


Abstract
Disease gene identification is still a challenge despite modern high-throughput methods. Many diseases are very rare or lethal and thus cannot be investigated with traditional methods. Several in silico methods have been developed but they have some limitations. We introduce a new method that combines information about protein-interaction network properties and Gene Ontology terms. Genes with high-calculated network scores and statistically significant gene ontology terms based on known diseases are prioritized as candidate genes. The method was applied to identify novel primary immunodeficiency-related genes, 26 of which were found. The investigation uses the protein-interaction network for all essential immunome human genes available in the Immunome Knowledge Base and an analysis of their enriched gene ontology annotations. The identified disease gene candidates are mainly involved in cellular signaling including receptors, protein kinases and adaptor and binding proteins as well as enzymes. The method can be generalized for any disease group with sufficient information.
INTRODUCTION
Although genes related to diseases have already been determined over several decades and thousands of such genes are already known, it is still very difficult to find genes underlying a specific disorder. Traditionally, most studies have been based on linkage analysis of several affected families, which generally results a large locus or a few loci with numerous, even hundreds, of genes. Still, the identification of the disease gene is difficult and laborious as several difficulties can impede this approach. Many diseases are so rare that it is impossible to find enough families for linkage analysis. For example, in the recently discovered primary immunodeficiency (PID) caused by defective endosomal adaptor protein p14, only a single patient is known (1). A similar case arises when the symptoms of the disease are so severe that hardly any patients have siblings, such as in severe combined immunodeficiencies (SCIDs) (2).
Even if the disease-related locus is identified, the gene identification can fail, especially if the genomic region is large. Similar problems also impede modern genome-wide association studies (3). Due to all these challenges, several attempts have been made to identify candidate genes using in silico methods (4–6). These approaches have been grouped in three categories (7). Some of the methods are sequence-based or they use other intrinsic properties of the genes like functional annotations (8). Others are based on gene expression patterns or other phenotypic properties (9). In the third category, the methods use some kind of interaction between the genes or their products as the basis of the predictions (10). The individual disease gene predictions usually apply one or two of these approaches, such as in the PhenoPred system (11) or SUSPECTS (12).
Most of the algorithms have been implemented locally, but there are also Internet-based services dedicated to disease gene predictions, and some are parts of more general servers that can help the process. For example, GFINDER works on a user-defined gene list (13). Among the results are overrepresented gene ontology (GO) terms and diseases, which have significant relatedness to the provided genes. Many of the disease gene prediction tools are denoted for prioritization, i.e. they try to identify genes in a starter set, which has a higher likelihood of being related to the target disease(s) than the rest of the set. Certain systems are open for the scientific community (14) and have been applied to cancers (15) and fetal alcohol syndrome (5), for example.
Primary immunodeficiencies impair the function of the immune system. Patients with these intrinsic defects have increased susceptibility to recurrent and persistent infections and may also have autoimmune and cancer-related symptoms (16–18). Most PIDs are rare and the diagnosed patients for a condition are often randomly spread around the world, and thus linkage or genome-wide association studies analysis in these rare diseases is barely possible.
There are numerous different mechanisms behind PIDs. Several PIDs affect T- or B-cell functions and, when both cell types are affected, lead to SCIDs. Other PIDs affect the major histocompatibility complex (MHC), antibody production, lymphocyte apoptosis, phagocytosis, the complement cascade or the innate immune system. Detailed information about PIDs is available from the ImmunoDeficiency Resource (IDR, http://bioinf.uta.fi/idr; 19). Altogether, there are over 200 PIDs (16) and 144 PID genes listed in the IDR.
With an exhaustive analysis of literature and databases, we identified altogether 847 genes and proteins that are crucial for the immune system and thus form the essential human immunome (20). These genes and proteins are involved in numerous functions, including cell surface recognition, transcription factors, DNA processing and adaptor proteins. System level analysis has been performed, e.g. for the evolutionary history of these genes (21), the emergence of immunity-related domains and gene ontologies (21), and the development of the interaction network of the immunome proteins during the evolution of the immune system (22,23).
GO terms (24) are used for systematic annotation of genes on three levels, namely biological processes (BP), cellular components (CC) and molecular functions (MF). GOs have been used, e.g. for grouping genes by their common properties and for elucidating the biological meaning of results in high-throughput experiments (25), such as with microarrays (26,27).
