

Abstract
Airway epithelial cells act as the first barrier against pathogens. These cells recognize conserved structural motifs expressed by microbial pathogens via Toll-like receptors (TLRs) expressed on the surface. In contrast to the level of expression in lymphoid cells, the level of expression of TLR2 and TLR4 in airway epithelial cells is low under physiological conditions. Here we explored whether Klebsiella pneumoniae upregulates the expression of TLRs in human airway epithelial cells. We found that the expression of TLR2 and TLR4 by A549 cells and human primary airway cells was upregulated upon infection with K. pneumoniae. The increased expression of TLRs resulted in enhancement of the cellular response upon stimulation with Pam3CSK4 and lipopolysaccharide, which are TLR2 and TLR4 agonists, respectively. Klebsiella-dependent upregulation of TLR expression occurred via a positive IκBα-dependent NF-κΒ pathway and via negative p38 and p44/42 mitogen-activated protein kinase-dependent pathways. We showed that Klebsiella-induced TLR2 and TLR4 upregulation was dependent on TLR activation. An isogenic capsule polysaccharide (CPS) mutant did not increase TLR2 and TLR4 expression. Purified CPS upregulated TLR2 and TLR4 expression, and polymyxin B did not abrogate CPS-induced TLR upregulation. Although no proteins were detected in the CPS preparation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and colloidal gold staining, we could not rule out the possibility that traces of protein in our CPS preparation could have been responsible, at least in part, for the TLR upregulation.
The lung is constantly exposed to potential pathogens. To cope with this, the lung has sophisticated defense mechanisms designed to clear pathogens without perturbing functionality. Mechanical defenses, such as coughing and mucociliary movement, are important for removing particulate matter from the tracheobronchial tree (39). Resident and recruited phagocytes in the lower respiratory tract and the alveoli eliminate particulate matter and pathogens that escape the first line of defense. In this scenario, airway epithelial cells detect pathogens and produce mediators important for the activation of innate and adaptive immune responses (35).
Airway epithelial cells recognize conserved structural motifs expressed by microbial pathogens, the so-called pathogen-associated molecular patterns (PAMPs). Toll-like receptors (TLRs), which are the major epithelial PAMP receptors (3), are a set of germ line-encoded receptors belonging to the pattern recognition receptor family (33). Activation of TLRs leads to induction of direct antimicrobial pathways, expression of costimulatory molecules, and release of cytokines and chemokines that influence airway inflammation (6, 7). TLR2 and TLR4 are the best-studied TLRs. While TLR4 seems to be involved mainly in the detection of lipopolysaccharide (LPS), TLR2 responds to a variety of gram-positive products (33, 70). Studies using TLR2 or TLR4 knockout mice have clearly shown the importance of both of these receptors for clearance of pulmonary infections (14, 37, 38, 44, 53, 61, 78).
Although under physiological conditions TLRs are expressed at low levels in airway epithelial cells, their expression is upregulated under inflammatory conditions (31, 60, 67) and during infection with pathogens such as nontypeable Haemophilus influenzae (67). These findings might have important implications in lung defense. First, the cellular responses to pathogens could be controlled by regulating the amount of TLR protein expressed. There have been several reports demonstrating that there is a correlation between the amount of TLR produced by a given cell and the amount of inflammatory mediators secreted (1, 2, 32, 47, 52). Second, the elevated levels of TLRs could contribute to the accelerated immune response of airway epithelial cells and also to resensitization of cells to pathogens, which may trigger an excessive inflammatory response.
Klebsiella pneumoniae causes a wide range of infections from urinary tract infections to pneumonia and is particularly devastating in immunocompromised patients, whose mortality rates are between 25 and 60% (59). The high prevalence of multidrug-resistant strains further complicates treatment of these infections (73). Capsule polysaccharide (CPS) is recognized as one of the most important virulence factors of this bacterium. CPS-deficient mutants do not colonize the mouse bladder as well as the wild-type strain (68), and various studies have shown that CPS-deficient mutants are unable to colonize pulmonary and systemic tissues (20, 41). In vitro studies have shown that the presence of CPS inhibits deposition of the complement component C3 onto the bacterium (4, 19, 21), mediates resistance to antimicrobial peptides (15), and reduces adhesion and phagocytosis of the bacterium by macrophages and epithelial cells (19, 20, 25, 50). In a recent study we showed that a CPS mutant activates cellular responses and that CPS might prevent this activation through blockage of bacterial adhesion and uptake (56). Altogether, these findings suggest that CPS plays an important role in the interaction between K. pneumoniae and the innate immune system.
Here we explored the possibility that K. pneumoniae upregulates the expression of TLRs in human airway epithelial cells via activation of specific signaling pathways. Our results show that the expression of TLR4 and TLR2 is upregulated by K. pneumoniae via a positive NF-κB signaling pathway and via negative p38 and p44/42 mitogen-activated protein (MAP) kinase pathways. Furthermore, Klebsiella-induced expression of TLRs is dependent on TLR activation. Finally, we analyzed whether CPS could be the bacterial component triggering the upregulation of TLRs.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and reagents.
K. pneumoniae 52145 is a clinical isolate (serotype O1:K2) that has been described previously (48). The isogenic mutant 52K10, which does not express CPS, was described recently (20). Bacteria were grown in Luria-Bertani medium at 37°C. When appropriate, antibiotics were added to the growth medium at the following concentrations: chloramphenicol, 25 μg/ml; and kanamycin, 20 μg/ml.
Blocking antibodies against TLR2 (clone TLR2.1 [43]) and TLR4 (clone HTA125 [64]) were purchased from Hycult Biotechnology. CAPE, an NF-κB inhibitor, and SB203580, an p38 MAP kinase inhibitor, were purchased from Sigma. U0126, a p44/42 MAP kinase inhibitor, was purchased from Calbiochem. LPS from Escherichia coli conjugated to Alexa488 was purchased from Molecular Probes. LPS purified from E. coli O111:B4 (Sigma Chemical Co.) was repurified exactly as previously described (30). The procedure used resulted in enterobacterial LPS preparations that utilized TLR4, but not TLR2, for signaling (30). Pam3CSK4 was purchased from InvivoGen.
Cell culture and infection.
Monolayers of A549 human lung carcinoma cells (ATCC CCL185) derived from type II pneumocytes were grown to 80% confluence in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum plus penicillin and streptomycin in 24-, 6-, or 96-well tissue culture plates at 37°C in a water-saturated atmosphere consisting of 95% air and 5% CO2. Primary human airway epithelial cells (NHBE) (Lonza) were maintained in bronchial epithelial basal medium (Lonza) by following the manufacturer's instructions. Before infection A549 or NHBE cells were washed three times with phosphate-buffered saline (PBS), and infection was performed using a multiplicity of infection of 100:1 unless otherwise indicated. Cell viability was assessed by trypan blue dye exclusion, and it was >95% even at 8 h postinfection.
Flow cytometry.
Monolayers of epithelial cells were detached by incubation with trypsin-EDTA and washed with 0.1% sodium azide in PBS. To analyze the expression of TLR2 and TLR4, nonpermeabilized cells were incubated with anti-TLR2 (clone TL2.1; 10 μg/ml; eBioscience) and anti-TLR4 (clone HTA125; 10 μg/ml; eBioscience) phycoerythrin-conjugated or immunoglobulin G2a isotype κ-labeled antibodies. Cells were incubated with the antibodies at room temperature (22 to 25°C) for 15 min. Analyses were performed using a Cultek Epics XL flow cytometer. At least 9,000 cells were acquired in every experiment. The levels of TLR2 and TLR4 were expressed as the relative mean fluorescence intensity (rmfi) in arbitrary units (AU), and the nonspecific binding was corrected by subtraction of the mean fluorescence intensity (mfi) values for isotype-matched antibodies. Experiments were performed in duplicate and repeated four times.
Preparation of nuclear and cytosolic extracts.
Nuclear and cytosolic proteins were extracted from approximately 5 × 106 cells. Briefly, A549 cells were washed with ice-cold PBS and suspended in hypotonic buffer (10 mM HEPES, 10 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, 0.1 mM EDTA, 0.2 mM NaF, 0.2 mM Na3VO4, 1 μg/ml leupeptin, 0.4 mM phenylmethylsulfonyl fluoride). The cells were left on ice for 15 min, and a cytosolic preparation was obtained by addition of Nonidet P-40 (final concentration, 0.1%), followed by centrifugation (4,000 × g, 3 min, 4°C). The supernatant was collected and used as the cytosolic fraction. The remaining pellet was resuspended in extraction buffer (50 mM HEPES, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, 10% glycerol, 0.2 mM NaF, 0.2 mM Na3VO4, 0.1 mM phenylmethylsulfonyl fluoride) and incubated on ice for 20 min with agitation. The nuclear proteins in the supernatant were recovered after centrifugation (12,000 × g, 20 min, 4°C) to remove nuclear debris. Proteins present in the extracts were quantified using the Coomassie blue protein assay (Pierce) with bovine serum albumin as a standard and were stored in aliquots at −80°C.
