

Abstract
Anaplasma phagocytophilum, the etiologic agent of human granulocytic anaplasmosis (HGA), has genes predicted to encode three sensor kinases, one of which is annotated PleC, and three response regulators, one of which is PleD. Prior to this study, the roles of PleC and PleD in the obligatory intracellular parasitism of A. phagocytophilum and their biochemical activities were unknown. The present study illustrates the relevance of these factors by demonstrating that both pleC and pleD were expressed in an HGA patient. During A. phagocytophilum development in human promyelocytic HL-60 cells, PleC and PleD were synchronously upregulated at the exponential growth stage and downregulated prior to extracellular release. A recombinant PleC kinase domain (rPleCHKD) has histidine kinase activity; no activity was observed when the conserved site of phosphorylation was replaced with alanine. A recombinant PleD (rPleD) has autokinase activity using phosphorylated rPleCHKD as the phosphoryl donor but not with two other recombinant histidine kinases. rPleCHKD could not serve as the phosphoryl donor for a mutant rPleD (with a conserved aspartic acid, the site of phosphorylation, replaced by alanine) or two other A. phagocytophilum recombinant response regulators. rPleD had diguanylate cyclase activity to generate cyclic (c) di-GMP from GTP in vitro. UV cross-linking of A. phagocytophilum lysate with c-di-[32P]GMP detected an ∼47-kDa endogenous protein, presumably c-di-GMP downstream receptor. A new hydrophobic c-di-GMP derivative, 2′-O-di(tert-butyldimethylsilyl)-c-di-GMP, inhibited A. phagocytophilum infection in HL-60 cells. Our results suggest that the two-component PleC-PleD system is a diguanylate cyclase and that a c-di-GMP-receptor complex regulates A. phagocytophilum intracellular infection.
Anaplasma phagocytophilum is an obligatory intracellular bacterium and a member of the order Rickettsiales that infects granulocytes in various mammals (8). Infection of endothelial cells has been recently shown in vivo and in vitro (11, 17). A. phagocytophilum causes human granulocytic anaplasmosis (HGA), an acute febrile disease that is potentially fatal, especially in elderly or immunocompromised individuals (3, 7). Under an electron microscope, A. phagocytophilum is a pleomorphic bacterium 0.2 to 2.0 μm in diameter, replicating in the membrane-bound inclusion in the host cell cytoplasm. The developmental cycle of the bacterium consists of two forms: small dense-cored cells (DC) with cell binding activity and the ability to enter host cells and large reticulate cells (RC) that are differentiated from DC. RC again mature into DC, which are released upon host cell lysis (16, 21, 31). However, little is known regarding the bacterial factors regulating A. phagocytophilum intracellular growth and development.
The bacterial two-component system (TCS) is a ubiquitous signal transduction paradigm that controls response, adaptation, and resistance to a variety of environmental conditions (18). TCSs are typically composed of a sensor kinase (SK) and a cognate response regulator (RR). In the cytoplasm, dimerization and intermolecular autophosphorylation of the His residue in the kinase domain occurs when the SK senses a particular environmental signal through the periplasmic sensor domain. The phosphoryl group is then transferred to an Asp residue in the phospho-receiver domain (hereafter called the receiver domain) of a cognate RR (18). This transfer, in turn, activates the output domain of the RR, which generally has DNA binding activity to regulate target gene transcription or an enzymatic activity, such as diguanylate cyclase (DGC), associated with the GGDEF (Gly-Gly-Asp-Glu-Phe) domain containing the sequence motif GGDEF within the RR (9). Genes predicted to encode three SKs and three RRs are found in A. phagocytophilum, and an inhibitor of histidine kinases prevents A. phagocytophilum infection of mammalian host cells (4). Thus, TCSs are considered essential for A. phagocytophilum infection. In a related bacterium, Ehrlichia chaffeensis, orthologs of these SKs and RRs were shown to constitute three functional pairs of TCSs (14); however, a TCS has yet to be definitively identified for A. phagocytophilum.
One of the proven TCS pairs of E. chaffeensis (14) and one of the predicted TCS pairs of A. phagocytophilum comprise the SK (PleC) and the RR (PleD), based on similarities of these proteins to the PleC and PleD produced by the aquatic free-living bacterium Caulobacter crescentus (4). C. crescentus PleD has DGC activity to generate a bacterial second messenger, cyclic (c) di-GMP, from GTP (20). DGC activity has not been shown in any predicted PleD proteins containing a GGDEF motif in the order Rickettsiales, including A. phagocytophilum and E. chaffeensis (4). Using mutation and reconstitution studies of C. crescentus and other bacteria, c-di-GMP was found to regulate bacterial cell surface-associated traits and community behavior, such as cell-cell signaling, biofilm formation, motility, differentiation, and virulence (22, 29).
Here, we demonstrate that during A. phagocytophilum development in human promyelocytic HL-60 cells, PleC and PleD were synchronously upregulated at the exponential growth stage and downregulated prior to extracellular release. A. phagocytophilum PleC has histidine autokinase activity, and phosphotransfer occurs from PleC to PleD, implying that PleC and PleD constitute a functional TCS. We also show evidence that A. phagocytophilum PleD has DGC activity.
MATERIALS AND METHODS
Sequence analysis.
The DNAStar Protean program, PSORT analysis (http://psort.ims.u-tokyo.ac.jp), NCBI BLAST searches (http://www.ncbi.nlm.nih.gov/BLAST.cgi), and Motif scans (http://myhits.isb-sib.ch/cgi-bin/motif_scan) were utilized to analyze deduced amino acid sequences. Protein motifs were determined by Motif scan (http://myhits.isb-sib.ch/cgi-bin/motif_scan) and ExPASy Scan Prosite of Redasoft visual-cloning software (Redasoft, Toronto, Canada).
Bacterial strains and culture.
A. phagocytophilum HZ was propagated in HL-60 cells, a human promyelocytic leukemia cell line (ATCC, Manassas, VA), in RPMI 1640 medium supplemented with 5% fetal bovine serum and 2 mM l-glutamine at 37°C in 5% CO2/95% air. Escherichia coli strains NovaBlue (Novagen, Madison, WI), DH5α (Invitrogen, Carlsbad, CA), and BL21(DE3) (Novagen) were cultured in Luria-Bertani broth (24) supplemented with either ampicillin (50 μg/ml) or kanamycin (50 μg/ml) as required.
Reverse transcription-PCR.
Total RNA was extracted from 5 × 106 A. phagocytophilum-infected HL-60 cells (80% infected cells) with the RNeasy minikit (Qiagen, Valencia, CA). Buffy coat specimens were prepared from the blood of HGA patient NY37 (15), and total RNA was extracted with Trizol reagent (Invitrogen) and further purified with the RNeasy minikit. The RNA was reverse transcribed, and PCR was conducted as described previously (4, 15). To ensure the absence of DNA contamination in the RNA preparation, control assays were included that lacked reverse transcriptase.
Construction of plasmids for expression of the histidine kinase domain (HKD) of SKs and full-length RRs.
