

Abstract
We report a DNA shuffling–based approach for developing cell type–specific vectors through directed evolution. Capsid genomes of adeno-associated virus (AAV) serotypes 1–9 were randomly fragmented and reassembled using PCR to generate a chimeric capsid library. A single infectious clone (chimeric-1829) containing genome fragments from AAV1, 2, 8, and 9 was isolated from an integrin minus hamster melanoma cell line previously shown to have low permissiveness to AAV. Molecular modeling studies suggest that AAV2 contributes to surface loops at the icosahedral threefold axis of symmetry, while AAV1 and 9 contribute to two- and fivefold symmetry interactions, respectively. The C-terminal domain (AAV9) was identified as a critical structural determinant of melanoma tropism through rational mutagenesis. Chimeric-1829 utilizes heparan sulfate as a primary receptor and transduces melanoma cells more efficiently than all serotypes. Further, chimeric-1829 demonstrates altered tropism in rodent skeletal muscle, liver, and brain including nonhuman primates. We determined a unique immunological profile based on neutralizing antibody (NAb) titer and crossreactivity studies strongly supporting isolation of a synthetic laboratory-derived capsid variant. Application of this technology to alternative cell/tissue types using AAV or other viral capsid sequences is likely to yield a new class of biological nanoparticles as vectors for human gene transfer.
INTRODUCTION
Adeno-associated virus (AAV) is a 20–25-nm nonpathogenic human parvovirus that has gained popularity as a vector for gene therapy applications.1-3 Among the 12 identified serotypes, AAV 1–9 are being developed as gene therapy vectors because of their capsid-associated tissue tropism for specific tissues.4-7 These naturally occurring AAV serotypes constitute a family of capsid backbones that can be exploited for generation of chimeric vectors to enable the transduction of tissues refractory to AAV infection or limited tropism to specific tissues. Strategies for retargeting AAV capsids include insertion of peptide ligands, conjugate-based targeting, and presentation of large protein ligands on the AAV capsid.8-11 The generation of mosaic vectors through the transcapsidation or marker rescue approach, respectively, is a notable strategy to expand the tropism of AAV serotypes.12-15 More recently, combinatorial strategies for engineering AAV vectors using error-prone PCR, DNA shuffling, and other molecular cloning techniques have been explored.16,17
DNA shuffling is a powerful process for directed evolution, which generates diversity by recombination, combining useful mutations from individual genes. Single and multigene traits that require many mutations for improved phenotypes can be evolved rapidly.18-21 Recently, Schaffer and colleagues have applied a staggered extension process (analogous to single-gene shuffling) combined with error-prone PCR to the AAV2 capsid gene and selected for variants that can escape neutralizing antibodies.16 In this study, we have extended the technique by using a family of multiple AAV genes with different serotype capsid sequences serving as templates. The combinatorial AAV capsid library generated thus was combined with directed evolution to select CS1 (hamster melanoma cell) cell line for novel cell type–specific AAV variants. The CS1 cell line, derived from chinese hamster melanoma is a well-characterized system for study of integrin–matrix interactions.22 Our lab has successfully used this cell line to demonstrate that αvβ5 is a coreceptor for AAV2 infection with low permissiveness.23 In addition, the number of deaths from melanoma worldwide is >50,000/y, of which as high as 54% are directly related to metastases to brain.24 Generating a chimeric vector with propensity for melanoma cells would provide a significant advancement to the field of cancer gene delivery. For these reasons, we used this cell line to generate a cell type–specific chimeric capsid variant that would transduce CS1 melanoma cells. The results described herein illustrate the tremendous potential of directed evolution in generating novel cell type–specific chimeric vectors and identifying the molecular determinants of tissue tropisms exhibited by different capsid motifs. When combined with rational site-directed mutagenesis and molecular modeling tools, the aforementioned combinatorial approach is expected to unravel structure–function correlates of the variant capsid down to the amino acid level and through rational design lead to controlled tropism by simply altering critical amino acids on a parental AAV serotype capsid. More important, the shuffling of AAV capsid information exchange between various parvovirus and nonparvovirus capsid sequences should lead to a new generation of novel biological nanoparticles with distinct immune profile as well as cell- and tissue-specific tropism.
RESULTS
Generation and characterization of the chimeric AAV library
The chimeric AAV capsid library was generated by DNA shuffling of genes encoding capsid sequences of AAV serotypes 1–9 (except AAV7) (Figure 1a). The diversity of the library was determined to be 2 × 106 clones and DNA sequencing of 20 randomly selected clones verified that the chimeric Cap genes were indeed recombinants from the Cap genes of different parental AAV serotypes. After establishing the plasmid library, the AAV capsid library was generated in two additional steps.8 We first partially packaged the library containing recombinant Cap genes into AAV2 capsids by cotransfection of pXR2 along with the AAV plasmid library. The resulting AAV library was then used to infect HEK293 cells [multiplicity of infection (MOI) = 1] and the cells superinfected with adenovirus (Ad) dl309, thus ensuring production of chimeric AAV capsids packaging the corresponding chimeric AAV genome.
Figure 1.
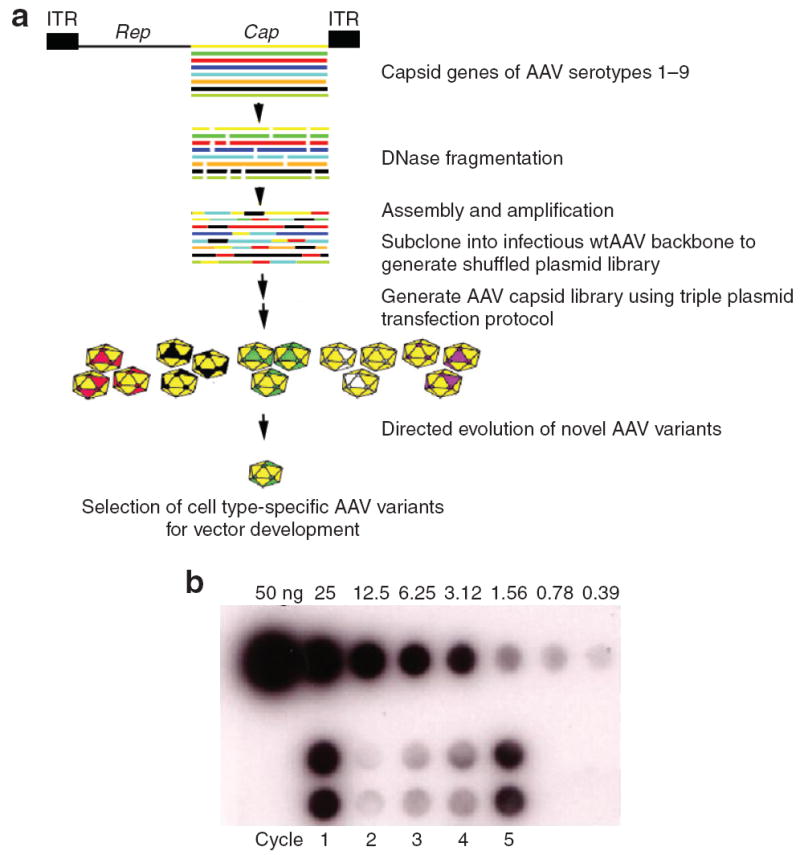
(a) Strategy for generating a combinatorial adeno-associated virus (AAV) library that will be subjected to directed evolution to isolate cell type/receptor-specific AAV mutants. AAV Cap genes were randomly fragmented and reassembled using cycles of denaturation, annealing, and extension using PCR. Following this reassembly reaction, PCR amplification with specific primers was used to generate full-length Cap chimeras for cloning into wild-type AAV2 backbone to generate shuffled plasmid library. AAV particle library was generated by plasmid transfection protocol. Specific cell line (hamster melanoma CS1 cells) previously shown to be restricted to transduction of AAVs will be tested with the chimeric AAV library for a premissive clone. Subsequent steps will involve development of novel AAV variants as targeted vectors for in vivo gene delivery applications. (b) Dot blot demonstrating the directed evolution of cell type–specific AAV variant from CS1 cells after five infectious cycles (n = 5). Data are shown in duplicate. ITR, inverted terminal repeat.
