie in energy wells, and they are separated by barriers of ∼12 kcal/mol and ∼9 kcal/mol, depending on the direction of the rotation. While this simulation was performed with a protonated dCTP+, which precludes formation of three WC hydrogen bonds, nevertheless the anti structure resides in an energy well. When the proton is removed from the dCTP the difference is ∼3.5 kcal/mol, but the anti structure has the lower energy, and the barriers are both ∼10 kcal/mol (Figure 4B). The presence of an anti energy well separated from the syn domain well by an ∼9-10 kcal/mol barrier regardless of the protonation state of the incoming nucleotide would preclude the interconversion of anti (WC) and syn (HG) structures within the time frame of our simulations.
Figure 4.

A. Free energy profile for rotation around the glycosidic torsion angle χ in the Hoogsteen control structure with incoming dCTP+. Minima correspond to the syn (33°) and anti (217°) domains, with syn ∼5 kcal/mol lower in energy.
B. Free energy profile for rotation around the glycosidic torsion angle χ in the Hoogsteen control structure with incoming unprotonated dCTP. Minima correspond to the syn(43°) and anti (211°) domains, with anti ∼3.5 kcal/mol lower in energy.
Discussion
The discovery that polι utilizes HG pairing in the active site to select the newly incorporated nucleotide showed that DNA polymerases can be more versatile than was previously believed. In light of previous work on accurate dG-AAF bypass by polι (49) we investigated how polι incorporated a correct partner opposite dG-AAF. Preliminary modeling showed that a syn conformation of the dG-AAF would place the bulky AAF fluorenyl rings on the cramped minor groove side in close contact with the enzyme, while an anti conformation was free of steric hindrance. Accordingly, we hypothesized that WC base pairing was necessary for selection and incorporation of dCTP opposite dG-AAF. A series of MD simulations based on an extensive search for optimal initial structures strongly supported this hypothesis.
Rationale for a WC conformation of dG-AAF in polι
Polι is unique in many ways. It is the least accurate, least processive polymerase discovered to date (8, 10, 11). In addition, it is the only polymerase observed to use a HG paired syn templating base in the active site with both undamaged and damaged DNA (13-15, 19). However, our results show WC base pairing with dG-AAF in the active site (Figure 3, S8, S9). In this case WC pairing is consistent with the mechanism of damage bypass proposed for polι based on its crystal structures..
Polι has been shown to enable nucleotide incorporation opposite minor-groove lesions or lesions that compromise WC hydrogen bonding via a HG pair (19-25); this places the damage away from the cramped minor groove side of the protein and into the spacious major groove side. Our results show that AAF, a bulky major groove lesion, is also readily placed into the same spacious major groove region in order to facilitate nucleotide incorporation opposite the damaged base (Figure 3D, S8, S9).
In addition, the series of binary and ternary crystal structures of polι (13-15) show that the templating base is in the anti conformation in the binary complex. The incoming nucleotide forces the templating base to rotate ∼180° around the glycosidic bond, in order to form a HG pair in the ternary complex. The energetic cost of this rotation is therefore less than the cost of adjusting the protein to accommodate the increased C1′-C1′ distance required for WC pairing. However, if there were a large, bulky major-groove lesion on the templating base, such as AAF, the cost of the rotation would increase greatly. This alters the energy landscape, and would plausibly make remaining in the anti conformation more energetically favorable than rotating to syn. The results observed by Choi and Guengerich with increasingly bulky adducts linked to N2 of guanine (25) may stem from a similar effect. Though polι can incorporate dCTP opposite these lesions, the efficiency of incorporation decreases with increasing size of the lesion, with strong effects seen even for N2-methyl-dG. This is hard to explain, as the syn conformation would place these adducts on the major groove side of the enzyme, which is spacious enough to accommodate adducts as large as AAF. However, the energetic cost of rotating the damaged dG through an area occupied by either the protein or the nascent DNA duplex would increase with the bulk of the damage. This could account for the observed decrease in efficiency for increasingly bulky dG N2 adducts, which could block incorporation if they remained on the cramped minor groove side.