Total DNA was extracted from A. phagocytophilum-infected HL-60 cells using a Qiaamp DNA minikit (Qiagen). The DNA fragments encoding HKDs of SKs and full-length RRs were amplified by PCR with the primers shown in Table 1 using A. phagocytophilum chromosomal DNA as a template. The PCR products were purified with a PCR purification kit (Qiagen). The amplified DNA fragments were digested with restriction enzymes and ligated into the restriction enzyme-digested vector pET33b (Novagen) (for N-terminally His-tagged recombinant PleD [rPleDNHis], C-terminally His-tagged recombinant PleD [rPleDCHis], and rCckAHKD), pET11a (Novagen) (for rPleCHKD, rNtrX, and rCtrA), or pMALc2x (New England BioLabs, Ipswich, MA) (for rNtrYHKD). E. coli strain NovaBlue was transformed, plasmids were extracted, and the sequences of the cloned fragments were confirmed. Mutations that alter conserved His residues of rPleCHKD to Ala and conserved Asp residues of rPleDNHis to Ala were introduced using the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the primers shown in Table 1. The mutations were confirmed by DNA sequencing.
TABLE 1.
Oligonucleotides used for cloning and expression of A. phagocytophilum SKs and RRs and their mutants
| Gene | Direction | Primer sequencea | Target DNA size (bp) | Enzyme | Plasmid |
|---|---|---|---|---|---|
| pleC | Forward | GGGCTAGCGGTCACCATCACCATCACCATGGAAAAAATGTAAAGTTGCTTAATAGCC | 834 | NheI | pET-11a(+) |
| Reverse | GGGATCCAAAGGCCTTTTCGCTCTC | BamHI | |||
| cckA | Forward | AACCATGGGTCACCACCACCACCACCACGGATTAGAAACTCAATTGGAACAATCGC | 1,347 | NcoI | pET-33b(+) |
| Reverse | TTCTCGAGTTTCTGGCCTTTGCACG | XhoI | |||
| ntrY | Forward | TGAATTCCGGGTTCAAGATGGGGATTTTGAT | 1,148 | EcoRI | pMALc2x |
| Reverse | TTAAGCTTCATTTCAAAAAAGTAATCCGTACCAC | HindIII | |||
| pleD | Forward | AGGATCCAGTGACTGCTAAAATACTAATAGTG | 1,388 | BamHI | pET-33b(+) |
| Reverse | AGCGGCCGCTTAGATTTTTTCAGACACAAC | NotI | |||
| ctrA | Forward | GGGCTAGCGGTCACCATCACCATCACCATGGAATGCGCATACTGTTAATAGA | 859 | NheI | pET-11a(+) |
| Reverse | GGGATCCGGCTTCTTCCGCATAATC | BamHI | |||
| ntrX | Forward | GGGCTAGCGGTCACCATCACCATCACCATGGATCTAAGGTAAAGAGATTTTACATA | 1,411 | NheI | pET-11a(+) |
| Reverse | GGGATCCGGTAAACAGCCCCAGCAT | BamHI | |||
| pleC mut | Forward | CAATTTCTTGCTAATGTCAGTGCTGAGTTACGCACTCCTCTAAAC | NAb | NA | NA |
| Reverse | GTTTAGAGGAGTGCGTAACTCAGCACTGACATTAGCAAGAAATTG | ||||
| pleD mut | Forward | CCAGATTTGATACTTCTCGCCGTCATGATGCCAGGAATGGACGGG | NA | NA | NA |
| Reverse | CCCGTCCATTCCTGGCATCATGACGGCGAGAAGTATCAAATCTGG | ||||
| pleD CHis | Forward | GGCCATGGGCACTGCTAAAATACTAATAGTGG | 1,385 | NcoI | pET-33b(+) |
| Reverse | GTGCGGCCGCGATTTTTTCAGACACAACCGAG | NotI |
Restriction enzyme cleavage sites are underlined.
NA, not applicable.
Expression and purification of recombinant proteins from E. coli.
Expression of recombinant proteins (rPleCHKD, rPleDNHis, rPleDCHis, rCckAHKD, rCtrA, and rNtrX) was induced in E. coli BL21(DE3), and rNtrYHKD fused with maltose binding protein was expressed in E. coli DH5α with 1 mM isopropyl-1-thio-β-d-galactopyranoside. For antibody production, all these proteins were purified from the E. coli insoluble fraction after solubilization with 6 M urea using a His-Select cartridge (Sigma-Aldrich, St. Louis, MO) or amylose resin (New England BioLabs). For biochemical assays, all these proteins except rCtrA were purified from the E. coli soluble fraction using a His-Select cartridge or amylose resin, and rCtrA was purified from the E. coli insoluble fraction, followed by refolding as described previously (14). Proteins were quantified with the BCA protein assay kit (Pierce, Rockford, IL) with bovine serum albumin as a standard.
Kinase and phosphotransfer assays.
Histidine autokinase activity of A. phagocytophilum rPleCHKD and phosphotransfer from rHKDs of SKs to recombinant RRs were assayed as described for E. chaffeensis (14).
Western blot analysis.
Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a sheet of the nitrocellulose membrane as described previously (14). The membrane was incubated with primary antibodies (1:500 dilution of antisera or 1:50 dilution of affinity-purified antibodies in Tris-buffered saline [20 mM Tris-HCl, pH 7.5, 0.15 M NaCl] with 1% skim milk), followed by incubation with a secondary antibody, horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (1:2,000) (KPL, Gaithersburg, MD). The membrane was then incubated with enhanced chemiluminescence Western blotting detection reagents (Pierce), and bands were visualized with the LAS-3000 luminescent-image analyzer (Fujifilm, Tokyo, Japan). Preimmune rabbit sera were used as negative controls for primary antibodies. Rabbit anti-rPleCHKD and anti-rPleD were prepared by ProSci (Poway, CA) and Proteintech (Chicago, IL), respectively. Antibodies were preabsorbed with the soluble fraction of uninfected HL-60 cells or affinity purified using rPleC or rPleD (500 μg per gel), separated by SDS-PAGE, and transferred to a sheet of nitrocellulose membrane as described previously (24).
Double-immunofluorescence labeling.
At 2 days postinfection (p.i.), A. phagocytophilum-infected HL-60 cells (60% infected cells) were cytocentrifuged onto a glass slide and fixed for 15 s in Diff-Quik Fixative (Dade Behring Inc., Newark, DE) at room temperature. The fixed infected cells were incubated with rabbit anti-rPleCHKD serum at 1:100 dilution in phosphate-buffered saline (PBS) (136.5 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 27.5 mM KH2PO4, pH 7.4) or rabbit anti-rPleD antiserum at 1:20 dilution in PBS and anti-P44 monoclonal antibody (Mab) 5C11 (IgG2b) at 1:100 dilution in PBS at 37°C for 1 h, followed by incubation with secondary antibodies (Alexa Fluor 488-conjugated goat anti-mouse IgG and Alexa Fluor 555-conjugated goat anti-rabbit IgG) (Molecular Probes, Eugene, OR) at a 1:100 dilution in PBS at 37°C for 1 h. As negative controls, A. phagocytophilum-infected HL-60 cells were incubated with preimmune rabbit serum or mouse IgG2b isotype control (30), as appropriate. Rabbit antisera (200 μl) were preabsorbed on ice with sonicated lysate from 2 × 106 uninfected HL-60 cells. The slides were analyzed using a Nikon Eclipse E400 fluorescence microscope with a xenon-mercury light source (Nikon Instruments Inc., Melville, NY).