Protein-interaction networks have been reconstructed for several organisms by using modern high-throughput methods (28–30). Analyses of these networks have revealed functionally important proteins (31,32). Different networks, ranging from social interactions (33,34) via protein–protein interactions (35,36) to the spreading of epidemics (37) as well as human made networks like the Internet (38,39), have been shown to share similar characteristics, which suggests common organizing principles for their emergence (40).
Here, we present a novel method for disease gene identification and prioritization. Our approach combines information about protein-interaction networks and GO terms. The method was applied to PIDs and human immunome data to suggest new genes that might have relevance to primary immunodeficiencies.
MATERIALS AND METHODS
Immunome genes and their interactions
Information about PID genes and immunodeficiencies was taken from the IDR (19). Human immune system-related proteins were collected from the Immunome database, a reference set of human immune system-related genes and proteins (21) recently integrated with our other immunome registries in the Immunome Knowledge Base (Ortutay and Vihinen, submitted for publication). Protein interactions were associated with the immunome proteins according to the Human Protein Reference Database (HPRD) (41). As only interactions between the immunome proteins were taken into account, no new nodes were added, but proteins without interactions were eliminated from the dataset. The final network contained 584 nodes of the 847 original ones, forming altogether 1349 interactions (22). Interactions that appeared more than once were simplified to single edges.
Degree, vulnerability and closeness centrality of the immunome proteins
Network property characteristics, vulnerability, closeness centrality and degree of the nodes were calculated using the igraph R library (42). The degree specifies the number of interactions of a protein.
Vulnerability is calculated using the global efficiency of the network. Efficiency quantifies the efficiency of the network in sending information between nodes, assuming that the efficiency between two nodes is proportional to the reciprocal of their distance (43). Global efficiency was calculated as follows:
where dij is the distance between the ith and jth nodes as the minimal number of edges on the shortest path between them and N is the total number of nodes in the network.
The vulnerability of a network was calculated using efficiency characteristics (44). The vulnerability, Vi, of a network associated with the ith node is Vi = (E − Ei)/E, where E is the global efficiency of the network while Ei is the global efficiency of the network without the node i and all of its interactions. The overall vulnerability of the network is the value of the most vulnerable node, i.e. the largest loss in performance when a node is deleted from the network.
Closeness centrality marks how far or close a certain node is to all the others, so that it can be interpreted as how central the position of the protein is in the network (45). It is defined by the inverse of the average length of the shortest paths to/from all the other vertices in the graph:
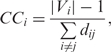 |
where |Vi| is the size of the reachable subnetwork from node i and dij is the distance between the ith and jth nodes.
To test the biological significance of these scores, we compared their distribution in essential immunome genes and for all the proteins of the immunome-interaction network. We used Mammalian Phenotype Ontologies (46) to define genes as essential when they caused embryonic, perinatal or neonatal lethality in mouse models (47) according to the Mouse Genome Database (48). The human orthologs of murine genes were considered as essential, when the murine gene was annotated with one of the following phenotypes: neonatal lethality (MP:0002058), embryonic lethality (MP:0002080), perinatal lethality (MP:0002081), postnatal lethality (MP:0002082), lethality-postnatal (MP:0005373), lethality-embryonic/perinatal (MP:0005374), embryonic lethality before implantation (MP:0006204), embryonic lethality before somite formation (MP:0006205) or embryonic lethality before turning of embryo (MP:0006206).
GO analysis
GO terms (24) were collected from the Immunome database for all the 847 genes in the essential immunome. Using topGO R library (49), we identified three sets of 50 genes from the immunome protein-interaction network, which had the highest degree, vulnerability or closeness centrality. These sets were compared to the whole dataset. We made independent analyses for vulnerability, closeness centrality and degree scores in combination with the ‘biological process’, ‘cellular component’ and ‘molecular function’ ontology terms, thus performing altogether nine analyses. We used Fisher's exact test for statistics and three methods for enrichment analysis of the GO terms in the significant group. The methods called ‘classic’, ‘elim’ and ‘weight’ have been described and compared in ref. (49). The ‘classic’ method tends to rank general terms highly, whereas methods ‘elim’ and ‘weight’ are more balanced and prefer more specific terms. This is especially true for the ‘weight’ method; therefore, in subsequent analyses, we used the P values from this method to evaluate the significance of the terms.