Structural features of AAF and unmodified control simulations suggest that WC pairing is preferred for incorporation opposite dG-AAF, but not undamaged dG
Our simulations for the unmodified control WC systems suggest that this pairing scheme is unlikely without the dG-AAF lesion. Our first unmodified control Watson-Crick structure was facilitated by a stacking interaction between His354 and the 5′ dT overhang on the templating strand that stabilized a WC compatible C1′-C1′ distance (Figure 3C). This 5′ overhang stacking appears to be infeasible with a longer single-stranded template region, as would be found in a replicative context. A second simulation with an unmodified WC pair derived from a WC dG-AAF simulation showed similar structural rearrangements, further supporting the idea that WC pairing is only possible with conformations placing the 5′ single stranded overhang on the major groove side of the templating base, away from the polymerase. The widened C1′-C1′ distance needed for WC pairing is stabilized by this abnormal position of the overhang. In polι the force on the nascent WC base pair due to the long C1′-C1′ distance is alleviated by a reorientation of the template backbone and the movement of the 5′ dT overhang to the major groove side. Addition of another base or bases on the 5′ side of the templating dG would not much alter this; however if the single-stranded overhang were very long, as in a replicative situation, this force would have to be alleviated in a different way, such as by rotating to a syn conformation.
Biochemical experiments with 7-deazaguanine as the templating base support the unlikelihood of Watson-Crick pairing in the unmodified case (26). Though the 7-deazaguanine is capable of WC pairing, HG pairing is disrupted. Nonetheless, nucleotide incorporation opposite the 7-deazaguanine by polι is severely impaired, indicating a strong dependence upon HG pairing.
Since the crystal structure (14) reveals a protonated syn dCTP+ the protonated state is clearly favored energetically for the syn conformation; the added hydrogen bond contributes to the energy needed for protonation, which likely occurs in the syn domain just as proper alignment for HG base pairing is achieved. The anti conformation, however, would disfavor protonation, since no structural advantage arises. Experimental investigations of the pKa of N3 protonated cytosine in triple helices containing HG base pairs (83-85) indicate that the N3 pKa is ∼3-5 units above that of free cytosine (∼4.3) (86). This corresponds to a stabilization energy range of ∼4-7 kcal/mol for the N3 in HG paired cytosine, with the higher value appearing more probable according to the pKa data (83-84). Clearly many factors (83-85) govern this value. Our energy analyses by umbrella sampling over the glycosidic torsion angle χ for dCTP+ and dCTP (Figure 4) were notable in indicating energy wells for the anti conformation in both cases. With the pKa data and the concept that the protonation and the pKa change occur just in time to facilitate HG pairing in the syn conformation one could propose that the syn well in Figure 4B is lowered by ∼4-7 kcal/mol by protonation. This would give an energy difference between syn dG-dCTP+ and anti dG-dCTP ranging between 0.5 and 3.5 kcal/mol, in favor of syn dG-dCTP+, with the higher value being more likely. Thus the WC conformation is unlikely to occur in the unmodified case, consistent with the ternary crystal structures and biochemical data (13-15, 19, 26, 27).
However, our simulations suggest that with the presence of the AAF lesion WC pairing becomes favorable. In the WC dG-AAF simulations WC pairing accommodates the bulky AAF moiety on the spacious major groove side of the enzyme. The high near reaction-ready occupancies we observed for the WC-AAF simulations suggest that these structures are plausible (Table 1).