Time course experiment.
A. phagocytophilum-infected HL-60 cells were harvested, sonicated on ice, passed through a 2.7-μm filter, and centrifuged at 18,000 × g for 10 min at 4°C to harvest host cell-free bacteria. The viability of the bacteria was determined with a Live/Dead BacLight kit (Invitrogen). To synchronize the infection, 1.46 × 107 HL-60 cells were mixed with isolated host cell-free bacteria at 37°C for 3 h with constant shaking (40 rpm) on an orbital shaker (New Brunswick Scientific Inc., Edison, NJ) and then washed three times with 10 ml RPMI 1640 medium to remove unbound bacteria and resuspended in 72 ml RPMI 1640 medium with 5% fetal bovine serum. The infected cells at the indicated time point were cytocentrifuged onto a glass slide and stained with Diff-Quik stain. The slides were analyzed in a Nikon alphaphot-2 light microscope (Nikon Instruments, Inc.). Cells were harvested at each time point by centrifugation at 18,000 × g and 4°C for 10 min and stored at −80°C. To determine the amount of A. phagocytophilum in each sample, total DNA was extracted using the Qiaamp DNA minikit, and real-time PCR was performed using the primers in Table 2 and the Brilliant Syber Green QPCR core reagent kit (Stratagene) in real-time PCR equipment, MX3000P (Stratagene).
TABLE 2.
Oligonucleotides used for gene-specific real-time PCR in A. phagocytophilum
| Target gene | Direction | Sequence (5′-3′) | Amplicon size (bp) |
|---|---|---|---|
| pleC | Forward | TGATAAGGGCGACATTGGTTGTGC | 89 |
| Reverse | AGCTGCTTTGCTTAGCGTTCTGTC | ||
| pleD | Forward | ACATGCGTACAAACCCTGCCATTG | 109 |
| Reverse | AAATCATCTGCACCAGCACTCAGC |
DGC activity.
rPleDCHis was purified from the E. coli soluble fraction as described for E. chaffeensis (4). Protein (20 μg) was incubated with 20 mM acetyl phosphate for 30 min at room temperature and then with 50 nmol of GTP for 1 to 4 h at room temperature in reaction buffer (75 mM Tris-HCl, pH 8.0, 250 mM NaCl, 25 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol). After the sample was boiled at 95°C for 5 min, denatured proteins were pelleted by centrifugation, and the resulting supernatant was filtered through a 0.22-μm filter and loaded onto a Synergi Fusion-RP C18 column (Phenomenex, Torrance, CA) attached to an Äkta purifier (GE Healthcare, Piscataway, NJ). Proteins were eluted with a linear gradient of 3 to 20% methanol in 10 mM triethylammonium acetate buffer (pH 7.0), and the eluted proteins were detected at an optical density of 254 nm. Chemically synthesized c-di-GMP (Biolog, Bremen, Germany) was used as a high-performance liquid chromatography (HPLC) standard.
UV cross-linking with c-di-[32P]GMP.
Recombinant Pseudomonas aeruginosa His6-WspR, which has high DGC activity (13), was purified from the E. coli soluble fraction using a His-Select cartridge as described previously (14). c-di-[α-32P]GTP was prepared by incubating 20 μg of rWspR, 100 μCi of [α-32P]GTP (3,000 Ci/mmol; 10 mCi/ml; Perkin Elmer, Shelton, CT), and 80 pmol unlabeled GTP in 50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol for 4 h at room temperature and was then incubated with 10 U of calf intestine alkaline phosphatase (New England BioLabs) for 30 min at room temperature to hydrolyze unreacted GTP. c-di-GMP production and elimination of unreacted GTP in the reaction mixture were confirmed by reverse-phase HPLC chromatography as described above after 20 μg of rWspR and 100 pmol of unlabeled GTP were incubated under the same reaction conditions. To investigate A. phagocytophilum c-di-GMP binding proteins, the host cell-free A. phagocytophilum lysate (70 μg) and A. phagocytophilum rPleD (1 μg) were incubated with 0.1 μCi c-di-[32P]GMP with or without 2 μM of unlabeled c-di-GMP at room temperature for 10 min, UV cross-linked for 10 min, precipitated by 10% trichloroacetic acid, resuspended in SDS sample buffer, and subjected to SDS-PAGE. After electrophoresis, the gel was dried and exposed to a Storage Phosphor Screen (Molecular Dynamics, Sunnyvale, CA). The screen was scanned, and the bands were quantified with a PhosphorImager 445 Si (Molecular Dynamics).
Effect of the c-di-GMP analog 2′-O-TBDMS -c-di-GMP on A. phagocytophilum infection.
2′-O-(tert-butyldimethylsilyl) (TBDMS)-c-di-GMP (C32H58N12O14P2Si2: mass, 952.99 Da; >98% pure as determined by HPLC) is a c-di-GMP hydrophobic analog produced as an intermediate during chemical synthesis of c-di-GMP (12) and was dissolved in dimethyl sulfoxide (DMSO) to make the stock solution. Host cell-free A. phagocytophilum was incubated with 2′-O-TBDMS-c-di-GMP at a final concentration of 0.05 mM in RPMI medium or the same volume of DMSO (no more than 0.5% [vol/vol]) and incubated at 37°C for 2 h. After the treated bacteria were washed twice with RPMI 1640 medium, the bacteria were added to uninfected HL-60 cells and continuously incubated in RPMI 1640 medium with 5% fetal bovine serum and 2 mM l-glutamine at 37°C. The cells were harvested and stained with Diff-Quik, and the percentage of A. phagocytophilum-infected cells was scored in 100 HL-60 cells in triplicate culture wells. Images of Diff-Quik-stained cells were captured by a Spot RT digital camera (Diagnostic Instruments, Sterling Heights, MI) coupled to a Nikon microscope. Total DNA was extracted from an aliquot of samples, and quantitative-PCR was performed using the primer pairs in Table 2 and human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) RT-F and human GAPDH RT-R to determine the relative number of organisms per HL-60 cell.
RESULTS
Sequence analysis of PleC and PleD.
Using hydropathy plots and the PSORT program, A. phagocytophilum PleC (APH0944; 470 amino acid residues, after removal of the 27-residue signal peptide; calculated molecular mass, 50 kDa) was predicted to be an inner membrane protein with one transmembrane region and an HKD containing H, N, G1, G2, and G3 motifs (18). The putative A. phagocytophilum PleC kinase domain (residues 206 to 470) has 41.5% amino acid sequence identity to the C. crescentus PleC kinase domain. The identity between the A. phagocytophilum and E. chaffeensis PleC kinase domains is 67.9%, suggesting similar kinase activities. In contrast, the A. phagocytophilum PleC sensor domain (residues 1 to 205) has only 6.5% identity with the C. crescentus PleC sensor domain. While the stimulus for the C. crescentus PleC sensor domain is unknown, the higher identity (31.6%) between the A. phagocytophilum and E. chaffeensis PleC sensor domains probably reflects not only their close phylogenic relationship, but also their similar intracellular inclusion environments, which are distinct from the aquatic environment surrounding free-living C. crescentus.