Directed evolution of a melanoma-specific AAV vector
Directed evolution of a CS1-specific AAV variant was achieved by infecting CS1 cells at an MOI of 1,000 in the presence of Ad dl309 (MOI = 20). As shown in Figure 1b, repeated cycling (every 48 h, n = 5) of the AAV library in CS1 cells resulted in an initial decrease in the recovery of replication-competent AAV as determined by dot blot, followed by evolution of a novel capsid variant (chimeric-1829). DNA extracted from cell lysate obtained from the fifth cycle served as a template for PCR using primers P3 and P4. Sequence analysis helped identify the isolate as a chimera derived from fragments of AAV1, AAV2, AAV8, and AAV9 genomes, referred to as chimeric-1829 (Figure 2a). Such recombination likely resulted from crossover at different 5′ and 3′ locations along the capsid-coding region. The first 1,200 base pairs of the chimeric-1829 Cap gene is derived from the 5′ region of AAV1 Cap, followed by a crossover including 90 base pairs from AAV8 Cap, a fragment from AAV2 up to nucleotide (nt) 2045, and the remaining portion derived from the corresponding region in AAV9 Cap. In addition, three silent mutations at nt 763, 1285, and 2099 were observed.
Figure 2. Physical characterization of chimeric-1829.
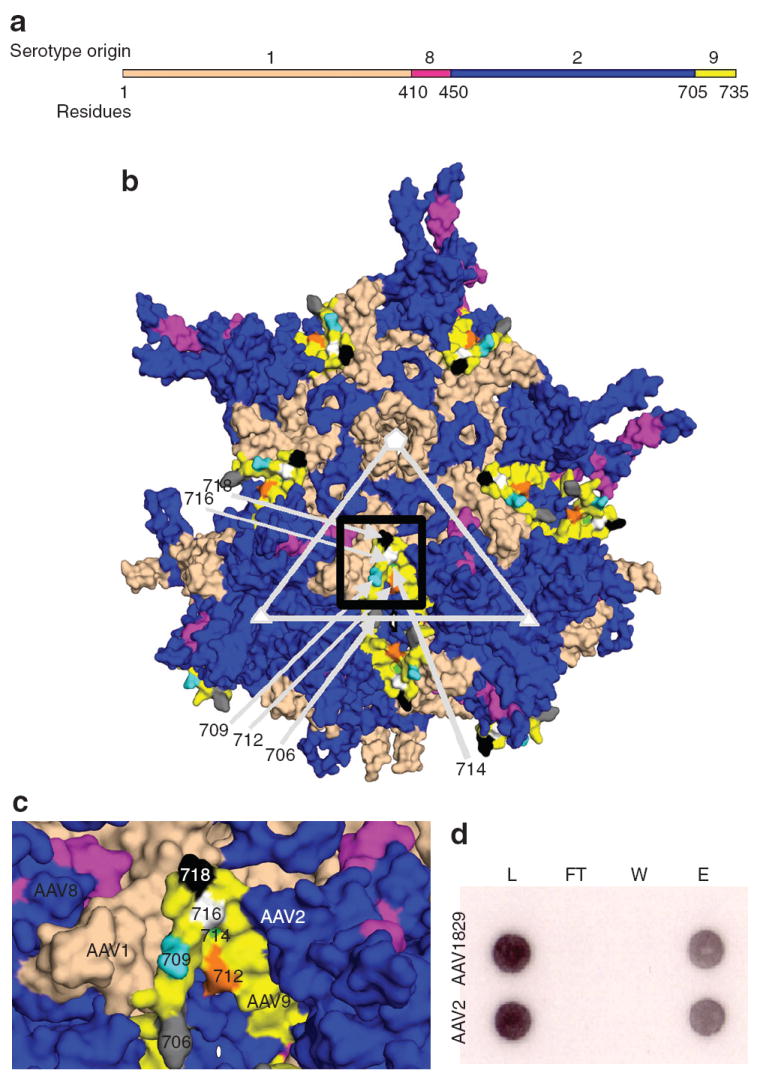
(a) Primary structure of novel adeno-associated virus (AAV) variant selected from CS1cell. (b) Three-dimensional model showing VP3 subunits of AAV1829 in relation to the fivefold (white pentagon); threefold (white triangle) and twofold (white oval) axes of symmetry. AAV1-derived residues are colored salmon, AAV2 in blue, AAV8 in dark pink, and AAV9 in yellow. (c) Black box region in 2. Panel b shown at higher magnification. Key residues in the C-terminal AAV9 region that differ from AAV2 are colored gray (706); cyan (709); orange (712); green (714); white (716); and black (718) using Vp1 numbering. (d) Heparin binding profiles of AAV1829 and AAV2. L, load; FT, flow through; W, wash; e, elution.
Structural analysis of chimeric-1829
As shown in Figure 2b, most surface-exposed variable loops in the threefold region of chimeric-1829 are derived from AAV2, including previously identified heparin-binding residues.25-27 The heparin-binding assay verified that chimeric-1829 and AAV2 share the same heparin-binding and elution profiles (Figure 2c). On the contrary, VP3 pentamer interactions at the fivefold axis of symmetry appear to be mainly derived from AAV1, although AAV2 contributes to the HI loop (located between βH and βI) that extends to overlap neighboring VP3 subunits. In addition, residues from AAV1 form part of the raised capsid surface region between the depressions at the two- and fivefold axes (Figure 2b). The residue stretch from 264 to 272 derived from AAV1 has been shown to be structurally variable between the AAV capsids,28,29 and modeling also predicted this variability for chimeric-1829. In addition, these residues have been shown to affect AAV2 cellular transduction dramatically and they form a minor part of the A20 epitope.30,31
Regions derived from AAV8 (residues 410–450) are located on the finger-like projection that forms the protrusions that surround the threefold axis and are mostly buried in the assembled capsid, with surface-exposed regions adjacent to the regions contributed by AAV1 (Figure 2b). The AAV9 residues (705–736) are located on the wall of the twofold dimple region and make contact with AAV1 residues that form the raised capsid region between the icosahedral two- and fivefold axes (Figure 2b). The AAV1 region between the two- and fivefold axes and the AAV8 and AAV9 regions, which differ from AAV1/AAV2 capsid sequences by <10 amino acids, might contribute to the melanoma-tropic trait seen in chimeric-1829 vectors. More important, the identification of such clusters of residues enables mapping specific regions on the AAV capsid that can be targeted for rational manipulation.