Examination of the MD ensembles for the WC-AAF simulations sheds some light on the changes necessary in the enzyme in order to accommodate a WC dG-AAF/dCTP pair in the active site. In their series of crystallographic studies of polι Nair et al. (13-15, 19) note the importance of three amino acids in the fingers domain (Gln59, Lys60, and Leu62) that hold the sugar of the templating base in a small hydrophobic cavity, leading to a short C1′-C1′ distance. This in turn forces the templating base to assume a syn conformation to avoid clashing with the incoming nucleotide. In the unmodified WC control structures the sugar of dG loses Van der Waals contacts with these three residues, allowing an increase in the C1′-C1′ distance (Figure 3C). However, in the WC-AAF simulations the sugar remains in the cavity, and the increased C1′-C1′ distance is enabled by a slight conformational change in the fingers domain of residues Gln59, Lys60, and Leu62 (Figure 3D). Residues Gln59, Lys60, and Leu62 move ∼2Å relative to the remainder of the fingers domain and the active site in order to accommodate a wider C1′-C1′ distance in the WC-AAF simulations. The active site is otherwise essentially unchanged. Thus, in the case of a damaged dG-AAF template, WC pairing is allowed without sacrificing other important contacts between the nascent base pair and the active site. Two additional hydrogen bonds in the active site may help compensate for the energetic cost of rearranging the enzyme to allow a wider C1′-C1′ distance: one additional hydrogen bond in the nascent base pair and a hydrogen bond between the anti templating dG and Gln59, not possible in the syn conformation (Figure 3C, D). The unmodified WC controls, while also manifesting these hydrogen bonds, assume a conformation unlikely in a replicative context and loses significant DNA/protein contacts (Figure 3C).
HG paired dG-AAF structures appear unlikely to lead to nucleotide incorporation due to numerous active site distortions
In contrast to the well-ordered active sites observed in the WC-AAF simulations the HG-AAF simulations show a disparate collection of distortions, all of which would impede incorporation of dCTP+ opposite the lesion (Figure 3B, S8, S9). As a result all of the HG-AAF structures have a near reaction-ready occupancy of 0% (Table 1). These distortions include loss of Mg2+ coordination, loss of hydrogen bonding between incoming dCTP+ and template as compared to the unmodified control HG simulation, dramatically increased Pα-O3′ distances, and displacement of the incoming nucleotide away from the template (Figure 3B, S8, S9, Tables S6, S8). None of the four initial simulations yielded a near reaction-ready structure, indicating that the observed incorporation of dCTP/dCTP+ opposite dG-AAF (49) would not likely result from an HG pair. Just as polι uses a syn conformation for the template to put bulky minor groove adducts on the spacious major groove side, our results suggest that it can use an anti conformation to place bulky major groove adducts such as dG-AAF on the major groove side as well.
Coordination of Mg2+ ions in polι is imperfect
All of our simulations, both with damaged and undamaged DNA, show somewhat impaired Mg2+ coordination of either the catalytic Mg2+ ion, MgA2+, or the nucleotide binding Mg2+ ion, MgB2+ (Figure S6, S7, Table S8). In addition, recent results show that polι has higher fidelity and processivity with Mn2+ (24, 87), which has a looser coordination requirement than Mg2+, as the catalytic ion. Together these results may indicate that the polι active site does not favor ideal coordination of the catalytic Mg2+ ions. This is consistent with polι's slow rate of nucleotide incorporation with Mg2+ ions(8, 10). Our simulations support this insofar as no simulation showed perfect octahedral coordination of both Mg2+ ions simultaneously at any time. However, current force field issues concerning proper treatment of Mg2+ ions is still a frontier issue (88). In this connection, simulations of large systems such as in the present work are necessarily subject to current approximations in the state of the art including force field and sampling considerations (89).
Conclusion
Our results elucidate how human DNA polymerase ι is able to incorporate a correct partner opposite a major-groove lesion. Primer extension results show that polι is capable of nucleotide incorporation opposite structurally diverse lesions, including a variety of minor groove adducts (20-22, 25) major groove adducts (90), as well as thymine dimers (30, 91-92), and abasic sites (10, 49, 93). Our results lend insight into this process, explaining in structural terms how polι correctly incorporates dCTP opposite a major-groove dG-AAF lesion. Though polι's function in vivo has been difficult to discern, recent results indicating that polι may be responsible for the dramatically increased cancer rate of XPV patients (94), who lack functional polη, show that understanding polι on a structural level may relate to human health. If polι must substitute for polη in XPV patients, it may be called upon to contribute to bypass of a variety of lesions, both major- and minor-groove, in cells that have lost the function of other, more reliable bypass polymerases. In conclusion, our study suggests an expanded role for polι in lesion bypass that could include both major and minor groove adducts, utilizing both WC and HG base pairing to place bulky lesions on the spacious major groove side.