C. crescentus PleD is an unorthodox RR consisting of a receiver domain (D1) with the phosphorylatable Asp, a receiver-like adaptor domain (D2), and the DGC domain (2, 9). Alignment of A. phagocytophilum PleD (APH0551; 456 residues; calculated molecular mass, 52 kDa) with C. crescentus PleD defined the three domains, D1, D2, and a GGDEF domain (Fig. 1). The D1 domain of A. phagocytophilum PleD contains the characteristic receiver residues (27), including the predicted phosphorylation site, Asp53 (Fig. 1). The D2 domain contains Asp207, which corresponds to Asp53 of the D1 domain, but it does not contain the other two characteristic residues of the receiver domain of RRs (a hydroxyl-containing residue and Lys). The C-terminal output domain of A. phagocytophilum PleD was found to contain four consensus regions characteristic of the GGDEF domain (10) associated with DGC activity (2) (Fig. 1).
FIG. 1.

Schematic representation of the domain structures of PleC and PleD in A. phagocytophilum and C. crescentus. The numbers represent amino acid residues. The horizontal bars above PleC and PleD indicate the regions cloned for the functional study. The percentages in parentheses between the two aligned proteins indicate sequence identity.
Expression of PleC and PleD by A. phagocytophilum.
We determined the mRNA expression of A. phagocytophilum pleC and pleD in a patient (NY37) who was previously confirmed to have HGA (15) and in cultured cells by reverse transcription-PCR. Both pleC and pleD mRNAs were expressed by A. phagocytophilum in the blood of patient NY37 and in HL-60 cells (Fig. 2). No amplicon was detected in the absence of reverse transcriptase.
FIG. 2.
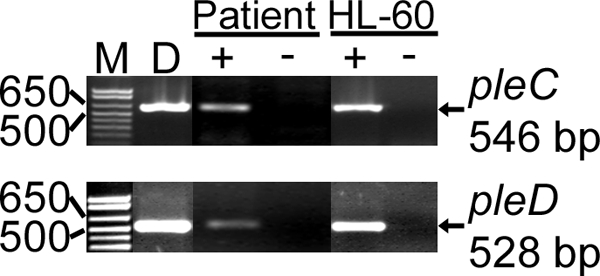
pleC and pleD are transcribed by A. phagocytophilum in the blood of an HGA patient and in human myelocytic leukemia HL-60 cells. M, molecular size marker; D, positive control (chromosomal DNA used as a template) for the PCR; + and − indicate the presence or absence of reverse transcriptase, respectively. Genes and base pair sizes of amplified products are indicated.
To assess PleC and PleD expression, localization, and function, DNA fragments encoding PleCHKD (Fig. 1, PleC, residues 206 to 470) and full-length PleD were cloned into E. coli expression vectors. The protein was expressed and purified to near homogeneity (Fig. 3A) from the insoluble inclusion bodies using nickel column chromatography. Rabbit antisera raised against rPleCHKD and rPleD specifically reacted with their respective recombinant proteins (Fig. 3A). The antisera also specifically recognized native proteins of A. phagocytophilum isolated from infected HL-60 cells (Fig. 3A). The size of each native protein was correlated with the size based on the respective amino acid sequences. These antisera did not react with proteins derived from uninfected HL-60 cells (Fig. 3A). Preimmune rabbit sera did not react with any proteins in uninfected HL-60 cells or A. phagocytophilum (data not shown).
FIG. 3.

PleC and PleD are expressed by A. phagocytophilum in human myelocytic leukemia HL-60 cells. (A) Lanes 1 and 2, nickel affinity-purified rPleC; lanes 5 and 6, nickel affinity-purified rPleD; lanes 3 and 7, A. phagocytophilum-infected HL-60 cells; lanes 4 and 8, uninfected HL-60 cells. Lanes 1, 2, 5, and 6, 0.5 μg protein per lane; lanes 3, 4, 7, and 8, 20 μg protein per lane. Lanes 1 and 5, Coomassie brilliant blue stain; lanes 2 to 4, Western blotting using anti-rPleC; lanes 6 to 8, Western blotting using anti-rPleD. Note rPleCHKD (30 kDa) and A. phagocytophilum native PleC (50 kDa) recognition by anti-rPleC and rPleD (56 kDa) and A. phagocytophilum native PleD (52 kDa) recognition by anti-PleD. (B) Infected HL-60 cells at 2 days p.i. (60% infected cells) were double labeled for immunofluorescence. The following antisera were used: anti-A. phagocytophilum P44 (anti-mouse IgG; green; P44) and anti-rPleC or anti-rPleD (anti-rabbit IgG; red; PleC or PleD) MAbs. The images on the right are superimposed images viewed with green and red filters (Merge) and a phase-contrast image to show the relative intracellular location of morulae. As controls for immunofluorescence labeling, A. phagocytophilum-infected HL-60 cells were incubated with preimmune rabbit serum (Preimmune) and secondary conjugated anti-rabbit IgG or with mouse isotype IgG2b control and secondary conjugated anti-mouse IgG. Scale bar, 5 μm.
Double-immunofluorescence labeling of A. phagocytophilum in HL-60 cells with rabbit anti-rPleC and anti-rPleD and mouse MAb 5C11 against A. phagocytophilum P44 confirmed that PleC and PleD were expressed by replicating A. phagocytophilum in microcolonies (morulae) in HL-60 cells (Fig. 3B). As negative controls for immunofluorescence labeling, A. phagocytophilum-infected HL-60 cells were incubated with the respective preimmune rabbit serum and secondary conjugated anti-rabbit IgG or with the isotype control mouse IgG2b and secondary conjugated anti-mouse IgG. There was no detectable labeling with these sera, indicating that labeling with both MAb 5C11 and rabbit anti-rPleC and anti-rPleD protein sera was specific (Fig. 3B).
Synchronous culture of A. phagocytophilum and temporal expression of PleC and PleD.
Host cell-free A. phagocytophilum (liberated from heavily infected HL-60 cells by mild sonication) was filtered to yield bacteria of relatively homogeneous size (0.2 to 0.5 μm) (Fig. 4A, Ap). Approximately 80% of the population was viable based on staining with a Live/Dead BacLight bacterial-viability kit (Fig. 4A, L/D). A synchronous culture of A. phagocytophilum was prepared using this preparation as the inoculum. At 0 h (0 h after washing to remove non-host-associated bacteria after 3 h of incubation of HL-60 cells with host cell-free A. phagocytophilum at 37°C), only a few bacteria per host cell were localized by Diff-Quik staining. At 12 h p.i., almost every infected HL-60 cell contained one or two small morulae (1 to 2 μm in diameter); at 24 h p.i., almost every infected HL-60 cell had one or two intermediate-size morulae (3 to 4 μm) loosely packed with small bacteria. At 36 h p.i., almost every infected HL-60 cell had one or two intermediate-size morulae (3 to 4 μm) plus several small morulae (1 to 2 μm) densely packed with bacteria. At 48 h p.i., almost every infected HL-60 cell had several large morulae (3 to 5 μm) densely packed with bacteria. At 60 h p.i., almost every infected cell had several large morulae (3 to 10 μm) with clumped bacteria, and at 72 h p.i., almost every infected HL-60 cell began to lyse, and the remaining intracellular morulae were dispersed with some clumped bacteria (Fig. 4A). Quantitative PCR using a pair of primers specific for A. phagocytophilum pleD showed the lag phase of A. phagocytophilum growth for approximately 0 to 36 h and the exponential growth phase from 36 to 72 h p.i. (Fig. 4B).