We also performed the analysis so that the 144 known PID genes from the IDR were considered as significant and were compared to all the immunome genes. We also identified the significant BP, CC and MF ontology terms for the PID-related genes.
Predicting PID-related candidate genes
The significant PID-related GO terms were combined with the results for the protein-interaction network-related scores in order to predict new PID-related genes. The lists of genes with the 50 highest vulnerability, closeness centrality or degree values were created. When combined, these lists contained 84 genes.
Another list was generated for genes containing significant PID-related GO terms. Significant GO terms (P < 0.05), which were annotated for at least three but not for more than 50 immunome genes, were chosen. This way, we managed to exclude too general and therefore uninformative terms like ‘cell part’ (GO:0044464) and terms that appeared to be significant just by chance because of their low frequency, like ‘cyclosporin A binding’ (GO:0016018), which was annotated for a single gene. The weight method was applied for enrichment analysis. This way, 54 significant GO terms were chosen from the three GO term categories. In total, 231 genes had these annotations.
To combine all the results, a Venn graph was drawn for the high-network score genes, the significant GO term-related genes and the known PID genes. Those genes that appeared among both the high score and the GO-related lists, but were not among the known PID-causing genes, were defined as PID candidate genes.
Performance evaluation of the method
To assess the performance of our approach to find PID genes, we have done the leave-one-out test. We rerun the method for 144 times (the number of known PID genes) by leaving one known PID gene out at time. First, the effect of the left out gene on the GO parameters was checked. Then, we tested whether the left out known PID gene was predicted to be disease-related. The performance test was implemented using the R statistical environment.
RESULTS
We developed a novel method for candidate disease gene identification. The method combines information about an interaction network describing the relations of proteins under study with their GO annotations to prioritize disease gene candidates. The method, which can be used for any disease gene group provided there is sufficient protein interaction and GO data available, was applied to the human immunome to search for novel PID candidates.
Protein-interaction network-related scores
Several parameters can be used to describe interaction networks. General but descriptive scores, degree, vulnerability and closeness centrality were calculated for 584 proteins present in the immunome protein-interaction network. Degree values, as expected, show a power-law distribution with a power-law exponent of 1.6. All three parameters show strong correlation, their pairwise Spearman's rank correlation coefficients are between 0.67 and 0.785. FYN oncogene related to SRC (FYN), lymphocyte-specific protein tyrosine kinase (LCK), Janus kinase 1 (JAK1), signal transducer and activator of transcription 1 (STAT1), receptor type protein tyrosine phosphatase C (PTPRC), CD4 antigen (CD4) and complement component 3 (C3) were among the top 15 proteins in all the three scorings.
The scores were used to describe the importance of the proteins in the interaction network. To test this hypothesis, we identified the genes in which mutations lead to early lethality in mouse models. We called these ‘essential genes’. Values for the three tested parameters are significantly higher for essential genes compared to other immunome genes (Figure 1). Of the 144 PID-related genes, 84 were among the top 50 genes when results for the individual scores were combined. Known PID-related proteins LCK, STAT1, PTPRC and C3 appeared in the top 15 in all three score lists. The PID-related proteins have significantly higher degrees compared to the whole dataset (P = 2.57 × 10−4).
Figure 1.

Protein-interaction network-related scores for essential and immunome genes. An immunome gene was considered essential if mutation in the mouse ortholog causes early lethality. P values for the Kruskal–Wallis rank sum test are shown on the plots. (A) Degree; (B) vulnerability; (C) connectivity.
Significant GO terms for the genes with the top 50 network scores
The enrichment of GO annotations for the immunome genes with the top 50 scores was identified. Signaling-related ontology terms dominated the ‘biological process’ ontologies. ‘Protein amino acid phosphorylation’ (GO:0006468) was the most significant in the case of all three scorings. Fifty-two genes in the protein-interaction network have this annotation. The calculated P values for those terms are between 4.6 × 10−4 and 8.2 × 10−9. Also, a high number of regulation-related ontology terms, such as ‘regulation of T-cell activation’ (GO:0050863) or ‘positive regulation of interleukin-12 biosynthetic process’ (GO:0045084) appear in the dataset.