Biological characterization of chimeric-1829 vectors in vitro
As shown in Figure 3a, chimeric-1829 vectors packaging the green fluorescent protein (GFP) transgene transduce CS1 cells with a higher efficiency than other AAV serotypes. In addition to the higher number of transduced cells, the intensity of GFP was also significantly higher for chimeric-1829. When tested at low MOI as previously described,23 chimeric-1829 transduced CS1 cells significantly more efficiently (data not shown). We tested the transduction efficiency of chimeric-1829 and parental serotypes on a large panel of human and murine melanoma cells of both recently or long-standing derivation. As shown in Table 1 and Figure 3b, AAV2 and chimeric-1829 could efficiently transduce human melanoma cell lines that have been in long-term culture, but neither efficiently infected recent primary cultures of two human melanomas (UNC Mel 1.2 and UNC Mel 3.1, both <14 days in culture). Interestingly, transduction efficiency of AAV2 in all mouse melanoma cell lines was markedly lower than that displayed by chimeric-1829. Of the mouse and human cell lines tested, AAV1 transduced only two human cell lines (A375 and WM-2664 at moderate and low efficiency, respectively), whereas AAV8 and AAV9 did not transduce any of these cell lines (Table 1 and Figure 3b). Heparin-inhibition studies were then carried out as described.12 As shown in Figure 3c, preincubation of chimeric-1829 capsids with soluble heparin (30 μg/ml; Sigma, St Louis, MO) blocked transduction in both CS1 hamster melanoma and HEK293 cells suggesting that chimeric capsid utilizes heparan sulfate as a primary receptor in vitro.
Figure 3. Biological characterization of chimeric-1829 compared with some parental serotypes.
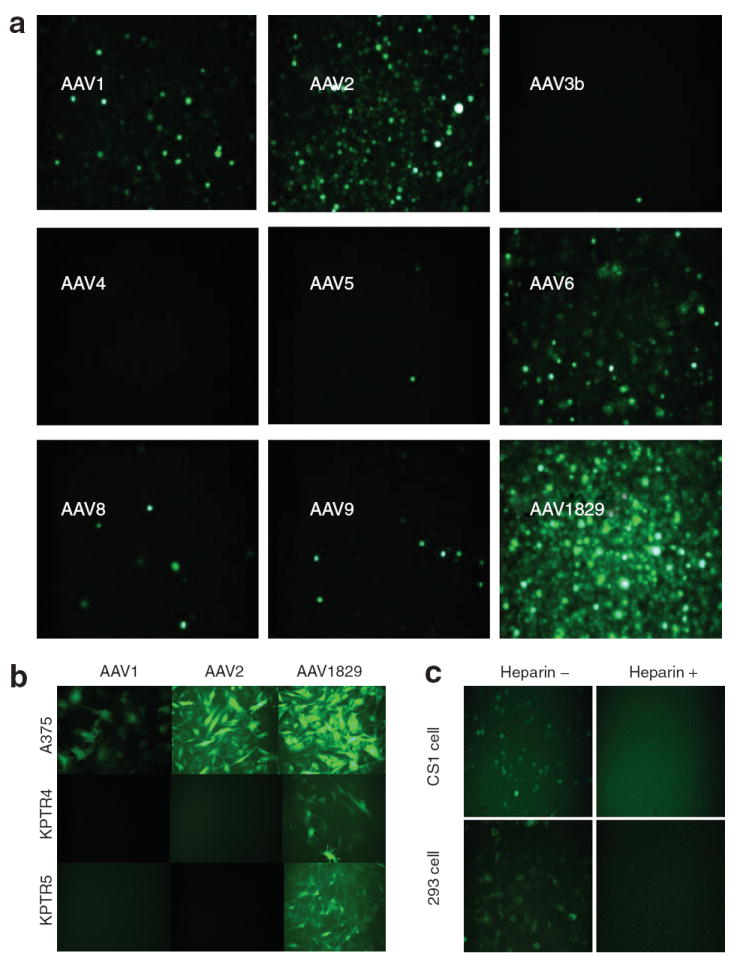
(a) Fluorescence micrographs of green fluorescent protein (GFP) transgene expression in CS1 cells transduced with AAV serotypes 1–9 (except 7) and the novel variant chimeric-1829 at a multiplicity of infection (MOI) of 1,000 for 48 hours. (b) Representative fluorescence micrographs of GFP transgene expression in different human and mouse melanoma cell lines infected with chimeric-1829 and parental serotypes at an MOI of 1,000 for 48 hours. (c) Heparin-inhibition assay. Top panels show CS1 cells, and bottom panels show 293 cells. Left and right columns are transduction profiles in the absence and presence of heparin, respectively.
Table 1.
The transduction efficiency of chimeric-1829 and its parental serotypes in human and mouse melanoma cell lines
| Cell line source | Cell line name | AAV1 | AAV2 | AAV8 | AAV9 | Chimeric-1829 |
|---|---|---|---|---|---|---|
| Human | Mel505 | − | + | − | − | + |
| UACC-257 | − | ++ | − | − | ++ | |
| A375 | +++ | ++++ | − | − | ++++ | |
| PMWK | − | ++ | − | − | ++ | |
| WM-2664 | + | ++++ | − | − | ++++ | |
| SKMEL-24 | − | +++ | − | − | +++ | |
| A2058 | ND | ++++ | ND | ND | ++++ | |
| UNC Mel 1. 2 | − | − | − | − | − | |
| UNC Mel 3.1 | − | − | − | − | − | |
| Mouse | KPTR1 | − | − | − | − | ++ |
| KPTR4 | − | − | − | − | ++ | |
| KPTR5 | − | − | − | − | +++ | |
| KPTR6 | − | − | − | − | +++ | |
| KR287T1 | − | − | − | − | +++ | |
| KR287T2 | ND | − | ND | ND | ++ | |
| KR389 | ND | − | ND | ND | + |
ND, no data.
Biological characterization of chimeric-1829 vectors in vivo
Although we initially selected our chimeric library on CS1 cells do to lack of AAV2 coreceptor and reduced susceptibility for vector transduction, we explored the ability of chimeric-1829 to transduce these tumor cells in vivo using severe combined immunodeficiency mice and CS1 derived tumors. We observed a three-and fivefold increase in transduction in vivo when comparing chimeric-1829 with serotypes 1 and 2 (Supplementary Materials and Methods). Types 8 and 9 did not transduce the CS1 target cells in this model (data not shown). Although we observed the ability to transduce target cells after direct injection, we did not detect transduction of target cells after tail vein injection, but instead observed a unique transduction pattern compared to controls (data not shown) and therefore decided to further explore this observation in normal mice to ensure these results were not influenced by the mouse background (e.g., severe combined immunodeficiency). The general transduction profile of chimeric-1829 in vivo in C57BL/6 was carried out using CBA-Luc cassette and injected into mice through different routes. As indicated by the representative bioluminescent images (Figure 4a and b), transgene expression levels in skeletal muscle and liver, obtained by intramuscular or intravenous injection of chimeric-1829 were much lower in comparison with AAV1 and AAV2. Corresponding injections with AAV8 and AAV9 vectors result in widely disseminated and high transgene expression levels in contrast to that obtained with chimeric capsid (data not shown).
Figure 4. In vivo transduction ability compared with some parental serotypes.