FIG. 4.

PleC and PleD expression in synchronously cultured A. phagocytophilum. (A) Synchronously cultured A. phagocytophilum. Ap, host cell-free A. phagocytophilum used as inoculum; Diff-Quik stain. L/D, Live/Dead BacLight bacterial-viability test of the host cell-free A. phagocytophilum; green, live bacteria; red, dead bacteria. (0 to 72) Synchronously cultured A. phagocytophilum in HL-60 cells using host cell-free A. phagocytophilum harvested from 0 to 72 h p.i.; Diff-Quik stain. The bacteria or morulae are indicated by arrowheads. Scale bar, 10 μm. (B) Synchronous growth of A. phagocytophilum as determined by quantitative PCR. Total DNA was extracted at 0 to 72 h p.i. The amount of DNA was determined by quantitative PCR and is presented relative to the DNA content measured at 0 h p.i., which was defined as 1. The values are the means ± standard deviations of triplicate samples. (C) Temporal expression of PleC and PleD during A. phagocytophilum intracellular development. PleC and PleD expression was determined by Western blot analysis using antisera against A. phagocytophilum rPleCHKD and rPleD. The numbers of bacterial-genome equivalents loaded per lane were normalized by real-time PCR using the single-copy genomic pleD as a template. Note the stronger band densities of both PleC and PleD at 60 h p.i. (times in hours are indicated below the blot).
After normalization using bacterial DNA, both PleC and PleD were synchronously detected starting at 36 h p.i. and peaked at 60 h p.i. in the mid-exponential growth stage (Fig. 4C).
Autokinase activity of rPleCHKD and specific phosphotransfer from rPleCHKD to rPleD.
To characterize the biological activities of properly folded A. phagocytophilum rPleCHKD and rPleD, recombinant proteins were purified from E. coli soluble extract. By culturing bacteria at lower temperature (20 to 30°C), rPleCHKD and rPleD became partially soluble and could be purified from the soluble E. coli sonicated extract by nickel affinity chromatography. rPleCHKD displayed autokinase activity (Fig. 5A) when [γ-32P]ATP was used as the phosphate donor. By aligning the A. phagocytophilum PleC sequence with other bacterial SKs, we predicted that His244 of PleC would be phosphorylated (Fig. 1). To confirm this prediction, we expressed and purified mutant rPleCHKD with His244 replaced by Ala through site-directed mutagenesis. Expression of the resulting mutant protein (rPleCHKDH244A) was induced, and it was purified to near homogeneity using the same method as for the wild-type protein. No autokinase activity was detected for the mutant protein (Fig. 5A), indicating that the conserved His residue (His244 of PleC) is required for phosphorylation. rPleD alone did not have autokinase activity (Fig. 5A). When purified rPleD was mixed with autophosphorylated rPleCHKD, rPleD autokinase activity was detected by using phosphorylated rPleCHKD as the phosphoryl donor (Fig. 5A). When mutant recombinant PleD (rPleDD53A) was mixed with autophosphorylated rPleCHKD, no autokinase activity was detected (Fig. 5A), indicating that the conserved Asp53 is required for phosphorylation.
FIG. 5.
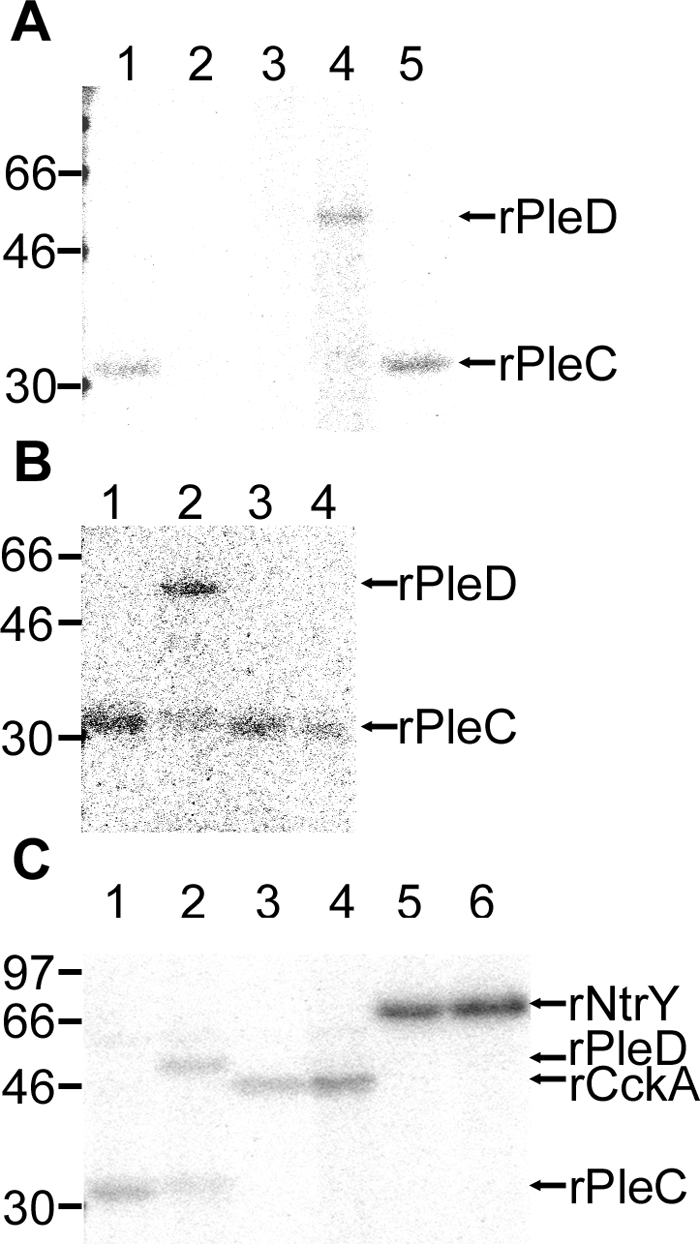
A. phagocytophilum PleC has specific His-dependent autokinase activity, and phosphotransfer from A. phagocytophilum PleC to PleD is dependent on a specific Asp of PleD. (A) Autoradiogram showing the specific His-dependent autokinase activity of rPleCHKD. Ten micrograms each of the wild-type rPleCHKD (lane 1) and rPleCHKDH244A (lane 2) were incubated with [γ-32P]ATP. Only the wild-type HKD was 32P phosphorylated. Lanes 4 and 5, autoradiogram showing the specific Asp-dependent phosphotransfer of 32P from rPleCHKD to rPleDs. rPleCHKD (2 μg) was incubated with [γ-32P]ATP, followed by incubation with 20 μg of rPleD (lane 4) or rPleDD53A (lane 5). rPleD alone (lane 3) was incubated with [γ-32P]ATP as a negative control. (B) Autoradiogram showing phosphotransfer of 32P from rPleCHKD to three recombinant RRs. rPleCHKD (10 μg) was incubated with [γ-32P]ATP (lane 1; PleC alone), followed by incubation with 20 μg wild-type rPleD (lane 2), rNtrX (lane 3), or rCtrA (lane 4). Phosphotransfer was evident only for rPleD (lane 2). (C) Autoradiogram showing phosphotransfer of 32P from three SKs to rPleD. rPleCHKD, rCckAHKD, and rNtrYHKD (10 μg each) were incubated with [γ-32P]ATP (lanes 1, 3, and 5; HKD alone), followed by incubation with rPleD (20 μg) (lanes 2, 4, and 6). Phosphotransfer occurred only from rPleC (lane 2). The numbers on the left indicate molecular masses in kDa.