NF-κB assay.
NF-κB activation was measured using a TransAM NF-κB enzyme-linked immunosorbent assay (ELISA) kit (Active Motif). A549 cells seeded into 60-mm tissue culture dishes were infected for different times. After infection, cells were washed twice with ice-cold PBS. To each well, 3 ml of cold PBS was added in order to collect the cells by scraping with a rubber policeman. Cells were centrifuged for 10 min at 1,000 rpm at 4°C, the supernatants were discarded, and nuclear and cytosolic extracts were prepared as described above. Samples of the nuclear fractions containing 15 μg of protein were analyzed by using the NF-κB ELISA. In this assay, activated NF-κB specifically bound to the NF-κB DNA binding site that was attached at the bottom of the ELISA plate. An antibody against the p65 subunit of NF-κB detected the NF-κB complex bound to the oligonucleotide, which in turn was recognized by a secondary antibody conjugated to horseradish peroxidase. The colorimetric reaction was read at 450 nm. A tumor necrosis factor alpha-stimulated HeLa cells extract, provided with the kit, was used as a positive control for NF-κB activation. The results were expressed as means ± standard deviations of three independent experiments.
Immunoblotting.
Cytoplasmic proteins (15 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), electrotransferred to a nitrocellulose membrane, and blocked with 3% skim milk in PBS. Immunostaining for IκBα was performed with polyclonal rabbit anti-IκBα antibody (Santa Cruz Biotechnology), whereas immunostaining to assess phosphorylation of p38 and p44/42 was performed with polyclonal rabbit anti-phospho-p38 antibody and anti-phospho-p44/42 antibody (Cell Signaling), respectively. Immunoreactive bands were visualized by incubation with swine anti-rabbit immunoglobulins conjugated to horseradish peroxidase (Dako P0217) using the SuperSignal West-dura system (Pierce). Blots were reprobed with anti-human tubulin, anti-p38, or anti-p44/42 polyclonal antibody to ensure that equal amounts of proteins were loaded in the lanes. Images were recorded with a GeneGnome HR imaging system (Syngene) as jpeg files, and they were exported to a personal computer for densitometry analysis using the ImageJ software (http://rsb.info.nih.gov/ij/download.html). Bands in each lane were selected and analyzed using the Histogram analysis tool, recording the mean intensity. The results were expressed as relative levels of protein (mean intensity of protein/mean intensity of tubulin × 100).
qRT-PCR.
A549 cells seeded into 60-mm tissue culture dishes were infected for different times. After infection, A549 cells were washed with PBS, and total RNA was purified using a Nucleospin RNAII kit (Macherey-Nagel) exactly as recommended by the manufacturer. RNA integrity was verified using a formaldehyde-agarose gel, quantified spectrophotometrically with a NanoDrop, and stored at −80°C. cDNA was obtained by retrotranscription of 5 μg of total RNA using a commercial RT2 First Strand kit as recommended by the manufacturer (Superarray Bioscience Corporation). The reaction included one step to eliminate traces of genomic DNA. Real-time PCR analyses were performed with a Smart Cycler real-time PCR instrument (Cepheid, Sunnyvale, CA). To amplify human TLR2 and TLR4, 500 ng of cDNA was used as a template in a 25-μl reaction mixture containing 1× Quantitect SYBR green PCR mixture (Qiagen) and QuantiTect primer assays (catalog numbers QT00236131 [TLR2] and QT00035238 [TLR4]; Qiagen). As an internal control we amplified the human glyceraldehyde-3-phosphate dehydrogenase housekeeping gene (GAPDH) using 50 ng of cDNA and the following intron-spanning primers: sense primer 5′-GAAGGTGAAGGTCGGAGTC-3′ and antisense primer 5′-GAAGATGGTGATGGGATTTC-3′). For detection of the GAPDH, TLR2, and TLR4 genes, the following thermocycling protocol was used: 95°C for 15 min for hot-start polymerase activation, followed by 45 cycles of denaturation at 95°C for 30 s, annealing at 60°C for the TLR genes or at 54°C for the GADPH gene for 30 s, and extension at 72°C for 30 s. SYBR green dye fluorescence was measured at 521 nm during the annealing phase. The threshold cycle (CT) value reflects the cycle number at which the fluorescence generated in a reaction crosses a given threshold. The CT value assigned to each well thus reflects the point during the reaction at which a sufficient number of amplicons have been accumulated. The relative amount of mRNA in each sample was calculated based on its CT value in comparison with the CT value of GAPDH. The results were expressed as changes in gene expression as determined using the following formula: 2 − [(CT of TLR genes − CT of GAPDH gene of infected cells)/(CT of TLR genes − CT of GAPDH gene of control cells)] (with CT values in arbitrary units). The specificity of the PCR products was determined by performing a melting curve analysis, and amplification products were resolved on a 1.5% agarose gel to confirm the correct sizes of the amplicons (92, 102, and 226 bp for TLR2, TLR4, and GAPDH, respectively). cDNAs were obtained from three independent extracts of mRNA, and each cDNA was amplified by quantitative reverse transcription (qRT)-PCR on two independent occasions.
Cytokine stimulation assay.
Epithelial cell monolayers were stimulated with different concentrations of either purified LPS or Pam3CSK4, and after 18 h supernatants were carefully removed from the wells, cell debris was removed by centrifugation, and samples were frozen at −80°C. The levels of interleukin-8 (IL-8) in the supernatants were determined by performing an ELISA with a commercial kit (Endogen) with a sensitivity of <2 pg/ml. The results were expressed as means ± standard deviations. Experiments were performed in duplicate and were repeated at least three times.
CPS purification.
Cell-bound CPS was purified by the phenol-water method (77). Briefly, bacteria were grown in Luria-Bertani medium in 2-liter flasks (1 liter per flask) on an orbital shaker (180 rpm) for 24 h at 37°C. Cells were removed by centrifugation and washed once with PBS. The pellet was extracted with phenol, and the polysaccharides present in the aqueous phase were precipitated by adding 5 volumes of methanol plus 1% (vol/vol) of a saturated solution of sodium acetate in methanol. After incubation for 24 h at −20°C, a pellet was recovered by centrifugation, dissolved in distilled water, dialyzed against water, and freeze-dried. To purify this preparation, it was dispersed (final concentration, 10 mg/ml) in 0.8% NaCl-0.05% NaN3-0.1 M Tris-HCl (pH 7) and digested with nucleases (50 mg/ml of DNase II type V and RNase A [Sigma Chemical Co., St. Louis, MO]) for 18 h at 37°C. Proteinase K (50 mg/ml; E. Merck, Darmstadt, Germany) was added, and the mixture was incubated for 1 h at 55°C and for 24 h at room temperature. The proteinase K digestion procedure was repeated twice, and the polysaccharides were precipitated as described above. The pellet was recovered by centrifugation and dissolved in distilled water. The LPS was removed by ultracentrifugation (105,000 × g, 16 h, 4°C), and samples were freeze-dried. The enzymatic treatment and ultracentrifugation steps were repeated one more time.
CPS was quantified by determining the concentrations of uronic acid in the samples, using a modified carbazole assay (11) exactly as described by Rahn and Whitfield (55). The protein in the preparation was quantified using the Coomassie blue protein assay (Pierce) with bovine serum albumin as a standard. The presence of LPS was determined by measuring the 3-deoxy-d-manno-2-octulosonic acid content by the thiobarbituric acid method, which was modified to correct for interference due to deoxy sugars (22). The protein content was less than 0.5%, and the 3-deoxy-d-manno-2-octulosonic acid content was less than 0.1%.
Statistical methods.
Statistical analyses were performed using analysis of variance, the two-sample t test, or, when the requirements were not met, by the Mann-Whitney U test. A P value of <0.05 was considered statistically significant. These analyses were performed using Prism4 for PC (GraphPad Software).