Supplementary Material
Acknowledgements
We thank Dr. Lei Jia for careful reading of this manuscript and helpful suggestions. We also wish to acknowledge excellent suggestions provided by one of the reviewers. Figures were created with Pymol (DeLano Scientific LLC) and Matlab (The Mathworks, Inc).
Abbreviations
- Polι
Human DNA polymerase iota
- dCTP+
2′-deoxycytidine 5′-triphosphate protonated at N3
- dG-AAF
N-(deoxyguanosin-8-yl)-2-acetylaminofluorene
- HG
Hoogsteen
- WC
Watson-Crick
- dNTP
deoxyribonucleotide 5′-triphosphate
- AAF
2-acetylaminofluorene
- dG-AF
N-(deoxyguanosin-8-yl)-2-aminofluorene
- MD
molecular dynamics
- PMF
potential of mean force
- ns
nanoseconds
- ps
picoseconds
- fs
femtoseconds
- WHAM
weighted histogram analysis method
Footnotes
This research is supported by NIH CA75449 to S.B. and R.S., and by the National Science Foundation through TeraGrid resources provided by the San Diego Supercomputer Center. Support for computational infrastructure and systems management was also provided by NIH CA28038 to S.B. and R.S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Supporting Information Available. Figures: S1- Initial dG-AAF conformations; S2-RMSD vs. time plot; S3- Torsion angles vs. time plot; S4- C1′-C1′ and Pα-03′ distances vs. time plot; S5- Base stacking; S6 & S7- Mg2+ coordination distances vs. time plots; S8- Most representative structure active site stereo views; S9- Most representative structure whole enzyme stereo views; S10- WC-AAF3 active site with hydrogen bonds; S11- Stereo views of the active site structures shown in Figure 3. Tables: S1- Initial torsions for all structures; S2-S4- AMBER parameters; S5-S7- Hydrogen bond occupancies; S8- Average Mg2+ coordination distances; S9- Torsion angle ensemble average values. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nat. Rev. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 2.Luch A. The mode of action of organic carcinogens on cellular structures. EXS. 2006:65–95. doi: 10.1007/3-7643-7378-4_4. [DOI] [PubMed] [Google Scholar]
- 3.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol. Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann AR. Translesion synthesis in mammalian cells. Exp. Cell Res. 2006;312:2673–2676. doi: 10.1016/j.yexcr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amsterdam) 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 6.McDonald JP, Rapic-Otrin V, Epstein JA, Broughton BC, Wang X, Lehmann AR, Wolgemuth DJ, Woodgate R. Novel human and mouse homologs of Saccharomyces cerevisiae DNA polymerase η. Genomics. 1999;60:20–30. doi: 10.1006/geno.1999.5906. [DOI] [PubMed] [Google Scholar]
- 7.Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-family of DNA polymerases. Mol. Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 8.Tissier A, McDonald JP, Frank EG, Woodgate R. Polι, a remarkably error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650. [PMC free article] [PubMed] [Google Scholar]
- 9.Vaisman A, Lehmann AR, Woodgate R. DNA polymerases η and ι. Adv. Protein Chem. 2004;69:205–228. doi: 10.1016/S0065-3233(04)69007-3. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase ι. Mol. Cell. Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Replication by human DNA polymerase ι occurs by Hoogsteen base-pairing. Nature. 2004;430:377–380. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- 14.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Human DNA polymerase ι incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure. 2005;13:1569–1577. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase ι active site. Structure. 2006;14:749–755. doi: 10.1016/j.str.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 17.Lone S, Townson SA, Uljon SN, Johnson RE, Brahma A, Nair DT, Prakash S, Prakash L, Aggarwal AK. Human DNA polymerase κ encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Alt A, Lammens K, Chiocchini C, Lammens A, Pieck JC, Kuch D, Hopfner KP, Carell T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase η. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 19.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase ι. Nat. Struct. Mol. Biol. 2006;13:619–625. doi: 10.1038/nsmb1118. [DOI] [PubMed] [Google Scholar]