Of the three A. phagocytophilum RRs (rPleD and rNtrX purified from the E. coli soluble fraction and rCtrA purified from the E. coli insoluble fraction and refolded), only rPleD autokinase activity was detected by using phosphorylated rPleCHKD as a phosphoryl donor (Fig. 5B). Of three autophosphorylated A. phagocytophilum rHKDs (rPleCHKD, rNtrYHKD, and rCckAHKD, purified from the E. coli soluble fraction), rPleD autokinase activity was detected only using phosphorylated rPleCHKD as a phosphoryl donor (Fig. 5C). These results demonstrate specific phosphotransfer from PleC to PleD.
DGC activity of rPleD.
To demonstrate DGC activity of A. phagocytophilum PleD, a C-terminal His-tagged recombinant PleD (rPleDCHis) was phosphorylated by acetylphosphate. Upon incubation of rPleDCHis with GTP, c-di-GMP was detected by HPLC using synthetic c-di-GMP as a standard (Fig. 6).
FIG. 6.
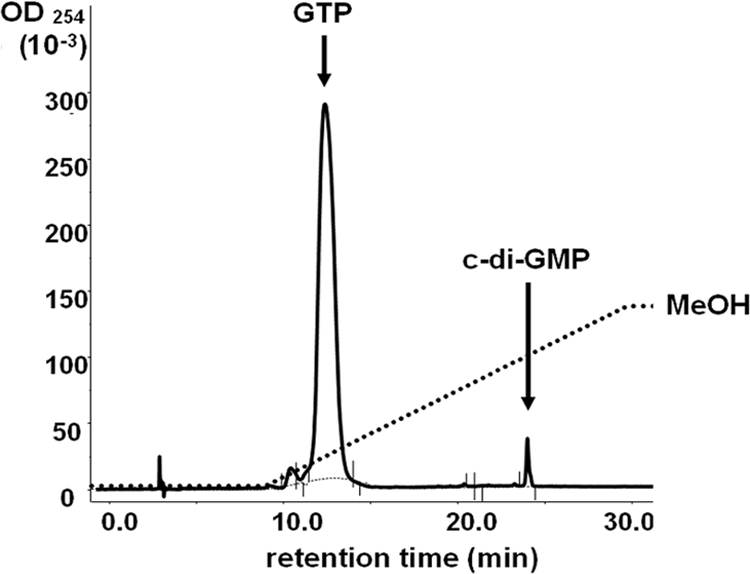
A. phagocytophilum rPleD has diguanylate cyclase activity. rPleDCHis (20 μg) was incubated with 50 nmol GTP for 1 h at room temperature; then, the sample was boiled, centrifuged, filtered, loaded onto an HPLC reverse-phase RC18 column, and eluted with a methanol gradient (dashed line). The solid line is a representative elution profile, and the retention times of GTP and a chemically synthesized c-di-GMP standard are indicated. OD254, optical density at 254 nm.
A. phagocytophilum c-di-[32P]GMP receptor.
DgrA, a c-di-GMP receptor that controls flagellar-motor function in C. crescentus, was recently detected using UV cross-linking with c-di-[33P]GMP (6). The UV cross-linking assay with c-di-[32P]GMP detected an ∼47-kDa band of A. phagocytophilum native protein at the mid-exponential stage of growth (Fig. 7). Binding of c-di-[32P]GMP was specific, because a 1,000-fold excess of unlabeled c-di-GMP competitively blocked c-di-[32P]GMP binding (Fig. 7). A. phagocytophilum PleD has an amino acid sequence similar to that of the binding site for allosteric product inhibition, as described for C. crescentus PleD (17, 20). Specific binding of c-di-[32P]GMP was also demonstrated with A. phagocytophilum rPleDNHis, suggesting allosteric inhibition of A. phagocytophilum PleD DGC by c-di-GMP (Fig. 7).
FIG. 7.
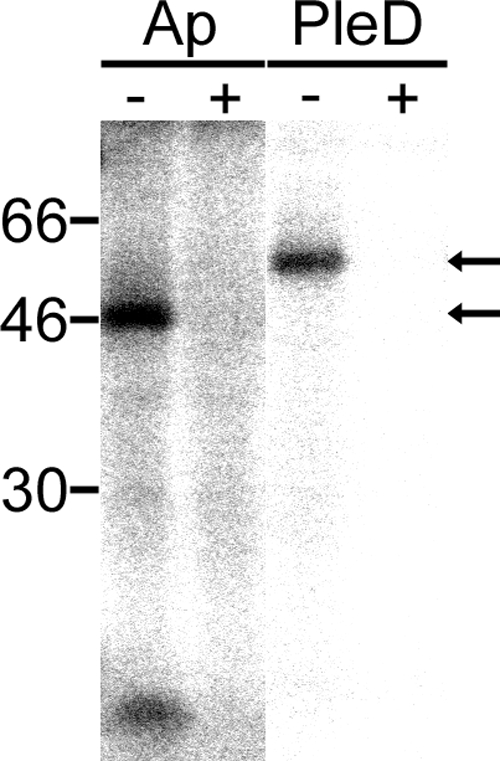
A. phagocytophilum produces a c-di-GMP binding protein. Autoradiogram of host cell-free A. phagocytophilum lysate (AP) (70 μg) and nickel affinity-purified rPleD (PleD) (1 μg) incubated with c-di-[32P]GMP, followed by UV cross-linking without (−) or with (+) 1,000-fold excess unlabeled c-di-GMP (2 μM). An ∼47-kDa protein band was detected (arrows), and the c-di-[32P]GMP binding was blocked by unlabeled c-di-GMP.
Inhibitory effect of a hydrophobic derivative of c-di-GMP.
We used a pharmacological approach to investigate the function of c-di-GMP in A. phagocytophilum. A. phagocytophilum was preincubated with a newly developed hydrophobic derivative of c-di-GMP, 2′-O-TBDMS-c-di-GMP, and added to uninfected HL-60 cells. Pretreatment with 0.05 mM (final concentration) 2′-O-TBDMS-c-di-GMP for 2 h significantly inhibited A. phagocytophilum infection in HL-60 cells compared to a DMSO control, as determined by real-time PCR and scoring of infected cells (Fig. 8). At 0 and 36 h p.i., both 2′-O-TBDMS-c-di-GMP- and DMSO-treated bacteria were detected at similar levels on or in HL-60 cells by Diff-Quik staining (Fig. 8A). In contrast, at 84 h p.i., approximately 16 and 1% of HL-60 cells were infected with A. phagocytophilum in the DMSO- and 2′-O-TBDMS-c-di-GMP-treated groups, respectively (Fig. 8B). At 132 h p.i., approximately 20% and 1% of HL-60 cells were infected with A. phagocytophilum in the DMSO- and 2′-O-TBDMS-c-di-GMP-treated groups, respectively, and some heavily infected HL-60 cells in the DMSO-treated group were lysed (Fig. 8).