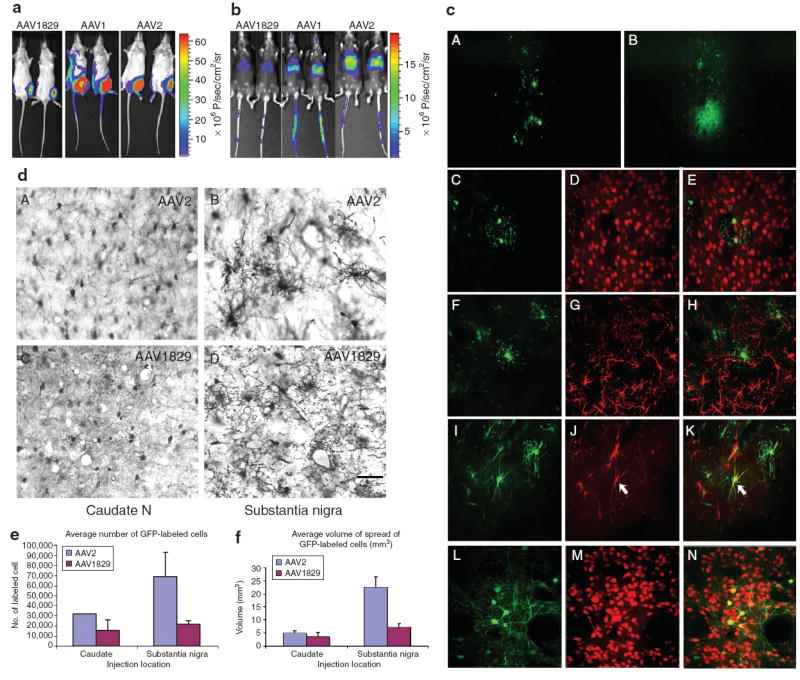
(a) Luciferase transgene expression in the hindlimbs of Balb/C mouse injected with 1010 vector genomes of the novel adeno-associated virus (AAV) variant and parental AAV1, 2. Bioluminescent images were obtained at the end of 1 month using a Xenogen IVIS system. (b) Luciferase transgene expression in the liver through tail vein injecton of 1011 vector genomes of the novel AAV variant and parental AAV1, 2. Bioluminescent images were obtained at the end of 1 month using a Xenogen IVIS system. (c) Patterns of transduction in the rat striatum 1 month after a 1 μl infusion of either AAV1829 or AAV2. (d) Comparison of the transduction efficiency of AAV2 and chimeric-1829 after injections into the (a and c) caudate nucleus and (b and d) substantia nigra of monkeys after immunostaining for green fluorescent protein (GFP). Scale bar = 50 μm. (e,f) Stereological counts of GFP-labeled cells and area of spread after each vector show that AAV2 shows greater transduction efficiency over a larger volume than chimeric-1829 in these areas.
We extended this analysis to rodent brain as melanocytes and neuronal precursors share a common lineage. In rat brain, extensive studies have shown that AAV serotypes 1, 2, 8, and 9 exhibited a predominant neuronal tropism,32-34 but this common tropism did not prove to be the case for chimeric-1829. As seen in Figure 4c, 1 month after infusion of AAV1829, the general pattern of transduction proved quite distinct from that of AAV2 (Figure 4c, panel A versus panel B, respectively). The vast majority of chimeric-1829-transduced cells did not colocalize with the neuronal marker NeuN (Figure 4c, panels C–E) or the astrocyte marker glial fibrillary acidic protein (Figure 4c, panels F–H) as previously shown for serotypes 1, 2, 8, and 9.32-34 However, a number of GFP positive cells did colocalize with nestin, indicative of neuronal fated progenitor cells, reactive astrocytes, or type I astrocytes.35,36 In marked contrast, the vast majority of AAV2-transduced cells colocalized with NeuN, a marker for neurons (Figure 4c, L–M). These studies were further corroborated by in vivo studies in monkey brains following injection into the caudate nucleus or the substantia nigra using AAV2 and chimeric-1829. As shown in Figure 4d, chimeric-1829 demonstrates a markedly reduced tropism for primate neurons in comparison with control. Figure 4e and f describe the reduced efficiency of transduction and spread of chimeric-1829 compared to AAV2 in caudate and substantia nigra further demonstrating the unique tropism of this vector.
Neutralizing antibody titers and crossreactivity
Based on modeling residue, the amino acid stretch from 264 to 272 derived from AAV1 forms a minor part of the A20 epitope.30,31 We tested chimeric-1829 in an enzyme-linked immunosorbent assay to see whether these structural amino acids contributed or interfered with A20 recognition. Figure 5a shows complete loss of A20 binding to chimeric-1829 when compared to AAV2 control suggesting a change in this structural epitope. In order to understand the immunological profile of chimeric-1829 with parental serotypes, neutralizing antibody (NAb) titers were analyzed by assessing the ability of serum antibody to inhibit vector transduction. NAb titers shown are reciprocals of the highest serum dilution leading to a 50% decrease in transduction levels. Sera from mice that received the booster injection exhibited NAb titers ranging from 1,000 to 10,000 (Figure 5b and c). As shown in Figure 5b, the antisera from AAV1-, AAV8-, and AAV9-immunized mice do not crossreact with chimeric-1829. Antisera from AAV2-immunized mice display some crossreactivity, albeit at a 25-fold lower NAb titer. In corollary, antisera against chimeric-1829 do not crossreact with AAV1, AAV8, or AAV9, while displaying crossreactivity with AAV2 capsids at a 25-fold lower NAb titer (Figure 5c). It is noteworthy to mention that AAV1 and AAV9 residues that differ from AAV2 are located in a minor A20 epitope and an antigenic region, respectively, previously identified on the AAV2 capsid.30 It is likely that such location of residues derived from other serotypes might play a role in the differential antigenic reactivity exhibited by chimeric-1829 in comparison with parental serotypes.
Figure 5. Neutralizing antibody (Nab) titer and crossreactivity.
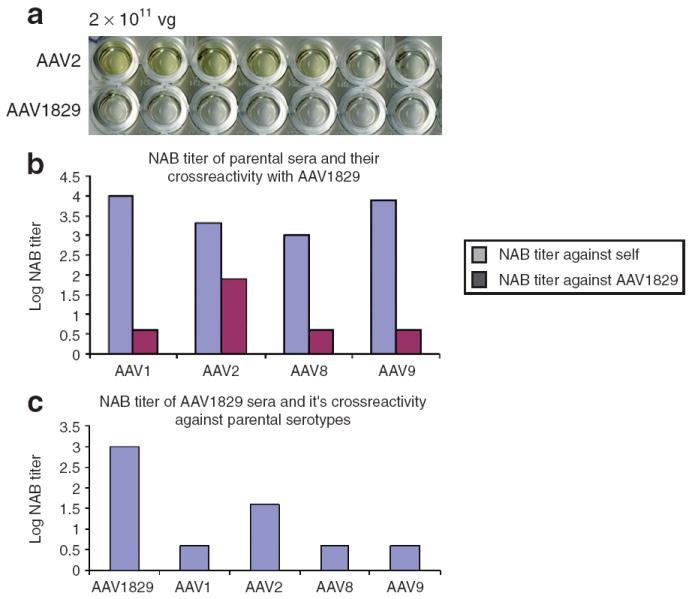
(a) Enzyme-linked immunosorbent semi-quantitative assessment of the affinity of chimeric-1829 and adeno-associated virus-2 (AAV2) to A20 (AAV2 molyclonal antibody), for details see Materials and Methods. (b) NAb titer of parental serotypes sera and their crossreactivity against AAV1829. (c) NAb titer of chimeric-1829 sera and it’s crossreactivity against parental serotypes.