RESULTS
K. pneumoniae upregulates the expression of TLR2 and TLR4 in human airway epithelial cells.
It has been shown previously that human airway epithelial cells express TLR2 and TLR4 on their surfaces, albeit at lower levels than monocytes and macrophages (5, 31). We examined whether K. pneumoniae 52145 upregulates the expression of TLR2 and TLR4 mRNA in human airway epithelial cells. As shown in Fig. 1A, the expression of TLR2 and TLR4 mRNA by A549 cells increased upon infection with K. pneumoniae. A time course experiment revealed that the expression of TLR2 peaked 4 h postinfection, whereas the expression of TLR4 peaked 2 h postinfection (Fig. 1A).
FIG. 1.

K. pneumoniae increases the expression of TLR2 and TLR4 in human airway epithelial cells. (A) A549 cells were infected with K. pneumoniae 52145 for different times, and the TLR2 and TLR4 mRNA levels were assessed by qRT-PCR. CON, noninfected cells (n = 3). (B) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells after infection with K. pneumoniae 52145 for 5 h. Iso, histogram showing the fluorescence intensity of A549 cells incubated with isotype-matched antibodies; CON, histogram showing the fluorescence intensity of A549 cells incubated with anti-TLR antibodies; Kpn, histogram showing the fluorescence intensity of infected A549 cells incubated with anti-TLR antibodies. (C) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells after infection with K. pneumoniae 52145 for 5 h. A549 cells were not infected (CON) or were infected with K. pneumoniae 52145 (Kpn). The mfi values for A549 cells incubated with isotype-matched antibodies were 10 ± 3 AU (n = 4). (D) Analysis of NHBE cells that were not infected (CON) or were infected with K. pneumoniae 52145 (Kpn). The mfi values for NHBE cells incubated with isotype-matched antibodies were 16 ± 5 AU (n = 4). The results (means and standard deviations) are expressed (in AU) as rmfi values (mfi values obtained after staining with anti-TLR antibodies − mfi values obtained after staining with isotype-matched antibodies). *, results are significantly different from the results for noninfected cells (P < 0.05).
To determine whether upregulation of TLR2 and TLR4 mRNA was accompanied by elevated protein levels, flow cytometry analyses were carried out. Figures 1B and C show that upregulation of TLR2 and TLR4 in A549 cells was observed at the protein level. Infection also upregulated the TLR2 and TLR4 levels in NHBE cells (Fig. 1D).
Increased expression of TLRs by human airway cells enhances the cell response to microbial stimuli.
Having established that K. pneumoniae infection increases the expression of TLR2 and TLR4 by airway epithelial cells, we asked whether the elevated TLR protein levels resulted in increased cellular responses to TLR agonists. To evaluate this, A549 cells were infected with K. pneumoniae, the bacteria were washed off, and gentamicin was added to kill the extracellular bacteria. After 2 h of treatment, the medium was removed, purified LPS (a TLR4 agonist) or Pam3CSK4 (a TLR2 agonist) was added to the cells, and the amount of IL-8 secreted by the cells was used as a cellular readout. Figure 2A shows that LPS induced secretion of a larger amount of IL-8 on K. pneumoniae-infected cells than on noninfected cells. These results are consistent with the higher level of binding of fluorescently labeled LPS to K. pneumoniae-infected cells than to noninfected cells (Fig. 2B). The TLR2-specific agonist also induced secretion of a larger amount of IL-8 on K. pneumoniae-infected cells than on noninfected cells (Fig. 2C). Noninfected cells and infected cells (with no LPS or Pam3CSK4 added) secreted similar amounts of IL-8 (85 ± 50 and 100 ± 60 pg/ml, respectively; P > 0.05).
FIG. 2.

K. pneumoniae infection increases TLR agonist responsiveness in A549 cells. (A) Levels of IL-8 secreted into the culture medium by A549 cells stimulated with LPS. Cells were not infected or were infected with K. pneumoniae 52145 for 5 h. After this, bacteria were washed off, and gentamicin was added to all wells. After 2 h, the medium was removed, and LPS was added. After 18 h supernatants were collected, and IL-8 was analyzed. The data are means ± standard deviations. ○, noninfected cells; •, infected cells. *, P < 0.05 (results are significantly different from the results for nontreated cells; n = 4). (B) K. pneumoniae infection increases the binding of LPS to A549 cells. Cells were not infected or were infected with K. pneumoniae 52145 for 5 h. After this, bacteria were washed off, and gentamicin was added to all wells. After 2 h, the medium were removed, and the cells were exposed to LPS conjugated to the fluorophore Alexa488 (5 μg/ml) for 1 h at 37°C. LPS binding was analyzed by flow cytometry. *, P < 0.05 (results are significantly different from the results for noninfected cells exposed to LPS); Δ, P < 0.05 (results are significantly different from the results for noninfected cells; n = 4). (C) Levels of IL-8 secreted into the culture medium by A549 cells stimulated with Pam3CSK4. The conditions were identical to those described above for panel A (n = 3). Before the agonists were added, the expression of TLR2 and TLR4 in representative wells was checked by flow cytometry, and it was significantly higher in K. pneumoniae-infected cells (TLR2 rmfi, 30 ± 7 AU; TLR4 rmfi, 24 ± 7 AU) than in noninfected cells (TLR2 rmfi, 12 ± 5 AU; TLR4 rmfi, 9 ± 3 AU). *, P < 0.05 (results are significantly different from the results for noninfected cells).
Together, these data indicate that infection of A549 cells with K. pneumoniae increases the cellular response to TLR2 and TLR4 agonists, and this is related to the infection-dependent increased expression of both TLRs.
IκBα-dependent activation of NF-κB is required for K. pneumoniae-induced TLR2 and TLR4 upregulation.
We sought to determine which intracellular pathways are involved in K. pneumoniae-induced TLR upregulation. Based on the key role of NF-κB in controlling the expression of genes involved in inflammation and immunity, we asked whether NF-κB is behind the K. pneumoniae-induced upregulation of TLRs. We studied whether infection of A549 cells with strain 52145 activated NF-κB. Figure 3A shows that nuclear fractions obtained from infected cells after 60 and 90 min contained larger amounts of active NF-κB than nuclear fractions obtained from noninfected cells (control) or from infected cells after 30 and 120 min. Next, we asked whether CAPE, a chemical inhibitor used to block the NF-κB signaling pathway (49), altered the K. pneumoniae-induced TLR upregulation. As shown in Fig. 3B, CAPE (15 μg/ml) reduced K. pneumoniae-induced TLR2 and TLR4 upregulation of protein levels. A lower concentration of the inhibitor (5 μg/ml) reduced only K. pneumoniae-induced TLR2 protein levels. CAPE-treated cells (15 μg/ml) expressed amounts of TLR2 and TLR4 (rmfi, 11 ± 7 and 10 ± 5 AU, respectively) similar to the amounts expressed by untreated cells (TLR2 rmfi, 17 ± 5 AU; TLR4 rmfi, 15 ± 3 AU). Addition of dimethyl sulfoxide (DMSO) (the vehicle used for CAPE) to K. pneumoniae-infected cells did not affect the expression of either TLR2 (rmfi in the presence of DMSO, 29 ± 7 AU; rmfi in the absence of DMSO, 30 ± 4 AU) or TLR4 (rmfi in the presence of DMSO, 23 ± 7 AU; rmfi in the absence of DMSO, 32 ± 6 AU).
FIG. 3.

IκBα-dependent activation of NF-κB is required for K. pneumoniae-induced TLR2 and TLR4 upregulation. (A) K. pneumoniae infection induced the translocation of NF-κB to the nuclei of A549 cells. The presence of NF-κB in nuclear extracts of infected cells was analyzed by using the NF-κB ELISA described in Materials and Methods (n = 3). OD450 nm, optical density at 450 nm. (B) The increased expression of TLR2 and TLR4 by A549 cells infected with K. pneumoniae 52145 was inhibited by CAPE, an inhibitor of NF-κB. Flow cytometry analysis was used to examine TLR2 and TLR4 expression by A549 cells that were not infected (CON) or were infected in the presence of different concentrations of the inhibitor, which was added 1 h before the cells were infected. The results (means and standard deviations) are expressed as rmfi values. The mfi values for A549 cells incubated with isotype-matched antibodies were 9 ± 2 AU. *, P < 0.05 (results are significantly different from the results for noninfected cells); Δ, P < 0.05 (results are significantly different from the results for infected cells in the absence of inhibitor; n = 4). (C) (Upper panel) Immunoblot showing IκBα levels in cytoplasmic extracts of A549 cells infected with K. pneumoniae for different times. (Lower panel) Immunoblot showing tubulin levels under the same conditions. The results are representative of four independent experiments. The relative ratios of IκBα (means ± standard deviations) obtained after densitometry analysis of gels are indicated below the lower panel (n = 4). α-IκBα, anti-IκBα; α-tubulin, anti-tubulin.