FIG. 8.

2′-O-TBDMS-c-di-GMP inhibits A. phagocytophilum infection in human myelocytic leukemia HL-60 cells. A. phagocytophilum bacteria were pretreated with DMSO solvent control or 0.05 mM 2′-O-TBDMS-c-di-GMP and, after being washed, incubated with HL-60 cells in the absence of the compound. (A) Representative Diff-Quik-stained images of HL-60 cells infected with A. phagocytophilum pretreated with 0.05 mM 2′-O-TBDMS-c-di-GMP or DMSO at 0, 36, 84, or 132 h p.i. The arrowheads indicate bacterial inclusions or bacteria. Scale bar, 10 μm. (B) Relative numbers of organisms per HL-60 cell at 84 and 132 h p.i. determined by real-time PCR in triplicate wells. The percentages of infected HL-60 cells at 84 and 132 h p.i. were determined in 100 HL-60 cells in triplicate wells. *, significantly different from the DMSO solvent control by Student's t test (P < 0.05; n = 3).
DISCUSSION
The present study demonstrated A. phagocytophilum PleCHKD autokinase activity and direct and specific phosphotransfer from A. phagocytophilum PleC to A. phagocytophilum PleD. In C. crescentus, two SKs, PleC and DivJ, phosphorylate PleD in vitro (20), and PleC kinase activity is required for PleD DGC activity in vivo (19). Amino acid identity between C. crescentus PleCHKD and DivJHKD is 43.0%. We determined that one of three A. phagocytophilum SKs is PleC, but not DivJ, since the HKD of A. phagocytophilum PleC has higher sequence identity to the HKD of C. crescentus PleC (37.4% identity) than to the HKD of C. crescentus DivJ (32.1% identity). These results are similar to those for E. chaffeensis PleC and PleD (14) and perhaps can be extended to pleC and pleD orthologs found in other members of the order Rickettsiales (4). There is no functional homolog of DivJ in A. phagocytophilum, since there was no observed cross-reaction with other putative A. phagocytophilum TCSs, as no phosphotransfer was detected from A. phagocytophilum rPleCHKD to other RRs (rNtrX and rCtrA) or from other HKDs (rNtrYHKD and rCckAHKD) to rPleD.
In the present study, the amount of PleD per bacterium drastically changed during A. phagocytophilum growth. Although pleC and pleD do not constitute an operon, the synchronized pattern of PleC and PleD expression in cultured HL-60 cells (37°C) demonstrates that they are coregulated in A. phagocytophilum during intracellular development. In C. crescentus, the temporal expression of PleC and PleD levels during bacterial development has not been reported; however, temporal changes in PleD-green fluorescent protein distribution have been reported: during stalk formation, PleD localizes to the cell pole, where the stalk is developed, whereas in swarmer cells, PleD is distributed diffusely throughout the cytoplasm (20). Activated PleD at the stalk pole of C. crescentus has been hypothesized to be responsible for the morphological swarmer-to-stalk cell differentiation via the action of the messenger molecule c-di-GMP (20). In A. phagocytophilum, PleC and PleD expression peaked during the mid-exponential growth phase, and thus, c-di-GMP might control the transition between reticulate cells and DC. If the A. phagocytophilum PleC-PleD system functions as a bacterial developmental-cycle regulator, these proteins might be essential for obligatory intracellular parasitism (and thus survival) of A. phagocytophilum. Mutation of either pleC or pleD abrogates normal cell differentiation; however, PleC and PleD are not essential for C. crescentus viability (26).
In the present study, we demonstrated that A. phagocytophilum rPleD has DGC activity. We found that the DGC activity of A. phagocytophilum rPleD was much lower than that of P. aeruginosa rWspR (data not shown). This result is consistent with the fact that no A. phagocytophilum protein has been predicted that contains an EAL (Glu-Ala-Leu) (http://www.sanger.ac.uk/Software/Pfam) (25) or HD-GYP (a subgroup of the HD superfamily of metal-dependent phosphohydrolases that contain an additional GYP motif) (23) domain associated with c-di-GMP-specific phosphodiesterase activity. While there may be some other types of phosphodiesterase lurking about in the background waiting to be discovered, it is possible that A. phagocytophilum PleD DGC activity is low to avoid c-di-GMP overproduction. Additionally, A. phagocytophilum PleD contains a predicted interactive site similar to the C. crescentus PleD interactive site, which allows allosteric noncompetitive feedback inhibition by c-di-GMP (5). This feedback inhibition may maintain PleD DGC activity at a low level in A. phagocytophilum. In support of this hypothesis, the c-di-GMP cross-linking study showed c-di-GMP binding to A. phagocytophilum rPleD. A hydrophobic derivative of c-di-GMP, 2′-O-TBDMS-c-di-GMP, inhibited A. phagocytophilum infection in HL-60 cells. These data suggest that c-di-GMP levels need to be temporarily regulated during A. phagocytophilum development inside host cells.
The inhibitory mechanisms of 2′-O-TBDMS-c-di-GMP warrant further investigation, as this is the first c-di-GMP analog shown to have antimicrobial effects. This c-di-GMP derivative may potentially competitively block the A. phagocytophilum c-di-GMP receptors and uncouple c-di-GMP from downstream events. Alternatively, when a large amount of the unregulated c-di-GMP derivative is taken up by A. phagocytophilum, it may dysregulate the global c-di-GMP signaling pathway.
Although c-di-GMP regulates bacterial cell surface-associated traits and community behavior, such as cell-cell signaling, biofilm formation, motility, differentiation, and virulence (22, 29), little is known about the downstream effector molecules. Recently, the c-di-GMP binding domain PilZ was identified in proteins from several bacteria, and PilZ-containing proteins are suggested to serve as adaptor molecules to downstream effectors (1). A PilZ-containing protein, DgrA, was recently shown to indirectly control flagellar-motor function in C. crescentus (6). Our c-di-GMP cross-linking study suggests that A. phagocytophilum expresses c-di-GMP binding proteins of ∼47 kDa. Recently, it was reported that c-di-GMP may be sensed by a riboswitch to regulate downstream genes (28). These observations suggest c-di-GMP might play a versatile role in bacterial signal transduction pathways. Further characterization of c-di-GMP binding proteins and the mechanisms by which 2′-O-TBDMS-c-di-GMP inhibits A. phagocytophilum infection would provide new insights into the roles of c-di-GMP and TCSs in obligatory intracellular parasitism.
Acknowledgments
This work was supported by National Institutes of Health grant R01 AI054476.
We thank Stephen Lory, Harvard Medical School (Boston, MA), for the E. coli strain overexpressing P. aeruginosa rWspR and Matthias Christen, University of Basel (Basel, Switzerland), for providing detailed methods for the c-di-GMP binding assay.
Footnotes
Published ahead of print on 31 October 2008.