Most of the significant ‘molecular function’ ontology terms were related to kinase activity and receptor activity. Significant ‘cellular component’ terms were less informative, being either too general or annotated for just a few proteins. Sometimes, they were obviously important for immunology, like ‘immunological synapse’ (GO:0001772) or ‘interleukin-1 receptor complex’ (GO:0045323).
Significant GO terms for the PID-related genes
GO enrichment analysis was performed for the known PID genes. The most significant terms had close relationships with immunology. In the case of MF, proteolysis-related terms, like ‘chymotrypsin activity’ (GO:0004263) and ‘endopeptidase inhibitor activity’ (GO:0004866) are dominant. The significant CC ontology terms point to structures important for immunology, such as ‘membrane attack complex’ (GO:0005579), where all seven proteins annotated with this term are PID genes, and thus the significance of this term is high (P value 3.6 × 10−7 with the weight method).
Genes with high-network scores and significant GO terms as predicted disease genes
We combined the gene lists for the high-network score genes and the significant PID-related GO terms in order to identify new potential PID genes (Figure 2). Altogether, 84 genes were among the 50 highest scores, when results for degree, closeness centrality and vulnerability were combined. Twenty-two of these genes were already-known PID genes. Two hundred and thirty-one genes had GO terms significantly related to PID genes, and 74 of them were PID genes. Altogether, 83 of the 144 PID-related genes were selected either by the GO terms or high-network scores. Thirty-nine genes had both high scores and significant terms, 13 of which were PID-related. So, finally, we have a list of 26 suspected PID candidate genes (Table 1).
Figure 2.

Identification of novel PID candidate genes by using information about the network properties, GO terms and known immunodeficiency genes. The grey area indicates the 26 candidate genes that have high-protein network scores and GO terms enriched in PID genes and which are not among already known PID genes.
Table 1.
Identified PID candidate genes
| Symbol | Full name | GeneID | Known diseases |
|---|---|---|---|
| CD4 | CD4 antigen (p55) | 920 | 186 940 CD4+ lymphocyte deficiency |
| CD9 | CD9 antigen (p24) | 928 | |
| CTSG | cathepsin G | 1511 | |
| FADD | Fas-associating protein with death domain | 8772 | |
| FYN | Protein-tyrosine kinase fyn | 2534 | |
| GZMB | Granzyme B | 3002 | |
| IGF1R | Insulin-like growth factor 1 receptor | 3480 | 147 370 Intrauterine and postnatal growth retardation |
| IL2RB | Interleukin 2 receptor, β | 3560 | |
| INSR | Insulin receptor | 3643 | 610 549 Diabetes mellitus, insulin-resistant, with acanthosis nigricans |
| 609 968 Hyperinsulinemic hypoglycemia | |||
| 246 200 Leprechaunism | |||
| 262 190 Rabson–Mendenhall syndrome | |||
| IRAK1 | Interleukin-1 receptor-associated kinase 1 | 3654 | |
| ITGB1 | β 1 integrin | 3688 | |
| JAK1 | Janus kinase 1 | 3716 | |
| JAK2 | Janus kinase 2 | 3717 | 600 880 Budd–Chiari syndrome |
| 601 626 Leukemia, acute myelogenous | |||
| 254 450 Myelofibrosis, idiopathic | |||
| 263 300 Myeloproliferative disorder with erythrocytosis—polycythemia vera | |||
| 187 950 Thrombocythemia, essential | |||
| KIT | v-Kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | 3815 | 606 764 Gastrointestinal stromal tumor, somatic |
| 273 300 Germ cell tumors | |||
| 273 300 Mast cell leukemia—mastocytosis with associated hematologic disorder—piebaldism | |||
| LRP1 | Low-density lipoprotein-related protein 1 | 4035 | |
| MAPK14 | Mitogen-activated protein kinase 14 | 1432 | |
| PDGFRB | Platelet-derived growth factor receptor, β-polypeptide | 5159 | 131 440 Myelomonocytic leukemia, chronic—myeloproliferative disorder with eosinophilia |
| RELA | v-Rel reticuloendotheliosis viral oncogene homolog A | 5970 | |
| RIPK1 | Receptor (TNFRSF)-interacting serine-threonine kinase 1 | 8737 | |
| SOCS1 | Suppressor of cytokine signaling 1 | 8651 | |
| STAT3 | Signal transducer and activator of transcription 3 | 6774 | |
| THY1 | Thy-1 cell surface antigen | 7070 | |
| TRAF1 | TNF receptor-associated factor 1 | 7185 | |
| TRAF2 | TNF receptor-associated factor 2 | 7186 | |
| TYK2 | Tyrosine kinase 2 | 7297 | |
| XRCC5 | X-ray repair complementing defective repair in Chinese hamster cells 5 | 7520 |
For each genes, the HGNC approved symbol, full name, Entrez GeneID and OMIM code for known diseases is provided.