Because one of the major pathways for NF-κB activation involves the phosphorylation of IκBα followed by degradation of the protein, we analyzed the levels of IκBα expression in cytoplasmic extracts by immunoblotting. IκBα degradation was apparent in extracts from cells infected with K. pneumoniae 52145 (Fig. 3C).
Collectively, these findings demonstrated that IκBα-dependent activation of NF-κB is required for K. pneumoniae-induced TLR2 and TLR4 upregulation in A549 cells.
Activation of p38 and p44/42 MAP kinases is negatively involved in K. pneumoniae-induced TLR2 and TLR4 upregulation.
Besides the NF-κB pathway, many cellular stimuli also activate MAP kinase pathways (23). Therefore, we asked whether MAP kinases are also involved in K. pneumoniae-induced TLR upregulation. Activation of p38, p44/42, and JNK occurs through phosphorylation of serine and threonine residues (23, 51). Western blot analysis showed that K. pneumoniae induced activation of p38 and p44/42 (Fig. 4A) but not activation of JNK (data not shown).
FIG. 4.

Activation of p38 and p44/42 MAP kinase pathways is negatively involved in K. pneumoniae-induced TLR2 and TLR4 upregulation. (A) Immunoblots showing phospho-p38 (Pp38), total p38, phospho-p44/42 (Pp44/42), and total p44/42 levels in cytoplasmic extracts of A549 cells infected with K. pneumoniae for different times. The results are representative of three independent experiments. The relative ratios of phospho-p38 (means ± standard deviations) obtained after densitometry analysis of gels are indicated below the p38 panel (n = 3). (B) The expression of TLR2 and TLR4 by A549 cells infected with K. pneumoniae 52145 is increased by SB203580, an inhibitor of p38. Flow cytometry analysis was used to examine TLR2 and TLR4 expression by A549 cells that were not infected (CON) or were infected in the absence or presence (5 μM) of the inhibitor, which was added 2 h before the cells were infected. The results (means and standard deviations) are expressed as rmfi values (n = 4). (C) The expression of TLR2 and TLR4 by A549 cells infected with K. pneumoniae 52145 is increased by U0126, an inhibitor of p44/42. Flow cytometry analysis was used to examine TLR2 and TLR4 expression by A549 cells that were not infected (CON) or were infected in the absence or presence (10 μM) of the inhibitor, which was added 1 h before the cells were infected. The results (means and standard deviations) are expressed as rmfi values (n = 4). The mfi values for A549 cells incubated with isotype-matched antibodies were 9 ± 2 AU. *, results are significantly different from the results for noninfected cells (P < 0.05); Δ, results are significantly different from the results for infected cells in the absence of inhibitors (P < 0.05).
Next we explored the role of p38 in K. pneumoniae enhancement of TLR expression by using SB203580, a specific inhibitor of p38 MAP kinase. Figure 4B shows that the p38 inhibitor increased K. pneumoniae-induced TLR2 and TLR4 upregulation. Control experiments showed that SB203580-treated cells expressed amounts of TLR2 and TLR4 (rmfi, 15 ± 7 and 13 ± 5 AU, respectively) similar to the amounts expressed by untreated cells (TLR2 rmfi, 16 ± 5 AU; TLR4 rmfi, 13 ± 3 AU). To study the role of p44/42 in K. pneumoniae induction of TLR2 and TLR4 expression, we used the specific inhibitor U0126. The p44/42 inhibitor also enhanced K. pneumoniae-dependent TLR2 and TLR4 expression (Fig. 4C). U0126-treated cells expressed amounts of TLR2 and TLR4 (rmfi, 13 ± 4 and 14 ± 3 AU, respectively) similar to the amounts expressed by untreated cells (TLR2 rmfi, 17 ± 7 AU; TLR4 rmfi, 13 ± 3 AU). Addition of DMSO (the vehicle used for SB203580 and U0126) to K. pneumoniae-infected cells did not affect the expression of TLR2 (rmfi in the presence of DMSO, 24 ± 5 AU; rmfi in the absence of DMSO, 27 ± 5 AU) and TLR4 (rmfi in the presence of DMSO, 22 ± 8 AU; rmfi in the absence of DMSO, 19 ± 6 AU).
In summary, the results indicate that the p38 and p44/42 MAP kinase pathways may be negatively involved in K. pneumoniae-induced TLR upregulation.
K. pneumoniae-induced TLR2 and TLR4 upregulation is dependent on TLR activation.
In most cases, the activation of NF-κB and MAP kinase signaling pathways triggered by a bacterial infection is dependent on the activation of TLRs (70, 71). Therefore, we asked whether TLR-dependent signaling plays a role in K. pneumoniae-induced TLR2 and TLR4 upregulation. To study this, A549 cells were incubated for 30 min with anti-TLR2 antibody (clone TL2.1 [43]) or anti-TLR4 antibody (clone HTA125 [64]) and then infected with K. pneumoniae, and the expression of TLR2 and TLR4 was determined by qRT-PCR (Fig. 5). It has been shown previously that the concentrations of antibodies used block the activation of TLR2 and TLR4 signaling-dependent pathways (43, 56, 64). The anti-TLR2 and anti-TLR4 antibodies reduced the expression of TLR2 and TLR4 increased by K. pneumoniae infection (Fig. 5).
FIG. 5.
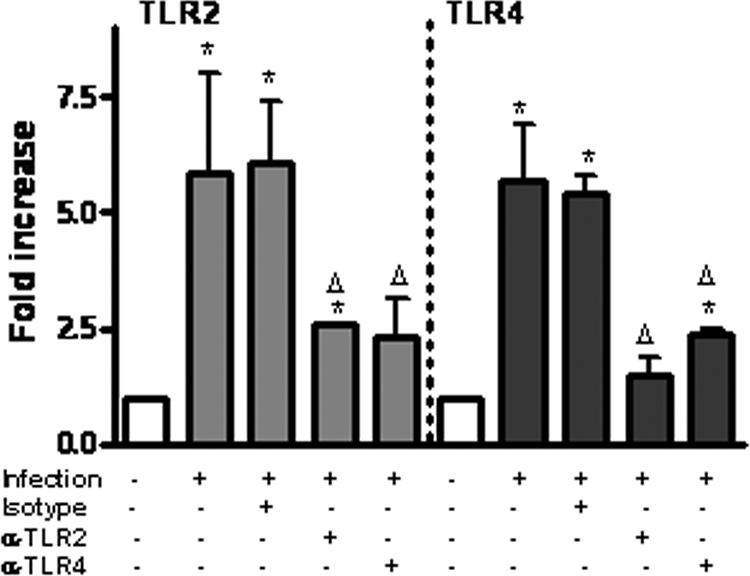
K. pneumoniae-induced TLR2 and TLR4 upregulation is dependent on TLR activation. A549 cells were not infected or were infected with K. pneumoniae 52145 in the presence or absence of the blocking antibodies anti-TLR2 (α-TLR2) (clone TL2.1; 10 μg/ml) and anti-TLR4 (α-TLR4) (clone HTA125; 10 μg/ml) for 3 h. The antibodies were added 30 min before the cells were infected. The TLR2 and TLR4 mRNA levels were assessed by qRT-PCR. The results (means and standard deviations) are representative of three independent experiments. *, results are significantly different from the results for noninfected cells (P < 0.05); Δ, results are significantly different from the results for infected cells in the absence of antibodies (P < 0.05).
CPS upregulates the expression of TLR2 and TLR4.