REFERENCES
- 1.Amikam, D., and M. Y. Galperin. 2006. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics 223-6. [DOI] [PubMed] [Google Scholar]
- 2.Ausmees, N., R. Mayer, H. Weinhouse, G. Volman, D. Amikam, M. Benziman, and M. Lindberg. 2001. Genetic data indicate that proteins containing the GGDEF domain possess diguanylate cyclase activity. FEMS Microbiol. Lett. 204163-167. [DOI] [PubMed] [Google Scholar]
- 3.Bakken, J. S., J. S. Dumler, S. M. Chen, M. R. Eckman, L. L. Van Etta, and D. H. Walker. 1994. Human granulocytic ehrlichiosis in the upper Midwest United States. A new species emerging? JAMA 272212-218. [PubMed] [Google Scholar]
- 4.Cheng, Z., Y. Kumagai, M. Lin, C. Zhang, and Y. Rikihisa. 2006. Intra-leukocyte expression of two-component systems in Ehrlichia chaffeensis and Anaplasma phagocytophilum and effects of the histidine kinase inhibitor closantel. Cell. Microbiol. 81241-1252. [DOI] [PubMed] [Google Scholar]
- 5.Christen, B., M. Christen, R. Paul, F. Schmid, M. Folcher, P. Jenoe, M. Meuwly, and U. Jenal. 2006. Allosteric control of cyclic di-GMP signaling. J. Biol. Chem. 28132015-32024. [DOI] [PubMed] [Google Scholar]
- 6.Christen, M., B. Christen, M. G. Allan, M. Folcher, P. Jeno, S. Grzesiek, and U. Jenal. 2007. DgrA is a member of a new family of cyclic diguanosine monophosphate receptors and controls flagellar motor function in Caulobacter crescentus. Proc. Natl. Acad. Sci. USA 1044112-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demma, L. J., R. C. Holman, J. H. McQuiston, J. W. Krebs, and D. L. Swerdlow. 2005. Epidemiology of human ehrlichiosis and anaplasmosis in the United States, 2001-2002. Am. J. Trop. Med. Hyg. 73400-409. [PubMed] [Google Scholar]
- 8.Dumler, J. S., A. F. Barbet, C. P. Bekker, G. A. Dasch, G. H. Palmer, S. C. Ray, Y. Rikihisa, and F. R. Rurangirwa. 2001. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol Microbiol. 512145-2165. [DOI] [PubMed] [Google Scholar]
- 9.Galperin, M. Y., A. N. Nikolskaya, and E. V. Koonin. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 20311-21. [DOI] [PubMed] [Google Scholar]
- 10.Hecht, G. B., and A. Newton. 1995. Identification of a novel response regulator required for the swarmer-to-stalked-cell transition in Caulobacter crescentus. J. Bacteriol. 1776223-6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herron, M. J., M. E. Ericson, T. J. Kurtti, and U. G. Munderloh. 2005. The interactions of Anaplasma phagocytophilum, endothelial cells, and human neutrophils. Ann. N. Y. Acad. Sci. 1063374-382. [DOI] [PubMed] [Google Scholar]
- 12.Kawai, R., R. Nagata, A. Hirata, and Y. Hayakawa. 2003. A new synthetic approach to cyclic bis(3′→5′)diguanylic acid. Nucleic Acids Res. Suppl. 3103-104. [DOI] [PubMed] [Google Scholar]
- 13.Kulasakara, H., V. Lee, A. Brencic, N. Liberati, J. Urbach, S. Miyata, D. G. Lee, A. N. Neely, M. Hyodo, Y. Hayakawa, F. M. Ausubel, and S. Lory. 2006. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc. Natl. Acad. Sci. USA 1032839-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumagai, Y., Z. Cheng, M. Lin, and Y. Rikihisa. 2006. Biochemical activities of three pairs of Ehrlichia chaffeensis two-component regulatory system proteins involved in inhibition of lysosomal fusion. Infect. Immun. 745014-5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin, Q., N. Zhi, N. Ohashi, H. W. Horowitz, M. E. Aguero-Rosenfeld, J. Raffalli, G. P. Wormser, and Y. Rikihisa. 2002. Analysis of sequences and loci of p44 homologs expressed by Anaplasma phagocytophila in acutely infected patients. J. Clin. Microbiol. 402981-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munderloh, U. G., S. D. Jauron, V. Fingerle, L. Leitritz, S. F. Hayes, J. M. Hautman, C. M. Nelson, B. W. Huberty, T. J. Kurtti, G. G. Ahlstrand, B. Greig, M. A. Mellencamp, and J. L. Goodman. 1999. Invasion and intracellular development of the human granulocytic ehrlichiosis agent in tick cell culture. J. Clin. Microbiol. 372518-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munderloh, U. G., M. J. Lynch, M. J. Herron, A. T. Palmer, T. J. Kurtti, R. D. Nelson, and J. L. Goodman. 2004. Infection of endothelial cells with Anaplasma marginale and A. phagocytophilum. Vet. Microbiol. 10153-64. [DOI] [PubMed] [Google Scholar]
- 18.Parkinson, J. S., and E. C. Kofoid. 1992. Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 2671-112. [DOI] [PubMed] [Google Scholar]
- 19.Paul, R., T. Jaeger, S. Abel, I. Wiederkehr, M. Folcher, E. G. Biondi, M. T. Laub, and U. Jenal. 2008. Allosteric regulation of histidine kinases by their cognate response regulator determines cell fate. Cell 133452-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul, R., S. Weiser, N. C. Amiot, C. Chan, T. Schirmer, B. Giese, and U. Jenal. 2004. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 18715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rikihisa, Y. 1991. The tribe Ehrlichieae and ehrlichial diseases. Clin. Microbiol. Rev. 4286-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Römling, U., M. Gomelsky, and M. Y. Galperin. 2005. C-di-GMP: the dawning of a novel bacterial signalling system. Mol. Microbiol. 57629-639. [DOI] [PubMed] [Google Scholar]
- 23.Ryan, R. P., Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y. W. He, L. H. Zhang, S. Heeb, M. Camara, P. Williams, and J. M. Dow. 2006. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. USA 1036712-6717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Simm, R., M. Morr, A. Kader, M. Nimtz, and U. Römling. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol. Microbiol. 531123-1134. [DOI] [PubMed] [Google Scholar]
- 26.Sommer, J. M., and A. Newton. 1989. Turning off flagellum rotation requires the pleiotropic gene pleD: pleA, pleC, and pleD define two morphogenic pathways in Caulobacter crescentus. J. Bacteriol. 171392-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stock, A. M. 2003. Response regulator proteins and their interactions with histidine protein kinase, p. 237-271. In M. Inouye and R. Dutta (ed.), Histidine kinases in signal transduction. Academic Press, Piscataway, NJ.
- 28.Sudarsan, N., E. R. Lee, Z. Weinberg, R. H. Moy, J. N. Kim, K. H. Link, and R. R. Breaker. 2008. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science 321411-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamayo, R., J. T. Pratt, and A. Camilli. 2007. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 61131-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, X., T. Kikuchi, and Y. Rikihisa. 2006. Two monoclonal antibodies with defined epitopes of P44 major surface proteins neutralize Anaplasma phagocytophilum by distinct mechanisms. Infect. Immun. 741873-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woldehiwet, Z., and G. R. Scott. 1982. Stages in the development of Cytoecetes phagocytophila, the causative agent of tick-borne fever. J. Comp. Pathol. 92469-474. [DOI] [PubMed] [Google Scholar]