- 20.Rechkoblit O, Zhang Y, Guo D, Wang Z, Amin S, Krzeminsky J, Louneva N, Geacintov NE. Trans-lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 2002;277:30488–30494. doi: 10.1074/jbc.M201167200. [DOI] [PubMed] [Google Scholar]
- 21.Washington MT, Minko IG, Johnson RE, Wolfle WT, Harris TM, Lloyd RS, Prakash S, Prakash L. Efficient and error-free replication past a minor-groove DNA adduct by the sequential action of human DNA polymerases ι and κ. Mol. Cell. Biol. 2004;24:5687–5693. doi: 10.1128/MCB.24.13.5687-5693.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfle WT, Johnson RE, Minko IG, Lloyd RS, Prakash S, Prakash L. Human DNA polymerase ι promotes replication through a ring-closed minor-groove adduct that adopts a syn conformation in DNA. Mol. Cell. Biol. 2005;25:8748–8754. doi: 10.1128/MCB.25.19.8748-8754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson RE, Yu SL, Prakash S, Prakash L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol. Cell. Biol. 2007;27:7198–7205. doi: 10.1128/MCB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. Eukaryotic Y-family polymerases bypass a 3-methyl-2′-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JY, Guengerich FP. Kinetic evidence for inefficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase ι. J. Biol. Chem. 2006;281:12315–12324. doi: 10.1074/jbc.M600112200. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RE, Prakash L, Prakash S. Biochemical evidence for the requirement of Hoogsteen base pairing for replication by human DNA polymerase ι. Proc. Natl. Acad. Sci. U.S.A. 2005;102:10466–10471. doi: 10.1073/pnas.0503859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson RE, Haracska L, Prakash L, Prakash S. Role of Hoogsteen edge hydrogen bonding at template purines in nucleotide incorporation by human DNA polymerase ι. Mol. Cell. Biol. 2006;26:6435–6441. doi: 10.1128/MCB.00851-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potapova O, Chan C, DeLucia AM, Helquist SA, Kool ET, Grindley ND, Joyce CM. DNA polymerase catalysis in the absence of Watson-Crick hydrogen bonds: analysis by single-turnover kinetics. Biochemistry. 2006;45:890–898. doi: 10.1021/bi051792i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizukami S, Kim TW, Helquist SA, Kool ET. Varying DNA base-pair size in subangstrom increments: evidence for a loose, not large, active site in low-fidelity Dpo4 polymerase. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 30.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 31.Wilson RH, DeEds F, Cox AJ. The toxicity and carcinogenicity of 2-acetylaminofluorene. Cancer Res. 1941;1:595–608. [Google Scholar]
- 32.Heflich RH, Neft RE. Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat. Res. 1994;318:73–114. doi: 10.1016/0165-1110(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 33.Kriek E. Fifty years of research on N-acetyl-2-aminofluorene, one of the most versatile compounds in experimental cancer research. J. Cancer Res. Clin. Oncol. 1992;118:481–489. doi: 10.1007/BF01225261. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki N, Ohashi E, Hayashi K, Ohmori H, Grollman AP, Shibutani S. Translesional synthesis past acetylaminofluorene-derived DNA adducts catalyzed by human DNA polymerase κ and Escherichia coli DNA polymerase IV. Biochemistry. 2001;40:15176–15183. doi: 10.1021/bi010702g. [DOI] [PubMed] [Google Scholar]