We sought to pinpoint the bacterial factor responsible for K. pneumoniae-induced TLR upregulation. Taking into account the fact that several authors have demonstrated the important role of CPS in the interplay between K. pneumoniae and eukaryotic cells, we asked whether CPS might be the bacterial factor triggering TLR upregulation. We studied whether an isogenic CPS mutant, strain 52K10 (20), induced TLR2 and TLR4 upregulation upon infection of A549 and NHBE cells. Flow cytometry analysis revealed that 52K10 did not increase the protein levels of either TLR2 or TLR4 in A549 and NHBE cells (Fig. 6A and 6B, respectively), suggesting that CPS might be the bacterial factor involved in TLR upregulation. To study this hypothesis, we asked whether purified CPS increased TLR expression. Our results showed that CPS induced TLR2 and TLR4 upregulation in a dose-dependent manner in A549 and NHBE cells (Fig. 6C and D, respectively). To explore whether protein contaminants present in the CPS preparation could have been responsible for TLR upregulation, CPS was repurified by the method described by Hirschfeld and coworkers (30). This method is widely used to remove proteins from polysaccharide preparations. Analysis of the repurified CPS by SDS-PAGE followed by silver-staining revealed no traces of contaminant proteins (data not shown). SDS-PAGE-resolved preparations were transferred to a polyvinylidene difluoride membrane which was stained with colloidal gold to visualize proteins (46). As observed previously, no proteins were detected in the repurified CPS preparation. The repurified CPS (600 μg/ml) upregulated TLR2 (rmfi, 18 ± 1 AU; rmfi for A549 cells, 9 ± 0.5 AU [P < 0.05]) and TLR4 (rmfi, 15 ± 0.4 AU; rmfi for A549 cells, 10.5 ± 0.7 AU [P < 0.05]). To test whether LPS contamination was responsible for TLR upregulation, a widely used inhibitor of LPS-dependent signaling pathways, polymyxin B, was used. However, repurified CPS (600 μg/ml) in the presence of 10 μg/ml of polymyxin B upregulated TLR2 (rmfi, 19 ± 1 AU; rmfi of A549 cells treated with polymyxin B, 7 ± 0.7 AU [P < 0.05]) and TLR4 (rmfi, 16 ± 0.3 AU; rmfi of A549 cells treated with polymyxin B, 9.6 ± 0.4 AU [P < 0.05]).
FIG. 6.

CPS upregulates the expression of TLR2 and TLR4 in human airway epithelial cells. (A) Flow cytometry analysis of TLR2 and TLR4 expression by noninfected A549 cells (CON), cells infected with K. pneumoniae wild-type strain (52145), or cells infected with isogenic CPS mutant strain 52K10 for 5 h. The results (means and standard deviations) are expressed as rmfi values (n = 4). (B) Experiments similar to those described above for panel A except that NHBE cells were used instead of A549 cells (n = 4). (C) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells (CON) or A549 cells treated with different amounts of purified K. pneumoniae CPS for 5 h. The results (means ± standard deviations) are expressed as rmfi values (n = 4). (D) Experiments similar to those described above for panel C except that NHBE cells were used instead of A549 cells (n = 4). The mfi values for A549 cells and NHBE cells incubated with isotype-matched antibodies were 10 ± 3 and 16 ± 5 AU, respectively. *, P < 0.05 (results are significantly different from the results for untreated cells [CON]).
We asked whether NF-κB and p38 and p44/42 MAP kinase signaling pathways also control CPS-dependent upregulation of TLR2 and TLR4. Western blot studies revealed that repurified CPS induced the degradation of IκBα (Fig. 7A). CAPE (15 μg/ml) reduced CPS-induced TLR2 and TLR4 upregulation of protein levels to the levels of control cells (Fig. 7B). A 5-μg/ml concentration of CAPE reduced only TLR2 protein levels (Fig. 7B). Figure 7C shows that repurified CPS induced the phosphorylation of p38 and p44/42 MAP kinases. p38 and p44/42 MAP kinase inhibitors (SB203580 and U0126, respectively) enhanced CPS-induced TLR2 and TLR4 upregulation (Fig. 7D and E).
FIG. 7.

CPS-induced expression of TLR2 and TLR4 is regulated via a positive NF-κB pathway and via negative p38- and p44/42-dependent pathways. (A) (Upper panel) Immunoblot showing IκBα levels in cytoplasmic extracts of A549 cells treated with CPS (600 μg/ml) for different times. (Lower panel) Immunoblot showing tubulin levels under the same conditions. The results are representative of four independent experiments. The relative ratios of IκBα (means ± standard deviations) obtained after densitometry analysis of gels are indicated below the lower panel (n = 4). (B) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells that were not treated (CON) or treated with CPS (600 μg/ml) in the presence of different concentrations of the NF-κB inhibitor CAPE, which was added 1 h before the cells were treated. The results (means and standard deviations) are expressed as rmfi values (n = 4). (C) Immunoblots showing phospho-p38 (Pp38), phospho-p44/42 (Pp44/42), and tubulin levels in cytoplasmic extracts of A549 cells treated with CPS (600 μg/ml) for different times. The results are representative of three independent experiments. The relative ratios of phospho-p38 (means ± standard deviations) obtained after densitometry analysis of gels are indicated below the p38 panel (n = 3). (D) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells that were not treated (CON) or treated with CPS (600 μg/ml) in the absence or presence of the SB203580 p38 MAP kinase inhibitor (5 μM), which was added 2 h before the cells were treated. The results (means and standard deviations) are expressed as rmfi values (n = 4). (E) Flow cytometry analysis of TLR2 and TLR4 expression by A549 cells that were not treated (CON) or treated with CPS (600 μg/ml) in the absence or presence of the U0126 p44/42 MAP kinase inhibitor (10 μM), which was added 1 h before the cells were treated. The results (means and standard deviations) are expressed as rmfi values (n = 4). (F) A549 cells were not treated or treated with CPS (600 μg/ml) in the presence or absence of the blocking antibodies anti-TLR2 (α-TLR2) (clone TL2.1; 10 μg/ml) and anti-TLR4 (α-TLR4) (clone HTA125; 10 μg/ml) for 5 h. The antibodies were added 30 min before the cells were infected. The TLR2 and TLR4 mRNA levels were assessed by qRT-PCR. The results (means and standard deviations) are representative of three independent experiments. *, results are significantly different from the results for noninfected cells (P < 0.05); Δ, results are significantly different from the results for infected cells in the absence of inhibitors (P < 0.05).
We studied whether TLR-dependent signaling plays a role in CPS-dependent upregulation of TLR2 and TLR4. To do this, A549 cells were incubated for 30 min with anti-TLR2 and anti-TLR4 antibodies before CPS was added. qRT-PCR analysis showed that both antibodies reduced CPS-dependent increased expression of TLR2 and TLR4 (Fig. 7F).
Since K. pneumoniae 52145 expresses a serotype K2 CPS, we asked whether K. pneumoniae strains expressing CPS of other serotypes also upregulate TLRs. Indeed, strains expressing serotype K3 CPS (strain KD364), serotype K35 CPS (strain USA1555), and serotype K47 CPS (strain USA0352) upregulated TLR2 (rmfi, 19.5 ± 0.4, 30.4 ± 2, and 22 ± 0.9 AU, respectively; rmfi of A549 cells, 11 ± 0.5 AU [P < 0.05]) and TLR4 (rmfi, 15 ± 2, 12 ± 0.7, and 14 ± 0.4 AU, respectively; rmfi of A549 cells, 8.7 ± 0.3 AU [P < 0.05]).
DISCUSSION
In this work we showed that the expression of TLR2 and TLR4 by human airway epithelial cells is upregulated upon infection with K. pneumoniae. This occurs via a positive IκBα-dependent NF-κΒ pathway and via negative p38- and p44/42-dependent pathways. The increased expression of TLR2 and TLR4 results in enhancement of the cellular response upon stimulation with TLR2 and TLR4 agonists (Fig. 8). Finally, we obtained evidence indicating that CPS might be the bacterial factor inducing the upregulation of TLRs.
FIG. 8.

Schematic diagram of the signaling pathways involved in K. pneumoniae-induced expression of TLR2 and TLR4.
There is controversy over the surface expression of TLRs by airway epithelial cells (10, 47, 56, 75). Although the surface expression of TLR2 is not in dispute, some data indicate that TLR4 is not expressed in the cell surface but in the Golgi complex. In this study, we showed that K. pneumoniae increased the surface levels of TLR2 and TLR4 and also the mRNA levels of both genes. Supporting our results, it was shown recently that airway epithelial cells do express functional TLR2 and TLR4 on the surface and that inflammatory cytokines increase the surface levels as well as mRNA levels of both TLRs (5). Nevertheless, at present, we cannot rigorously rule out the possibility that upon infection TLR4 traffics from the Golgi complex to the cytoplasmic membrane of airway epithelial cells. In any case, K. pneumoniae infection increases TLR4 mRNA levels and surface levels of the protein.