Mapping domains contributing to the melanoma-specific tropism of chimeric-1829
In chimeric-1829, the AAV8 and AAV9 regions, which include amino acid residues 410–450 and 705–736, are close to the twofold interface and lie in regions potentially critical for AAV transduction.30 Moreover, both regions differ among serotypes by less than 10 amino acids or so, enabling rational mutagenesis of single amino acid residues within these critical regions. In the AAV8 region, only three amino acids are different from other serotypes. Among these, the 413T and 418D are similar to 413S and 418E seen in the VP3 subunit of AAV2. The Q411T mutant (Figure 6a, 9) demonstrated no significant change in transduction efficiency in comparison with chimeric-1829 (data not shown). To elucidate the role of the AAV9 region in melanoma tropism of chimeric-1829, we then generated a series of chimeric cap genes by swapping individual capsid domains from one serotype to the other as well as some point mutations through site-directed mutagenesis. As shown in Figure 6a, the AAV2 and AAV9 C-terminal residues (705–735) were swapped onto chimeric-1829 and AAV2, respectively (Figure 6a, 2 and 3). Four additional mutants, [chimeric-1829Y706N, A714T, N716D, and E718N (Figure 6a, 4–7)] were generated to identify the role of single residues in the C-terminal region. Two other residues (N709V and E712D) at positions in the AAV9 region were omitted because of high degree of similarity with the corresponding AAV2 C-terminal region. In addition, we swapped the AAV1 C terminus (705–735) onto chimeric-1829. However, this construct proved to be defective in packaging vector genomes and was not explored any further.
Figure 6. The transduction efficiency of chimeric-1829 and its mutants.
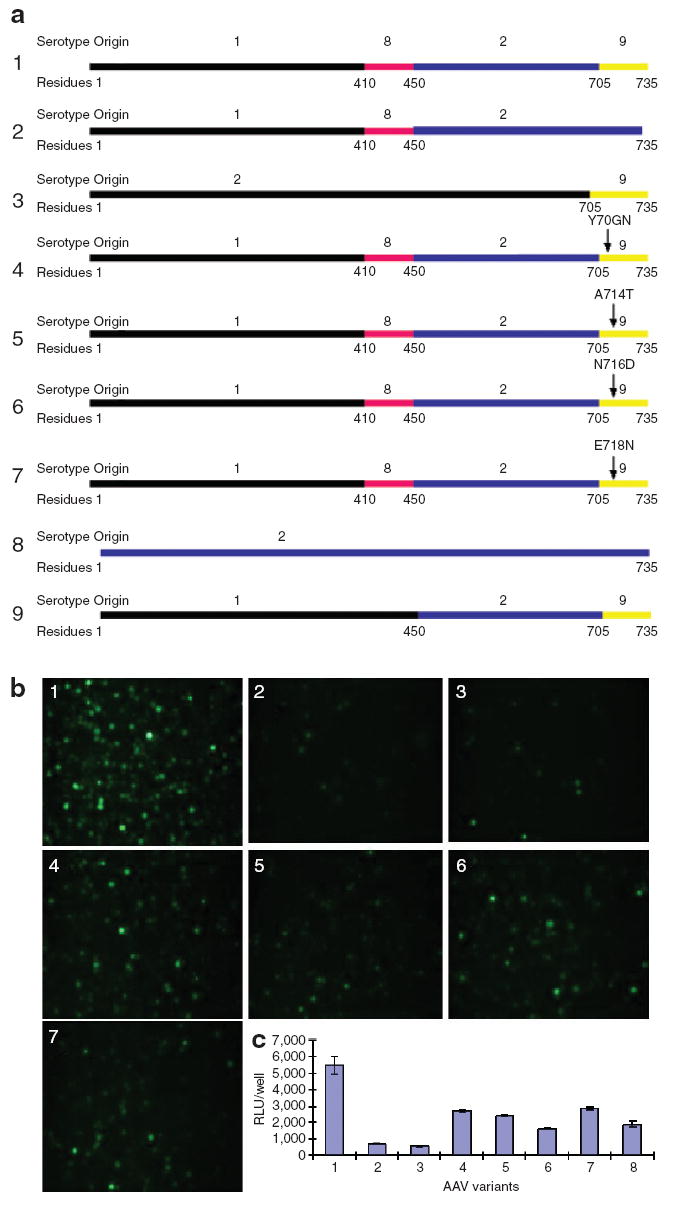
(a) Diagram of the chimeric capsid constructs and site point mutants. (b) Fluorescence micrographs of green fluorescent protein transgene expression in CS1 cells transduced with AAV1829 and its mutants at a multiplicity of infection (MOI) of 1,000 for 48 hours. (c) Luciferase transgene expression in CS1 cells transduced with AAV1829 and its mutants at an MOI of 1,000 for 48 hours. 1, Chimeric-1829; 2, AAV182; 3, AAV2–9; 4, chimeric-1829 Y706N; 5, Chimeric-1829 A714T; 6, Chimeric-1829 N716D; 7, Chimeric-1829 E718N; 8, AAV2; 9, AAV129. AAV, adeno-associated virus; RLU, relative luciferase unit.
As shown in Figure 6b, based on GFP transgene expression in CS1 cells, a spectrum of transduction efficiencies with the following hierarchy was seen: chimeric-1829 was most efficient in transducing CS1 cells, followed by E718N (no. 7), Y706N (no. 4), N716D (no. 6), A714T (no. 5), AAV29 (no. 3), and AAV182 (no. 2), in that order. This hierarchy is more or less consistent with the quantitative luciferase transgene expression obtained in CS1 cells with aforementioned mutants (Figure 6c). When the C-terminal residues (705–735) of chimeric-1829 were replaced with those of AAV2, transduction efficiency of the resulting chimeric vector decreased by nearly one order of magnitude, implicating this region as playing a critical role in melanoma cell tropism of chimeric-1829. However, in corollary, when the C-terminal domain from AAV9 was swapped onto AAV2, a modest decrease in transduction efficiency was seen.
DISCUSSION
In this study, we carried out DNA shuffling of AAV capsid genomes corresponding to AAV serotypes 1–9 to generate a chimeric AAV library. Until recently, attempts to elucidate structure–function attributes of the AAV capsid have primarily focused on site-directed and insertional mutagenesis, including epitope-tag or ligand-insertion mutants and epitope substitution mutants.14 However, such strategies are often biased toward regions previously identified as potential sites for manipulation. Starting with no prior knowledge of the best serotype to choose or the number of mutations needed to generate new phenotypes, random mutagenesis of any single parent would require libraries that are orders of magnitude larger. Thus, we rationalized that the application of molecular breeding to the AAV family in conjunction with rational mutagenesis should enable high-throughput characterization of critical domains on AAV serotype capsids as well as the accelerated evolution of novel tissue-specific AAV vectors.