- 35.Doisy R, Tang MS. Effect of aminofluorene and (acetylamino)fluorene adducts on the DNA replication mediated by Escherichia coli polymerases I (Klenow fragment) and III. Biochemistry. 1995;34:4358–4368. doi: 10.1021/bi00013a027. [DOI] [PubMed] [Google Scholar]
- 36.Belguise-Valladier P, Maki H, Sekiguchi M, Fuchs RP. Effect of single DNA lesions on in vitro replication with DNA polymerase III holoenzyme. Comparison with other polymerases. J. Mol. Biol. 1994;236:151–164. doi: 10.1006/jmbi.1994.1125. [DOI] [PubMed] [Google Scholar]
- 37.Shibutani S, Suzuki N, Grollman AP. Mutagenic specificity of (acetylamino)fluorene-derived DNA adducts in mammalian cells. Biochemistry. 1998;37:12034–12041. doi: 10.1021/bi981059+. [DOI] [PubMed] [Google Scholar]
- 38.Gillet LC, Alzeer J, Scharer OD. Site-specific incorporation of N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) into oligonucleotides using modified ‘ultra-mild’ DNA synthesis. Nucleic Acids Res. 2005;33:1961–1969. doi: 10.1093/nar/gki335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunz D, Hess MT, Naegeli H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J. Biol. Chem. 1996;271:25089–25098. doi: 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- 40.Shibutani S, Suzuki N, Tan X, Johnson F, Grollman AP. Influence of flanking sequence context on the mutagenicity of acetylaminofluorene-derived DNA adducts in mammalian cells. Biochemistry. 2001;40:3717–3722. doi: 10.1021/bi0027581. [DOI] [PubMed] [Google Scholar]
- 41.Fuchs RP, Fujii S. Translesion synthesis in Escherichia coli: lessons from the NarI mutation hot spot. DNA Repair (Amsterdam) 2007;6:1032–1041. doi: 10.1016/j.dnarep.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 42.Fuchs RP, Koffel-Schwartz N, Pelet S, Janel-Bintz R, Napolitano R, Becherel OJ, Broschard TH, Burnouf DY, Wagner J. DNA polymerases II and V mediate respectively mutagenic (−2 frameshift) and error-free bypass of a single N2-acetylaminofluorene adduct. Biochem. Soc. Trans. 2001;29:191–195. doi: 10.1042/0300-5127:0290191. [DOI] [PubMed] [Google Scholar]
- 43.Tan X, Suzuki N, Grollman AP, Shibutani S. Mutagenic events in Escherichia coli and mammalian cells generated in response to acetylaminofluorene-derived DNA adducts positioned in the Nar I restriction enzyme site. Biochemistry. 2002;41:14255–14262. doi: 10.1021/bi0202878. [DOI] [PubMed] [Google Scholar]
- 44.O'Handley SF, Sanford DG, Xu R, Lester CC, Hingerty BE, Broyde S, Krugh TR. Structural characterization of an N-acetyl-2-aminofluorene (AAF) modified DNA oligomer by NMR, energy minimization, and molecular dynamics. Biochemistry. 1993;32:2481–2497. doi: 10.1021/bi00061a005. [DOI] [PubMed] [Google Scholar]
- 45.Patel DJ, Mao B, Gu Z, Hingerty BE, Gorin A, Basu AK, Broyde S. Nuclear magnetic resonance solution structures of covalent aromatic amine-DNA adducts and their mutagenic relevance. Chem.Res.Toxicol. 1998;11:391–407. doi: 10.1021/tx9702143. [DOI] [PubMed] [Google Scholar]
- 46.Cho BP, Zhou L. Probing the conformational heterogeneity of the acetylaminofluorene-modified 2′-deoxyguanosine and DNA by 19F NMR spectroscopy. Biochemistry. 1999;38:7572–7583. doi: 10.1021/bi990182d. [DOI] [PubMed] [Google Scholar]
- 47.Cho BP. Dynamic conformational heterogeneities of carcinogen-DNA adducts and their mutagenic relevance. J. Environ. Sci. Health. 2004;22:57–90. doi: 10.1081/LESC-200038217. [DOI] [PubMed] [Google Scholar]