Most likely, the increased expression of TLRs by airway epithelial cells contributes to the clearance of invading pathogens. For example, it has been shown that there is a correlation between the levels of expression of TLRs and the phagocytosis of pathogens (13). Furthermore, it has been demonstrated that overexpression of TLRs is associated with an increase in the secretion of inflammatory mediators (1, 2, 9, 32, 36, 47, 52). Studies using animal models have shown that an early inflammatory response is important for the clearance of several pathogens (14, 27, 28, 37, 44, 53, 61, 78). It is tempting to postulate that the upregulation of TLRs could be one of the set of host defense systems induced by an infection, the so-called “common host response” (34). However, if this initial inflammatory response is not controlled, the early improvement in disease resistance is followed by extensive tissue damage (9, 74). In addition, the increased TLR expression could contribute to resensitization of cells to TLR agonists, which may trigger an excessive inflammatory response. On the other hand, there have been reports suggesting that abnormal elevated levels of TLRs are associated with systemic inflammation and worsening of the clinical status in various diseases (16, 52, 58, 65). Taken together, these findings suggest that the amount of TLRs expressed by a given cell should be tightly controlled and that by controlling the amount of TLRs expressed cells fine-tune their responsiveness.
It is interesting that nontypeable H. influenzae, a pathogen frequently associated with respiratory infections, also induces the upregulation of TLRs in vivo and in vitro (44, 67). The intracellular signaling pathways responsible are similar to those described in this paper (67). This may indicate that there is a common regulatory circuit regulating the expression of TLRs in airway epithelia and, furthermore, that the activation of this circuit is independent of the infecting microorganism. The molecular mechanism underlying p38 and p44/42 inhibition of TLR2 and TLR4 expression remains unclear. The human promoter of TLR2 and TLR4 contains multiple transcription factor binding elements, including Ets-1, which has been shown to be regulated by MAP kinase pathways (29, 45, 57, 72). Nevertheless, in both promoters there are some regulatory elements that have not been completely characterized (29, 57). The possibility that in addition to these transcription factors there are posttranscriptional mechanisms, like increased mRNA stability, cannot be ruled out.
Even though capsulated K. pneumoniae upregulated the expression of TLR2 and TLR4 in a TLR2- and TLR4-dependent fashion (this study), the upregulation was not accompanied by the secretion of inflammatory mediators (56), whereas an isogenic K. pneumoniae CPS mutant did not upregulate TLR expression (this study) but induced the secretion of inflammatory mediators in a TLR2- and TLR4-dependent manner (56). These findings suggest that TLR2 and TLR4 are involved in the recognition of K. pneumoniae but the cellular response is different depending on the presence of CPS. The results of in vivo experiments also support this notion. Thus, after intranasal infection of mice capsulated K. pneumoniae induces less inflammatory mediators and less infiltration of inflammatory cells than a capsule mutant induces (40, 81). All these results are reminiscent of those reported for Cryptococcus neoformans, a fungus with a thick anionic CPS (8). Infection with this fungus causes signaling through TLR2 and TLR4 in a mouse model (79, 80), and heavily capsulated strains generally elicited a poor inflammatory response in vitro and in vivo (12, 42, 76). The available evidence indicates that C. neoformans CPS has immunomodulatory properties and most likely is responsible for host immunosuppression (24). K. pneumoniae CPS is also anionic, like alginate expressed by Pseudomonas aeruginosa or the Vi polysaccharide expressed by Salmonella enterica serovar Typhi (18, 63, 69). Similar to capsulated Klebsiella, alginate- or Vi-expressing bacteria induce a poor inflammatory response, in contrast to isogenic CPS mutants (17, 54). Furthermore, Vi polysaccharide seems to suppress early inflammatory responses (62). When the data are considered together, it is tempting to postulate that anionic CPSs are PAMPs that trigger downregulation of inflammatory responses. Studies to examine this hypothesis are being performed now.
Two pieces of evidence suggest that CPS might be the bacterial factor responsible for K. pneumoniae-induced TLR upregulation. First, an isogenic CPS mutant, strain 52K10, did not upregulate TLR expression. Second, purified CPS increased the expression of TLRs. However, we are aware that despite extensive purification our CPS preparation still contained traces of proteins and LPS. In our efforts to rule out the possibility that protein contaminants have an effect, the CPS preparation was repurified using a method developed to remove proteins from polysaccharide preparations (30, 46). Although no proteins were detected in the repurified CPS preparation by SDS-PAGE and colloidal gold staining, we cannot rule out the possibility that protein traces could be responsible, at least in part, for TLR upregulation. In contrast, the fact that polymyxin B, an inhibitor of LPS-dependent signaling pathways, did not affect the CPS-dependent upregulation of TLRs allows us to rule out the possibility that traces of LPS are responsible for CPS-induced increases in TLR expression.
Finally, our findings suggest that purified CPS could upregulate TLR expression in a TLR-dependent fashion, thereby indicating that TLRs have a role as pattern recognition receptors for K. pneumoniae CPS. In support of this, two reports have suggested that TLRs can recognize CPSs from pathogens. C. neoformans CPS is recognized by TLR4 (66), whereas P. aeruginosa alginate activates macrophages via TLR2 and TLR4 (26). In any case, it is possible that K. pneumoniae CPS activates signaling pathways other than TLRs. However, our data indicate that these putative pathways are not involved in CPS-dependent TLR upregulation.
Acknowledgments
We are grateful to members of the Bengoechea lab for helpful discussions.
Fellowship support of M.A.C. provided by Govern Illes Balears is gratefully acknowledged. J.G. was the recipient of a “Contrato de Investigador Miguel Servet” from Instituto de Salud Carlos III. This work was funded by grants PI05/2311 and PI06/1629 from Fondo de Investigación Sanitaria and by grant GEN2006-27776-C2-2-E/PAT from ERA-NET Pathogenomics to J.A.B.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 17 November 2008.
REFERENCES
- 1.Abreu, M. T., E. T. Arnold, L. S. Thomas, R. Gonsky, Y. Zhou, B. Hu, and M. Arditi. 2002. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J. Biol. Chem. 27720431-20437. [DOI] [PubMed] [Google Scholar]
- 2.Abreu, M. T., P. Vora, E. Faure, L. S. Thomas, E. T. Arnold, and M. Arditi. 2001. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 1671609-1616. [DOI] [PubMed] [Google Scholar]
- 3.Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406782-787. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez, D., S. Merino, J. M. Tomas, V. J. Benedi, and S. Alberti. 2000. Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella pneumoniae clinical isolates. Infect. Immun. 68953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong, L., A. R. Medford, K. M. Uppington, J. Robertson, I. R. Witherden, T. D. Tetley, and A. B. Millar. 2004. Expression of functional Toll-like receptor (TLR-)2 and TLR-4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 31241-245. [DOI] [PubMed] [Google Scholar]
- 6.Beutler, B. 2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430257-263. [DOI] [PubMed] [Google Scholar]
- 7.Beutler, B., K. Hoebe, X. Du, and R. J. Ulevitch. 2003. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J. Leukoc. Biol. 74479-485. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharjee, A. K., J. E. Bennett, and C. P. Glaudemans. 1984. Capsular polysaccharides of Cryptococcus neoformans. Rev. Infect. Dis. 6619-624. [DOI] [PubMed] [Google Scholar]