As proof of principle, a chimeric virus composed of capsid sequences from AAV1, 8, 2, and 9 was obtained through selection in a hamster melanoma cell line (Figure 1). Selection of chimeric library on CS1 cell has preferentially selected a chimeric particle with enhanced tropism for CS1 cell line compared to parental donors (Figure 3a). However, this tropism is not unique to CS1 integrin minus cells but extends to all rodent melanoma cell lines tested regardless of absence or presence of integrin (see Figure 3, Table 1). Although chimeric-1829 transduced these cells readily, none of the parental donor capsids was able to infect the rodent melanoma cells demonstrating that in the case of this chimeric capsid, the whole is the best of the parts. We then expanded our study to characterize the transduction profile of chimeric-1829 in a range of human melanoma cell lines and cultures. Interestingly, chimeric-1829 displayed transduction efficiencies comparable to parental AAV2 vectors in all human cells, while parental serotypes 8 and 9 were negative and type 1 restrictive to transducing one of nine cell lines (Figure 3b, Table 1). It is tempting to speculate that such selective tropism for hamster/mouse melanoma cells displayed by chimeric-1829 could arise from the ability of the capsid to exploit a species-specific cellular factor (such as a cell surface coreceptor) for rodent melanoma cell entry. Although this critical finding highlights a potential caveat associated with application of this technique in developing AAV vectors for human gene therapy, it is important to note that the strategy allows elucidation of the determinants of species-specific differences in AAV serotypes. Obvious solutions to this problem are the use of human cell lines and “humanized” mouse models for directed evolution.
Following the isolation of such cell type–specific mutant(s), we utilized a battery of structural and molecular cloning tools to further investigate specific domains and residues that might potentially constitute determinants for melanoma cell–specific tropism in chimeric-1829. Structural analysis of a 3D model generated for the chimeric-1829 vector suggests that the heparin-binding regions are derived from AAV2, while antigenic regions were derived from more than one serotype capsid (Figure 2b and c). These results were further corroborated through heparin-inhibition studies (Figure 3c) and characterization of the immunological profile of chimeric-1829 (Figure 5a–c). In particular, the unique immunological profile of AAV1829 in comparison with its parental serotypes is an important finding reported in this study (Figure 5). We observed low-to-modest crossreactivity of NAbs against chimeric-1829 with AAV2 capsids (25-fold) and no crossreactivity to parental capsids 1, 8, or 9. The primary outcome of this analysis suggests that antigenic regions on the AAV capsid can be manipulated by grafting domains derived from other serotype capsids. This approach could aid in the generation of NAb escape mutants relevant for vector readministration in a clinical setting. The generation of chimeric mutants through rational mutagenesis was a critical component in establishing the molecular determinants for melanoma tropism displayed by chimeric-1829. A cumulative assessment of mutants generated through domain swapping and site-directed mutagenesis suggests that the C-terminal residues derived from AAV9 likely play a necessary, but not sufficient, role in conferring melanoma cell tropism to the chimeric-1829 capsid. In particular, we demonstrated that residues derived from AAV9, which form the wall of the twofold dimple, could play an important role in transducing CS1 hamster melanoma cells. This suggests that this region likely interacts with a yet unidentified cellular factor, or potentially involved in capsid structural/conformational transitions that are important for efficient rodent melanoma cell entry and transduction. It is worth noting that we recently generated a chimeric AAV capsid currently being used in a phase I clinical trial for Duchene muscular dystrophy. In this vector, three of the five amino acids that were altered to enhance skeletal muscle transduction were also located in the 705–735 amino acid region, independently confirming the importance of this domain in AAV infectivity (Bowles and R.J.S., unpublished results).
In addition to displaying selective tropism for melanoma cells in vitro, chimeric-1829 displays tropism for CS1 cells in severe combined immunodeficiency model (Supplementary Materials and Methods) and altered tropism for murine liver and skeletal muscle in vivo (Figure 4a and b). Although modest, such “detargeting” of capsids vectors, in conjunction with tropism for specific cell types, could afford greater control over the biodistribution of viral vectors, thereby avoiding potential side effects due to transduction of nontarget tissues in vivo. It is further corroborated through our vector-infusion studies in the rat brain (Figure 4). Previous studies have shown that the parental serotypes of chimeric-1829 exhibit a preferential tropism for neurons.32-34 In contrast, chimeric-1829 appears to exhibit reduced tropism for neurons in both rat and monkey brains and potentially, preferential tropism for glial fibrillary acidic protein/nestin positive non-neuronal cells of rodent origin. Besides the fact that most melanoma metastasizes to the brain (54%),24 neuronal precursors (nestin positive cells) in the central nervous system and melanocytes are derived from a common linage (neural crest). As a number of brain tumors demonstrate surface markers similar to progenitor cells (nestin positive), which suggests reversion back to an earlier precursor cell type, we realized that the ability of chimeric-1829 to transduce a selected cell population may suggest a common link between these central nervous system cell types and the fact that they may share a similar lineage pathway with melanocytes. Thus, DNA shuffling/directed evolution proves capable of dramatically shifting capsid in vivo tropism.
In summary, we have described the utilization of DNA shuffling of a family of AAV capsid genes and directed evolution to generate a chimeric capsid library and a cell type–specific chimeric particle isolate (i.e., 1829), respectively. Further exploitation of the capsid library for isolation of novel chimeric mutants in vitro and in vivo is currently in progress (airway, heart, and brain; W.L., unpublished results) and likely to yield new capsid vectors with altered tropisms and antigenic profiles. More important, we anticipate that the mutants generated through shuffling of AAV serotype–specific capsid motifs or capsid sequences from similar (B19) or divergent (SV40, Ad, etc.) viruses will lay the foundation for designing the next generation of novel vectors for human gene therapy applications namely “biological nanoparticles” that process unique tissue targeting, immune profiles, and biodistribution.
MATERIALS AND METHODS
Cells and viruses
HEK293 and CS1 cells (a gift from David Cheresh, University of California at San Diego) were maintained at 37 °C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10% fetal bovine serum and penicillin–streptomycin (100 U/ml). Ad dl309 has been described previously.37 For human melanoma cell lines UNC Mel 1.2 and 3.1, the primary melanomas were disaggregated in 0.25% trypsin–EDTA and then grown on uncoated plastic in Dulbecco’s modified Eagle’s medium with the addition of 10% fetal bovine serum, penicillin–streptomycin, and 50 μmol/l β-mercaptoethanol (Gibco, Grand Island, NY). Human melanoma cell lines were cultured at 37 °C in 5% CO2 atmosphere in Dulbeccos modified Eagle’s medium (Gibco), with the addition of 10% fetal bovine serum, penicillin–streptomycin, and 50 μmol/l β-mercaptoethanol. For human melanoma cell lines, A375, A2058, Mel505, UACC-257, PMWK, WM-2664, and SKMEL-24, the sources and handling of these lines were shown as described.38 For culturing of murine tumor cell lines, KPTR1, KPTR4, KPTR5, KPTR6, KR287T1, KR287T2, KR389, primary murine tumors were handled as described.39,40 All human studies were performed in accordance with the University of North Carolina (UNC) Institutional Review Board, and all murine studies in accord with the UNC Institutional Animal Care and Use Committee.