- 48.Evans FE, Miller DW, Levine RA. 1H NMR study of self-association and restricted internal rotation of the C8-substituted deoxyguanosine 5′-monophosphate adduct of the carcinogen 2-(acetylamino)fluorene. J. Biomol. Struct. Dyn. 1986;3:935–948. doi: 10.1080/07391102.1986.10508475. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z. Response of human DNA polymerase ι to DNA lesions. Nucleic Acids Res. 2001;29:928–935. doi: 10.1093/nar/29.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grunberger D, Nelson JH, Cantor CR, Weinstein IB. Coding and conformational properties of oligonucleotides modified with the carcinogen N2--acetylaminofluorene. Proc. Natl. Acad. Sci. U.S.A. 1970;66:488–494. doi: 10.1073/pnas.66.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuchs R, Daune M. Physical studies on deoxyribonucleic acid after covalent binding of a carcinogen. Biochemistry. 1972;11:2659–2666. doi: 10.1021/bi00764a017. [DOI] [PubMed] [Google Scholar]
- 52.Fuchs R, Daune M. Changes of stability and conformation of DNA following the covalent binding of a carcinogen. FEBS Letters. 1971;14:206–208. doi: 10.1016/0014-5793(71)80618-x. [DOI] [PubMed] [Google Scholar]
- 53.Milhe C, Dhalluin C, Fuchs RP, Lefevre JF. NMR evidence of the stabilisation by the carcinogen N2-acetylaminofluorene of a frameshift mutagenesis intermediate. Nucleic Acids Res. 1994;22:4646–4652. doi: 10.1093/nar/22.22.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Broyde S. A new anti conformation for N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (AAF-dG) allows Watson-Crick pairing in the Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) Nucleic Acids Res. 2006;34:785–795. doi: 10.1093/nar/gkj479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 58.van Meerssche M, Germain G, Declercq J-P, Touillaux R, Roberfroid M, Razzouk C. 2-(Acetylamino)fluorene, C15H13NO. Acta Crystallogr. C Cryst. Struct. Commun. 1980;9:515–518. [Google Scholar]
- 59.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 60.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 61.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 62.Cheatham TE, 3rd, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 63.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 64.Hehre WJ, Ditchfie R, Pople JA. Self-consistent molecular-orbital methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972;56:2257–2261. [Google Scholar]
- 65.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J, J. A. Vreven, T., Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Guassian 03. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 66.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 67.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the multimolecule and multiconformational RESP methodology to biopolymers-charge derivation for DNA, RNA, and proteins. J. Chem. Phys. 1995;16:1357–1377. [Google Scholar]
- 68.Perlow RA, Broyde S. Toward understanding the mutagenicity of an environmental carcinogen: structural insights into nucleotide incorporation preferences. J. Mol. Biol. 2002;322:291–309. doi: 10.1016/s0022-2836(02)00751-9. [DOI] [PubMed] [Google Scholar]
- 69.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 70.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 8. University of California; San Francisco: 2004. [Google Scholar]
- 71.Mezei M. Optimal position of the solute for simulations. J. Comput. Chem. 1997;18:812–815. [Google Scholar]
- 72.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 73.Darden T, York D, Pedersen L. Particle mesh Ewald-an nlog (n) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 74.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 75.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian equations of motion of a system with constraints-molecular-dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 76.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 77.Harvey SC, Tan RKZ, Cheatham TE. The flying ice cube: Velocity rescaling in molecular dynamics leads to violation of energy equipartition. J. Comput. Chem. 1998;19:726–740. [Google Scholar]