- 9.Bihl, F., L. Salez, M. Beaubier, D. Torres, L. Lariviere, L. Laroche, A. Benedetto, D. Martel, J. M. Lapointe, B. Ryffel, and D. Malo. 2003. Overexpression of Toll-like receptor 4 amplifies the host response to lipopolysaccharide and provides a survival advantage in transgenic mice. J. Immunol. 1706141-6150. [DOI] [PubMed] [Google Scholar]
- 10.Birchler, T., R. Seibl, K. Buchner, S. Loeliger, R. Seger, J. P. Hossle, A. Aguzzi, and R. P. Lauener. 2001. Human Toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur. J. Immunol. 313131-3137. [DOI] [PubMed] [Google Scholar]
- 11.Bitter, T., and H. M. Muir. 1962. A modified uronic acid carbazole reaction. Anal.Biochem. 4330-334. [DOI] [PubMed] [Google Scholar]
- 12.Blackstock, R., K. L. Buchanan, A. M. Adesina, and J. W. Murphy. 1999. Differential regulation of immune responses by highly and weakly virulent Cryptococcus neoformans isolates. Infect. Immun. 673601-3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blander, J. M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 3041014-1018. [DOI] [PubMed] [Google Scholar]
- 14.Branger, J., S. Knapp, S. Weijer, J. C. Leemans, J. M. Pater, P. Speelman, S. Florquin, and P. T. van der. 2004. Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect. Immun. 72788-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campos, M. A., M. A. Vargas, V. Regueiro, C. M. Llompart, S. Alberti, and J. A. Bengoechea. 2004. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 727107-7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Candia, L., J. Marquez, C. Hernandez, A. H. Zea, and L. R. Espinoza. 2007. Toll-like receptor-2 expression is upregulated in antigen-presenting cells from patients with psoriatic arthritis: a pathogenic role for innate immunity? J. Rheumatol. 34374-379. [PubMed] [Google Scholar]
- 17.Cobb, L. M., J. C. Mychaleckyj, D. J. Wozniak, and Y. S. Lopez-Boado. 2004. Pseudomonas aeruginosa flagellin and alginate elicit very distinct gene expression patterns in airway epithelial cells: implications for cystic fibrosis disease. J. Immunol. 1735659-5670. [DOI] [PubMed] [Google Scholar]
- 18.Corsaro, M. M., C. C. De, T. Naldi, M. Parrilli, J. M. Tomas, and M. Regue. 2005. 1H and 13C NMR characterization and secondary structure of the K2 polysaccharide of Klebsiella pneumoniae strain 52145. Carbohydr. Res. 3402212-2217. [DOI] [PubMed] [Google Scholar]
- 19.Cortes, G., D. Alvarez, C. Saus, and S. Alberti. 2002. Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect. Immun. 701075-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortes, G., N. Borrell, B. de Astorza, C. Gomez, J. Sauleda, and S. Alberti. 2002. Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 702583-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Astorza, B., G. Cortes, C. Crespi, C. Saus, J. M. Rojo, and S. Alberti. 2004. C3 promotes clearance of Klebsiella pneumoniae by A549 epithelial cells. Infect. Immun. 721767-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Díaz-Aparicio, E., V. Aragon, C. Marin, B. Alonso, M. Font, E. Moreno, S. Perez-Ortiz, J. M. Blasco, R. Diaz, and I. Moriyon. 1993. Comparative analysis of Brucella serotype A and M and Yersinia enterocolitica O:9 polysaccharides for serological diagnosis of brucellosis in cattle, sheep, and goats. J. Clin. Microbiol. 313136-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong, C., R. J. Davis, and R. A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 2055-72. [DOI] [PubMed] [Google Scholar]
- 24.Ellerbroek, P. M., A. M. Walenkamp, A. I. Hoepelman, and F. E. Coenjaerts. 2004. Effects of the capsular polysaccharides of Cryptococcus neoformans on phagocyte migration and inflammatory mediators. Curr. Med. Chem. 11253-266. [DOI] [PubMed] [Google Scholar]
- 25.Favre-Bonte, S., B. Joly, and C. Forestier. 1999. Consequences of reduction of Klebsiella pneumoniae capsule expression on interactions of this bacterium with epithelial cells. Infect. Immun. 67554-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flo, T. H., L. Ryan, E. Latz, O. Takeuchi, B. G. Monks, E. Lien, O. Halaas, S. Akira, G. Skjak-Braek, D. T. Golenbock, and T. Espevik. 2002. Involvement of Toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J. Biol. Chem. 27735489-35495. [DOI] [PubMed] [Google Scholar]
- 27.Greenberger, M. J., R. M. Strieter, S. L. Kunkel, J. M. Danforth, R. E. Goodman, and T. J. Standiford. 1995. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J. Immunol. 155722-729. [PubMed] [Google Scholar]
- 28.Greenberger, M. J., R. M. Strieter, S. L. Kunkel, J. M. Danforth, L. L. Laichalk, D. C. McGillicuddy, and T. J. Standiford. 1996. Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J. Infect. Dis. 173159-165. [DOI] [PubMed] [Google Scholar]
- 29.Haehnel, V., L. Schwarzfischer, M. J. Fenton, and M. Rehli. 2002. Transcriptional regulation of the human Toll-like receptor 2 gene in monocytes and macrophages. J. Immunol. 1685629-5637. [DOI] [PubMed] [Google Scholar]
- 30.Hirschfeld, M., Y. Ma, J. H. Weis, S. N. Vogel, and J. J. Weis. 2000. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J. Immunol. 165618-622. [DOI] [PubMed] [Google Scholar]
- 31.Homma, T., A. Kato, N. Hashimoto, J. Batchelor, M. Yoshikawa, S. Imai, H. Wakiguchi, H. Saito, and K. Matsumoto. 2004. Corticosteroid and cytokines synergistically enhance Toll-like receptor 2 expression in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 31463-469. [DOI] [PubMed] [Google Scholar]
- 32.Imasato, A., C. sois-Mouthon, J. Han, H. Kai, A. C. Cato, S. Akira, and J. D. Li. 2002. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of Toll-like receptor 2. J. Biol. Chem. 27747444-47450. [DOI] [PubMed] [Google Scholar]
- 33.Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20197-216. [DOI] [PubMed] [Google Scholar]
- 34.Jenner, R. G., and R. A. Young. 2005. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 3281-294. [DOI] [PubMed] [Google Scholar]
- 35.Kagnoff, M. F., and L. Eckmann. 1997. Epithelial cells as sensors for microbial infection. J. Clin. Investig 1006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalis, C., B. Kanzler, A. Lembo, A. Poltorak, C. Galanos, and M. A. Freudenberg. 2003. Toll-like receptor 4 expression levels determine the degree of LPS-susceptibility in mice. Eur. J. Immunol. 33798-805. [DOI] [PubMed] [Google Scholar]
- 37.Knapp, S., C. W. Wieland, S. Florquin, R. Pantophlet, L. Dijkshoorn, N. Tshimbalanga, S. Akira, and T. van der Poll. 2006. Differential roles of CD14 and Toll-like receptors 4 and 2 in murine Acinetobacter pneumonia. Am. J. Respir. Crit. Care Med. 173122-129. [DOI] [PubMed] [Google Scholar]
- 38.Knapp, S., C. W. Wieland, C. van't Veer, O. Takeuchi, S. Akira, S. Florquin, and P. T. van der. 2004. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J. Immunol. 1723132-3138. [DOI] [PubMed] [Google Scholar]
- 39.Knowles, M. R., and R. C. Boucher. 2002. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 9571-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawlor, M. S., S. A. Handley, and V. L. Miller. 2006. Comparison of the host responses to wild-type and cpsB mutant Klebsiella pneumoniae infections. Infect. Immun. 745402-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawlor, M. S., J. Hsu, P. D. Rick, and V. L. Miller. 2005. Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol. Microbiol. 581054-1073. [DOI] [PubMed] [Google Scholar]
- 42.Levitz, S. M., A. Tabuni, H. Kornfeld, C. C. Reardon, and D. T. Golenbock. 1994. Production of tumor necrosis factor alpha in human leukocytes stimulated by Cryptococcus neoformans. Infect. Immun. 621975-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lien, E., T. J. Sellati, A. Yoshimura, T. H. Flo, G. Rawadi, R. W. Finberg, J. D. Carroll, T. Espevik, R. R. Ingalls, J. D. Radolf, and D. T. Golenbock. 1999. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 27433419-33425. [DOI] [PubMed] [Google Scholar]
- 44.Lorenz, E., D. C. Chemotti, A. L. Jiang, and L. D. McDougal. 2005. Differential involvement of Toll-like receptors 2 and 4 in the host response to acute respiratory infections with wild-type and mutant Haemophilus influenzae strains. Infect. Immun. 732075-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma, W., W. Lim, K. Gee, S. Aucoin, D. Nandan, M. Kozlowski, F. az-Mitoma, and A. Kumar. 2001. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 27613664-13674. [DOI] [PubMed] [Google Scholar]