Construction of the chimeric plasmid library
The DNA shuffling procedure using the JBS DNA shuffling kit has been described previously.41 In brief, the sequence of helper plasmids pXR and two sets of primers were designed in a nested configuration. The outer primers, P1 and P2 were used to amplify DNA for the fragmentation reaction. The 4.4-kilobase fragment encompassing the entire Cap, part of the Rep gene and part of the plasmid backbone sequence was amplified from each of eight parental AAV helper plasmids (pXR1-6,8,9). The PCR products were then mixed together in equimolar amounts and subjected to random fragmentation by DNaseI. Fragments ranging from 400 to 1,200 base pair in size were resolved in an agarose gel and reassembled through cycles of denaturation, annealing, and extension in the absence of primers. Cycling was performed as follows: 96 °C, 90 seconds; 35 cycles of (94 °C, 30 seconds; 65 °C, 90 seconds; 62 °C, 90 seconds; 59 °C, 90 seconds; 56 °C, 90 seconds; 53 °C, 90 seconds; 50 °C, 90 seconds; 47 °C, 90 seconds; 44 °C, 90 seconds; 41 °C, 90 seconds; 72 °C, 4 minutes); 72 °C, 7 minutes; and finally 4 °C. The assembled fragments were then amplified using inner primers P3 and P4, digested with SfiI and XbaI, and ligated into the pSSV9 backbone digested with the same enzymes. Ligation mixtures were column purified and transformed into competent DH10B cells.18 Plasmid library diversity was determined by counting the number of clones obtained from a representative aliquot of transformed bacteria on agar plates containing 100 μg/ml ampicillin. The entire transformation mixture was plated on agar plates and plasmid DNA isolated from pooled clones without further growth in liquid culture. To assess the diversity of the generated library, 20 sample clones from the library were subjected to sequence analysis. Primer sequences are listed in the Table 2.
Table 2.
Primers used in this study
| Name | Sequence |
|---|---|
| P1 | 5′-CGAGATTGTGATTAAGGTCC-3′ |
| P2 | 5′-CAGGTTTCCCGACTGGAAAG-3′ |
| P3 | 5′-AATGGCGCCGTGTGAGTAAG-3′ |
| P4 | 5′-GCTCTAGACGACACCAAAGTTCAACTGA-3′ |
| P-Y706N | 5′-TCCAGTACACTTCCAACTATAACAAGTCTAATAATGTTGA-3′ |
| P-A714T | 5′-GTCTAATAATGTTGAATTTACTGTTAATACTGAAGGTGTA-3′ |
| P-N716D | 5′-ATAATGTTGAATTTGCTGTTGATACTGAAGGTGTATATAG-3′ |
| P-E718N | 5′-TTGAATTTGCTGTTAATACTAATGGTGTATATAGTGAACC-3′ |
| P-Q411T | 5′-GAACCGGCAACAACTTCACGTTTACTTACACCTTCG-3 |
Generation of AAV library transfer shuttles and the chimeric AAV capsid library
As described previously,8 the chimeric AAV library was generated using the plasmid library through a two-step process. First, AAV genomes from the shuffled plasmid library were partially packaged into AAV2 capsids. This was achieved by transfecting HEK293 cells grown in 10 15-cm plates with a 1:1:2 ratio of the pXR2 plasmid (containing AAV2 Rep and Cap genes without inverted terminal repeats), the plasmid library, and the pXX6-80 helper plasmid (60, 60, and 120 μg, respectively). The resulting AAV library transfer shuttles were harvested, and purified by CsCl gradient ultracentrifugation (~400,000 g/5 h) using a Beckman NVT65 rotor in a Sorvall Ultra 80 centrifuge. The chimeric AAV capsid library was then generated by infecting HEK293 cells with AAV library transfer shuttles (MOI = 1) followed by super-infection with Ad dl309 (4 plaque forming units/cell). Cells were then harvested after 36 hours and the chimeric AAV capsid library encapsidating their respective chimeric genomes were purified as described earlier.
Isolation of cell type–specific AAV vectors
Target CS1 cells were coinfected with the AAV capsid library (MOI = 1,000) and adenovirus dl309 (MOI = 20) and incubated at 37 °C for 48 hours. The cells were then harvested after 48 hours, rinsed with 5 ml phosphate-buffered saline, resuspended in 1 ml phosphate-buffered saline, and lysed through freeze–thaw cycles (3×). Cellular debris was then removed by centrifugation and a fraction of the supernatant used to infect the next batch of target cells for the second cycle of selection. After each round of selection, viral genomic DNA was purified from 50 μl aliquots of crude cell lysates using the DNeasy kit (Qiagen) for determination of viral genome titer by dot blot hybridization. DNA extracted from cell lysate after the fifth cycle served as a template for PCR using primers P3 and P4 described previously. The amplified PCR fragment was then subjected to sequence analysis and cloned into the pXR2 backbone.
Physical characterization of chimeric-1829
Sequence analysis and alignment of the variant sequence with parental serotype sequences was carried out using VECTOR NTI10. The 3D model of VP3 trimers and pentamers of the capsids of chimeric-1829 isolates were generated using SWISS MODEL with coordinates from the AAV2 crystal structure (PDB ID: 1lp3) serving as template. Subsequent surface rendering and mapping of regions derived from different AAV serotypes were performed using Pymol (http://www.pymol.org). Recombinant vectors encapsidating GFP or firefly luciferase transgenes driven by the cytomegalovirus or chicken β-actin (CBA) promoter, respectively were produced as described previously.42 Viral titers were determined using DNA dot blot hybridization. Heparin-binding studies with the chimeric-1829 isolate were carried out as described earlier.43-45
Biological characterization of chimeric-1829
Heparin-inhibition assay
The heparin-inhibition assay was performed as described previously.43 In brief, 24 hours before infection, chimeric-1829/GFP vectors (MOI = 5) were incubated in the presence of heparin at 30 μg/ml for 1 hour at 37 °C. Cells were then washed 2 hours later. GFP expression was monitored 48 hours after infection using a Nikon TE-300 inverted fluorescent microscope.
In vitro transduction assays
The transduction efficiency of parental AAV serotypes used in generating the AAV library and novel isolate, chimeric-1829, were determined in target CS1 melanoma cells. In brief, CS1 cells were transduced with chimeric-1829 carrying the GFP transgene cassette [1,000 vector genomes (vg)/cell] and transgene expression was monitored 48 hours after infection using a Nikon TE-300 inverted fluorescent microscope.
In vivo studies
The chimeric-1829 vector packaging the CBA-Luc transgene was administered to C57BL/6 mice through the intramuscular (hindlimb, 1010 vg) or intravenous (tail vein, 1011 vg) routes. One month later after administration, luciferase transgene expression levels were determined using the Xenogen IVIS system. For in vivo brain study materials, see Supplementary Materials and Methods.
NAb titers and crossreactivity
Balb/c mice were injected with the chimeric-1829 vector or corresponding parental vectors chimeric-1829 (5 × 1010 vg) by subcutaneous injection and a booster dose 2 weeks later. After another 2 weeks, sera from mice were analyzed for NAb titer and crossreactivity by assessing the ability of serum antibody to inhibit AAV vector transduction. In brief, various dilutions of antibodies were preincubated with reporter virus for 1 hour at 37 °C and added to Hela/U87MG cell cultures. GFP expression cells were then countered using fluorescent microscope. NAB titers were calculated as the highest serum dilution inhibiting transduction by 50% of that determined for control (vector plus serum from naive animals).
Enzyme-linked immunosorbent assay with monoclonal antibody A20
Serial dilutions of AAV variants (2 × 1011vg/100 μl) were added to each well of a 96-well micro-titre plate. Plates were incubated at 4 °C overnight in 100 μl blocking buffer (0.1 mol/l phosphate-buffered saline with 1% bovine serum albumin) followed by incubation at 37 °C for 1 hour. Plates were then washed three times with phosphate-buffered saline–Tween, 100 μl of A20 antibody was added (15× dilution) and incubated at 37 °C for 1 hour. After three washes and incubation with 100 μl of anti-mouse horseradish peroxidase–conjugated antibody (5,000 times dilution; Pierce), plates were washed three times, 100 μl of 3.3′, 5.5′-tetramethylbenzidine substrate solution was added (PBL) to each well followed by incubation in the dark for 15 minutes. Finally, 100 μl stop solution (PBL) was added to each well and pictures obtained with a scanner.