- 78.Simmerling C, Elber R, Zhang J, Pullman A, et al., editors. Modeling of Biomolecular Structure and Mechanisms. Kluwer; Netherlands: 1995. MOIL-View -A program for visualization of structure and dynamics of biomolecules and STO-A program for computing stochastic paths; pp. 241–265. [Google Scholar]
- 79.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang L, Yu X, Hu P, Broyde S, Zhang Y. A water-mediated and substrate-assisted catalytic mechanism for Sulfolobus solfataricus DNA polymerase IV. J. Am. Chem. Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shankar K, Djamal B, Robert HS, Peter AK, John MR. The weighted histogram analysis method for free-energy calculations on biomolecules. I: The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 82.Saenger W. Principles of nucleic acid structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 83.Plum GE, Breslauer KJ. Thermodynamics of an intramolecular DNA triple helix: a calorimetric and spectroscopic study of the pH and salt dependence of thermally induced structural transitions. J. Mol. Biol. 1995;248:679–695. doi: 10.1006/jmbi.1995.0251. [DOI] [PubMed] [Google Scholar]
- 84.Asensio JL, Lane AN, Dhesi J, Bergqvist S, Brown T. The contribution of cytosine protonation to the stability of parallel DNA triple helices. J. Mol. Biol. 1998;275:811–822. doi: 10.1006/jmbi.1997.1520. [DOI] [PubMed] [Google Scholar]
- 85.Wu P, Kawamoto Y, Hara H, Sugimoto N. Effect of divalent cations and cytosine protonation on thermodynamic properties of intramolecular DNA double and triple helices. J. Inorg. Biochem. 2002;91:277–285. doi: 10.1016/s0162-0134(02)00444-0. [DOI] [PubMed] [Google Scholar]
- 86.Zimmer C, Luck G, Venner H, Fric J. Studies on the conformation of protonated DNA. Biopolymers. 1968;6:563–574. doi: 10.1002/bip.1968.360060410. [DOI] [PubMed] [Google Scholar]
- 87.Frank EG, Woodgate R. Increased catalytic activity and altered fidelity of human DNA polymerase i in the presence of manganese. J. Biol. Chem. 2007;282:24689–24696. doi: 10.1074/jbc.M702159200. [DOI] [PubMed] [Google Scholar]
- 88.Oelschlaeger P, Klahn M, Beard WA, Wilson SH, Warshel A. Magnesium-cationic dummy atom molecules enhance representation of DNA polymerase beta in molecular dynamics simulations: improved accuracy in studies of structural features and mutational effects. J. Mol. Biol. 2007;366:687–701. doi: 10.1016/j.jmb.2006.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Adcock SA, McCammon JA. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frank EG, Sayer JM, Kroth H, Ohashi E, Ohmori H, Jerina DM, Woodgate R. Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase ι. Nucleic Acids Res. 2002;30:5284–5292. doi: 10.1093/nar/gkf643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tissier A, Frank EG, McDonald JP, Iwai S, Hanaoka F, Woodgate R. Misinsertion and bypass of thymine-thymine dimers by human DNA polymerase ι. EMBO J. 2000;19:5259–5266. doi: 10.1093/emboj/19.19.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vaisman A, Takasawa K, Iwai S, Woodgate R. DNA polymerase ι-dependent translesion replication of uracil containing cyclobutane pyrimidine dimers. DNA Repair (Amsterdam) 2006;5:210–218. doi: 10.1016/j.dnarep.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 93.Vaisman A, Frank EG, McDonald JP, Tissier A, Woodgate R. Polι-dependent lesion bypass in vitro. Mutat. Res. 2002;510:9–22. doi: 10.1016/s0027-5107(02)00248-8. [DOI] [PubMed] [Google Scholar]
- 94.Wang Y, Woodgate R, McManus TP, Mead S, McCormick JJ, Maher VM. Evidence that in xeroderma pigmentosum variant cells, which lack DNA polymerase η, DNA polymerase ι causes the very high frequency and unique spectrum of UV-induced mutations. Cancer Res. 2007;67:3018–3026. doi: 10.1158/0008-5472.CAN-06-3073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.