- 46.Manthey, C. L., P. Y. Perera, B. E. Henricson, T. A. Hamilton, N. Qureshi, and S. N. Vogel. 1994. Endotoxin-induced early gene expression in C3H/HeJ (Lpsd) macrophages. J. Immunol. 1532653-2663. [PubMed] [Google Scholar]
- 47.Monick, M. M., T. O. Yarovinsky, L. S. Powers, N. S. Butler, A. B. Carter, G. Gudmundsson, and G. W. Hunninghake. 2003. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol. Chem. 27853035-53044. [DOI] [PubMed] [Google Scholar]
- 48.Nassif, X., J. M. Fournier, J. Arondel, and P. J. Sansonetti. 1989. Mucoid phenotype of Klebsiella pneumoniae is a plasmid-encoded virulence factor. Infect. Immun. 57546-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natarajan, K., S. Singh, T. R. Burke, Jr., D. Grunberger, and B. B. Aggarwal. 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 939090-9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oelschlaeger, T. A., and B. D. Tall. 1997. Invasion of cultured human epithelial cells by Klebsiella pneumoniae isolated from the urinary tract. Infect. Immun. 652950-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ono, K., and J. Han. 2000. The p38 signal transduction pathway: activation and function. Cell Signal. 121-13. [DOI] [PubMed] [Google Scholar]
- 52.Pons, J., J. Sauleda, V. Regueiro, C. Santos, M. Lopez, J. Ferrer, A. G. Agusti, and J. A. Bengoechea. 2006. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir. Res. 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Power, M. R., Y. Peng, E. Maydanski, J. S. Marshall, and T. J. Lin. 2004. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J. Biol. Chem. 27949315-49322. [DOI] [PubMed] [Google Scholar]
- 54.Raffatellu, M., D. Chessa, R. P. Wilson, R. Dusold, S. Rubino, and A. J. Baumler. 2005. The Vi capsular antigen of Salmonella enterica serotype Typhi reduces Toll-like receptor-dependent interleukin-8 expression in the intestinal mucosa. Infect. Immun. 733367-3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahn, A., and C. Whitfield. 2003. Transcriptional organization and regulation of the Escherichia coli K30 group 1 capsule biosynthesis (cps) gene cluster. Mol. Microbiol. 471045-1060. [DOI] [PubMed] [Google Scholar]
- 56.Regueiro, V., M. A. Campos, J. Pons, S. Alberti, and J. A. Bengoechea. 2006. The uptake of a Klebsiella pneumoniae capsule polysaccharide mutant triggers an inflammatory response by human airway epithelial cells. Microbiology 152555-566. [DOI] [PubMed] [Google Scholar]
- 57.Rehli, M., A. Poltorak, L. Schwarzfischer, S. W. Krause, R. Andreesen, and B. Beutler. 2000. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 gene. J. Biol. Chem. 2759773-9781. [DOI] [PubMed] [Google Scholar]
- 58.Riordan, S. M., N. Skinner, A. Nagree, H. McCallum, C. J. McIver, J. Kurtovic, J. A. Hamilton, S. Bengmark, R. Williams, and K. Visvanathan. 2003. Peripheral blood mononuclear cell expression of Toll-like receptors and relation to cytokine levels in cirrhosis. Hepatology 371154-1164. [DOI] [PubMed] [Google Scholar]
- 59.Sahly, H., and R. Podschun. 1997. Clinical, bacteriological, and serological aspects of Klebsiella infections and their spondylarthropathic sequelae. Clin. Diagn. Lab Immunol. 4393-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakai, A., J. Han, A. C. Cato, S. Akira, and J. D. Li. 2004. Glucocorticoids synergize with IL-1beta to induce TLR2 expression via MAP kinase phosphatase-1-dependent dual inhibition of MAPK JNK and p38 in epithelial cells. BMC Mol. Biol. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schurr, J. R., E. Young, P. Byrne, C. Steele, J. E. Shellito, and J. K. Kolls. 2005. Central role of Toll-like receptor 4 signaling and host defense in experimental pneumonia caused by gram-negative bacteria. Infect. Immun. 73532-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma, A., and A. Qadri. 2004. Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc. Natl. Acad. Sci. USA 10117492-17497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sherbrock-Cox, V., N. J. Russell, and P. Gacesa. 1984. The purification and chemical characterisation of the alginate present in extracellular material produced by mucoid strains of Pseudomonas aeruginosa. Carbohydr. Res. 135147-154. [DOI] [PubMed] [Google Scholar]
- 64.Shimazu, R., S. Akashi, H. Ogata, Y. Nagai, K. Fukudome, K. Miyake, and M. Kimoto. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1891777-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shiraki, R., N. Inoue, S. Kobayashi, J. Ejiri, K. Otsui, T. Honjo, M. Takahashi, K. Hirata, M. Yokoyama, and S. Kawashima. 2006. Toll-like receptor 4 expressions on peripheral blood monocytes were enhanced in coronary artery disease even in patients with low C-reactive protein. Life Sci. 8059-66. [DOI] [PubMed] [Google Scholar]
- 66.Shoham, S., C. Huang, J. M. Chen, D. T. Golenbock, and S. M. Levitz. 2001. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J. Immunol. 1664620-4626. [DOI] [PubMed] [Google Scholar]
- 67.Shuto, T., A. Imasato, H. Jono, A. Sakai, H. Xu, T. Watanabe, D. D. Rixter, H. Kai, A. Andalibi, F. Linthicum, Y. L. Guan, J. Han, A. C. Cato, D. J. Lim, S. Akira, and J. D. Li. 2002. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J. Biol. Chem. 27717263-17270. [DOI] [PubMed] [Google Scholar]
- 68.Struve, C., and K. A. Krogfelt. 2003. Role of capsule in Klebsiella pneumoniae virulence: lack of correlation between in vitro and in vivo studies. FEMS Microbiol. Lett. 218149-154. [DOI] [PubMed] [Google Scholar]
- 69.Szu, S. C., and S. Bystricky. 2003. Physical, chemical, antigenic, and immunologic characterization of polygalacturonan, its derivatives, and Vi antigen from Salmonella typhi. Methods Enzymol. 363552-567. [DOI] [PubMed] [Google Scholar]
- 70.Takeda, K., and S. Akira. 2003. Toll receptors and pathogen resistance. Cell. Microbiol. 5143-153. [DOI] [PubMed] [Google Scholar]
- 71.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21335-376. [DOI] [PubMed] [Google Scholar]
- 72.Tanaka, K., N. Oda, C. Iwasaka, M. Abe, and Y. Sato. 1998. Induction of Ets-1 in endothelial cells during reendothelialization after denuding injury. J. Cell Physiol. 176235-244. [DOI] [PubMed] [Google Scholar]
- 73.Timko, J. 2004. Changes of antimicrobial resistance and extended-spectrum beta-lactamase production in Klebsiella spp. strains. J. Infect. Chemother. 10212-215. [DOI] [PubMed] [Google Scholar]
- 74.Togbe, D., S. Schnyder-Candrian, B. Schnyder, I. Couillin, I. Maillet, F. Bihl, D. Malo, B. Ryffel, and V. F. Quesniaux. 2006. TLR4 gene dosage contributes to endotoxin-induced acute respiratory inflammation. J. Leukoc. Biol. 80451-457. [DOI] [PubMed] [Google Scholar]
- 75.Tsutsumi-Ishii, Y., and I. Nagaoka. 2003. Modulation of human beta-defensin-2 transcription in pulmonary epithelial cells by lipopolysaccharide-stimulated mononuclear phagocytes via proinflammatory cytokine production. J. Immunol. 1704226-4236. [DOI] [PubMed] [Google Scholar]
- 76.Vecchiarelli, A., C. Retini, D. Pietrella, C. Monari, C. Tascini, T. Beccari, and T. R. Kozel. 1995. Downregulation by cryptococcal polysaccharide of tumor necrosis factor alpha and interleukin-1 beta secretion from human monocytes. Infect. Immun. 632919-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Westphal, O., and K. Jann. 1963. Bacterial lipopolysaccharides extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 583-91. [Google Scholar]
- 78.Wieland, C. W., S. Florquin, N. A. Maris, K. Hoebe, B. Beutler, K. Takeda, S. Akira, and P. T. van der. 2005. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Haemophilus influenzae from the mouse lung. J. Immunol. 1756042-6049. [DOI] [PubMed] [Google Scholar]
- 79.Yauch, L. E., M. K. Mansour, and S. M. Levitz. 2005. Receptor-mediated clearance of Cryptococcus neoformans capsular polysaccharide in vivo. Infect. Immun. 738429-8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yauch, L. E., M. K. Mansour, S. Shoham, J. B. Rottman, and S. M. Levitz. 2004. Involvement of CD14, Toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect. Immun. 725373-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida, K., T. Matsumoto, K. Tateda, K. Uchida, S. Tsujimoto, and K. Yamaguchi. 2000. Role of bacterial capsule in local and systemic inflammatory responses of mice during pulmonary infection with Klebsiella pneumoniae. J. Med. Microbiol. 491003-1010. [DOI] [PubMed] [Google Scholar]