Site-directed mutagenesis
Chimeric-1829 mutants were generated by performing QuikChange site-directed mutagenesis (Stratagene) on plasmids pXR1829 according to manufacturer instructions. In addition, domain-swapped chimeras were obtained using PCR and restriction digestion techniques to exchange fragments between chimeric-1829 and its parental serotypes. Primer sequences are listed in Table 2.
Structural analysis of the chimeric-1829 vector
A structural model of the chimeric-1829 virus was generated using the SWISS-MODEL online 3D model building server with the crystal structure of AAV2 supplied as template (23, PDB ID: 1lp3).46 The contributions of the four parental serotypes AAV1, 8, 2, and 9 to the chimeric vector were analyzed in the graphics program O and figures showing the serotype contributions were generated using the program Pymol.47,48
Supplementary Material
Figure S1. AAV transduction in mouse tumor model. Materials and Methods.
Materials and Methods.
Acknowledgments
We thank National Institutes of Health (grant nos. HL51818 and NS59518; to R.J.S) and National Institute of Neurological Disorders and Stroke(grant no. NS35633; to T.J.M) for research support.
References
- 1.Berns KI, Giraud C. Biology of adeno-associated virus. Curr Top Microbiol Immunol. 1996;218:1–23. doi: 10.1007/978-3-642-80207-2_1. [DOI] [PubMed] [Google Scholar]
- 2.Flotte TR. Recent developments in recombinant AAV-mediated gene therapy for lung diseases. Curr Gene Ther. 2005;5:361–366. doi: 10.2174/1566523054064986. [DOI] [PubMed] [Google Scholar]
- 3.Romano G. Current development of adeno-associated viral vectors. Drug News Perspect. 2005;18:311–316. doi: 10.1358/dnp.2005.18.5.917326. [DOI] [PubMed] [Google Scholar]
- 4.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 5.Virella-Lowell I, Zusman B, Foust K, Loiler S, Conlon T, Song S, et al. Enhancing rAAV vector expression in the lung. J Gene Med. 2005;7:842–850. doi: 10.1002/jgm.759. [DOI] [PubMed] [Google Scholar]
- 6.Smith A, Collaco R, Trempe JP. AAV vector delivery to cells in culture. Methods Mol Biol. 2004;246:167–177. doi: 10.1385/1-59259-650-9:167. [DOI] [PubMed] [Google Scholar]
- 7.Wang C, Wang CM, Clark KR, Sferra TJ. Recombinant AAV serotype 1 transduction efficiency and tropism in the murine brain. Gene Ther. 2003;10:1528–1534. doi: 10.1038/sj.gt.3302011. [DOI] [PubMed] [Google Scholar]
- 8.Muller OJ, Kaul F, Weitzman MD, Pasqualini R, Arap W, Kleinschmidt JA, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 9.Perabo L, Buning H, Kofler DM, Ried MU, Girod A, Wendtner CM, et al. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol Ther. 2003;8:151–157. doi: 10.1016/s1525-0016(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 10.Ponnazhagan S, Mahendra G, Kumar S, Thompson JA, Castillas M., Jr Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J Virol. 2002;76:12900–12907. doi: 10.1128/JVI.76.24.12900-12907.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warrington KH, Jr, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol. 2004;78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabinowitz JE, Bowles DE, Faust SM, Ledford JG, Cunningham SE, Samulski RJ. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol. 2004;78:4421–4432. doi: 10.1128/JVI.78.9.4421-4432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi VW, McCarty DM, Samulski RJ. AAV hybrid serotypes: improved vectors for gene delivery. Curr Gene Ther. 2005;5:299–310. doi: 10.2174/1566523054064968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Bowles DE, Rabinowitz JE, Samulski RJ. Marker rescue of adeno-associated virus (AAV) capsid mutants: a novel approach for chimeric AAV production. J Virol. 2003;77:423–432. doi: 10.1128/JVI.77.1.423-432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 17.Perabo L, Endell J, King S, Lux K, Goldnau D, Hallek M, et al. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J Gene Med. 2006;8:155–162. doi: 10.1002/jgm.849. [DOI] [PubMed] [Google Scholar]
- 18.Crameri A, Cwirla S, Stemmer WP. Construction and evolution of antibody-phage libraries by DNA shuffling. Nat Med. 1996;2:100–102. doi: 10.1038/nm0196-100. [DOI] [PubMed] [Google Scholar]
- 19.Crameri A, Whitehorn EA, Tate E, Stemmer WP. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 20.Crameri A, Raillard SA, Bermudez E, Stemmer WP. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- 21.Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 22.Nemerow GR, Stewart PL. Role of alpha(v) integrins in adenovirus cell entry and gene delivery. Microbiol Mol Biol Rev. 1999;63:725–34. doi: 10.1128/mmbr.63.3.725-734.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summerford C, Bartlett JS, Samulski RJ. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat Med. 1999;5:78–82. doi: 10.1038/4768. [DOI] [PubMed] [Google Scholar]
- 24.Skibber JM, Soong SJ, Austin L, Balch CM, Sawaya RE. Cranial irradiation after surgical excision of brain metastases in melanoma patients. Ann Surg Oncol. 1996;3:118–123. doi: 10.1007/BF02305789. [DOI] [PubMed] [Google Scholar]
- 25.Thomas L, Chan PW, Chang S, Damsky C. 5-Bromo-2-deoxyuridine regulates invasiveness and expression of integrins and matrix-degrading proteinases in a differentiated hamster melanoma cell. J Cell Sci. 1993;105:191–201. doi: 10.1242/jcs.105.1.191. [DOI] [PubMed] [Google Scholar]
- 26.Kern A, Schmidt K, Leder C, Muller OJ, Wobus CE, Bettinger K, et al. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini J, et al. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol. 2006;80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, et al. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81:12260–12267. doi: 10.1128/JVI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, et al. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97:3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 34.Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duggal N, Schmidt-Kastner R, Hakim AM. Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain Res. 1997;768:1–9. doi: 10.1016/s0006-8993(97)00588-x. [DOI] [PubMed] [Google Scholar]
- 36.Fukuda S, Kato F, Tozuka Y, Yamaguchi M, Miyamoto Y, Hisatsune T. Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J Neurosci. 2003;23:9357–9366. doi: 10.1523/JNEUROSCI.23-28-09357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones N, Shenk T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 38.Shields JM, Thomas NE, Cregger M, Berger AJ, Leslie M, Torrice C, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–1512. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- 39.Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997;11:2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene. 2003;22:5055–5059. doi: 10.1038/sj.onc.1206809. [DOI] [PubMed] [Google Scholar]
- 41.Joern JM, Meinhold P, Arnold FH. Analysis of shuffled gene libraries. J Mol Biol. 2002;316:643–656. doi: 10.1006/jmbi.2001.5349. [DOI] [PubMed] [Google Scholar]
- 42.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabinowitz JE, Xiao W, Samulski RJ. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology. 1999;265:274–285. doi: 10.1006/viro.1999.0045. [DOI] [PubMed] [Google Scholar]
- 45.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 48.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AAV transduction in mouse tumor model. Materials and Methods.
Materials and Methods.