We addressed the performance of the method by a statistical test. By using the leave-one-out method, all the 13 known PID genes with high-GO scores and network scores were identified as disease-related. The result is 100% correct for the high-score network and GO-enriched PID genes in Figure 2. Thus, it is very likely that also the 26 predicted PID genes, which have high-network and GO scores, have relevance to immunodeficiencies.
DISCUSSION
Protein-interaction networks have been recently used in several studies targeting diseases (47,50,51). The theory of scale-free networks and graph theoretical approaches has been applied to several areas including biological and medical networks. Networks for 867 human diseases and 1377 disease genes were constructed to study essential and disease genes and their role in the human interactome (47). Only 15 immunodeficiencies were included in this analysis. In another study based on a protein-interaction network (50), the clustering of disease genes was noticed in the interactome. A recent review (51) steps even further and discusses four possible areas where protein networks can be used in relationship with diseases, including identification of disease-related subnetworks and prediction of new disease genes.
In some studies, disease gene prioritization has been targeted by in silico methods. Protein-interaction networks have formed the basis for certain techniques. In these studies, the location of proteins in the interaction network has been revealed to determine if they are in a significant position in the network (10,52). Important proteins are essential and therefore frequently found among disease genes (47). Other approaches have integrated protein-interaction information with other data sources to select candidate disease genes (53–55). Our method combines the scores describing properties of a protein interaction network with GO terms, which provide information for protein functions, processes and localization.
We have shown that there is a relationship between the importance of proteins in the immunome-interaction network, as indicated by centrality scores, and GO terms. The vulnerability, closeness centrality and degree values were calculated for all 584 immunome proteins in the interaction network. The scores show strong correlations with each other. The scores were used as measures for the importance of proteins in the immunome network. As previously indicated (47), many important proteins have a high-degree value, their vulnerability is high and they also have high closeness centrality.
To test the biological relevance of the scores, we used them to analyze essential genes. Mutations in essential genes cause embryonic, perinatal or neonatal lethality in mouse models. Mutations in certain PID genes also cause lethality and, in some other PID genes, lead to very severe conditions (SCIDs). The three network scores were significantly higher for essential genes than for immunome genes in general.
In the next step, we identified GO terms that were enriched in the known PID genes. Many of these terms were related to signaling or regulation. In this analysis, we applied a recently introduced method (49), which has been shown to be capable of estimating the significance of overrepresented GO terms and was used to analyze microarray experiment data (56).
New PID candidate genes were defined as having ontology terms significantly associated with disease genes and the encoded proteins as having high-network scores. We identified 26 such genes that could lead to immunodeficiencies when mutated. Many of the detected candidate genes have numerous functions and several interaction partners. The biological functions and the relevance of the candidate genes for immunology are discussed in detail in Supplementary Data.
The power of our method became more apparent when two of the predicted candidate genes turned out to be known PID genes, but not yet annotated in the IDR version we used to identify known disease genes. The signal transducer and activator of transcription 3 (STAT3) (57) and tyrosine kinase 2 (TYK2) (58) are responsible for PIDs and involved in IL-6R-related responses to infections (59). In addition, genome-wide association studies have uncovered susceptibility genes (60). Among these is the TRAF1-C5 region (61), which contains one of our OID candidate genes TRAF1. Thus, experimental evidence supports our in silico predictions.
Our method uses information about interactions among the proteins of the human immunome and also their functional annotation. The immunome charts the genes and their products for immunological responses and thus includes the PID genes. In a similar way, this method can be generalized to any group of diseases and the related genes and proteins for which sufficient information is available.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
We thank the Medical Research Fund of Tampere University Hospital and Academy of Finland for financial support.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schaffer AA, Rathinam C, Taub N, Teis D, Zeidler C, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat. Med. 2007;13:38–45. doi: 10.1038/nm1528. [DOI] [PubMed] [Google Scholar]
- 2.Fischer A, Le Deist F, Hacein-Bey-Abina S, Andre Schmutz I, Basile Gde S, de Villartay JP, Cavazzana-Calvo M. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol. Rev. 2005;203:98–109. doi: 10.1111/j.0105-2896.2005.00223.x. [DOI] [PubMed] [Google Scholar]
- 3.Teo YY. Common statistical issues in genome-wide association studies: a review on power, data quality control, genotype calling and population structure. Curr. Opin. Lipidol. 2008;19:133–143. doi: 10.1097/MOL.0b013e3282f5dd77. [DOI] [PubMed] [Google Scholar]
- 4.Perocchi F, Mancera E, Steinmetz LM. Systematic screens for human disease genes, from yeast to human and back. Mol. Biosyst. 2008;4:18–29. doi: 10.1039/b709494a. [DOI] [PubMed] [Google Scholar]
- 5.Lombard Z, Tiffin N, Hofmann O, Bajic VB, Hide W, Ramsay M. Computational selection and prioritization of candidate genes for fetal alcohol syndrome. BMC Genomics. 2007;8:389. doi: 10.1186/1471-2164-8-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Iratxeta C, Bork P, Andrade-Navarro MA. Update of the G2D tool for prioritization of gene candidates to inherited diseases. Nucleic Acids Res. 2007;35:W212–W216. doi: 10.1093/nar/gkm223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oti M, Brunner HG. The modular nature of genetic diseases. Clin. Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 8.Shriner D, Baye TM, Padilla MA, Zhang S, Vaughan LK, Loraine AE. Commonality of functional annotation: a method for prioritization of candidate genes from genome-wide linkage studies. Nucleic Acids Res. 2008;36:e26. doi: 10.1093/nar/gkn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sultan M, Piccini I, Balzereit D, Herwig R, Saran NG, Lehrach H, Reeves RH, Yaspo ML. Gene expression variation in Down's syndrome mice allows prioritization of candidate genes. Genome Biol. 2007;8:R91. doi: 10.1186/gb-2007-8-5-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radivojac P, Peng K, Clark WT, Peters BJ, Mohan A, Boyle SM, Mooney SD. An integrated approach to inferring gene-disease associations in humans. Proteins Struct. Funct. Bioinformatics. 2008;72:1030–1037. doi: 10.1002/prot.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22:773–774. doi: 10.1093/bioinformatics/btk031. [DOI] [PubMed] [Google Scholar]
- 13.Ceresa M, Masseroli M, Campi A. A web-enabled database of human gene expression controlled annotations for gene list functional evaluation. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2007;2007:394–397. doi: 10.1109/IEMBS.2007.4352307. [DOI] [PubMed] [Google Scholar]
- 14.Thornblad TA, Elliott KS, Jowett J, Visscher PM. Prioritization of positional candidate genes using multiple web-based software tools. Twin Res. Hum. Genet. 2007;10:861–870. doi: 10.1375/twin.10.6.861. [DOI] [PubMed] [Google Scholar]
- 15.Higgins ME, Claremont M, Major JE, Sander C, Lash AE. Cancer genes: a gene selection resource for cancer genome projects. Nucleic Acids Res. 2007;35:D721–D726. doi: 10.1093/nar/gkl811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochs H, Puck J, Smith C. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. 2nd. Oxford: Oxford University Press; 2006. [Google Scholar]
- 17.Marodi L, Notarangelo LD. Immunological and genetic bases of new primary immunodeficiencies. Nat. Rev. Immunol. 2007;7:851–861. doi: 10.1038/nri2195. [DOI] [PubMed] [Google Scholar]
- 18.Morimoto Y, Routes JM. Immunodeficiency overview. Prim. Care. 2008;35:159–173. doi: 10.1016/j.pop.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Samarghitean C, Väliaho J, Vihinen M. IDR knowledge base for primary immunodeficiencies. Immunome Res. 2007;3:6. doi: 10.1186/1745-7580-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ortutay C, Vihinen M. Immunome: a reference set of genes and proteins for systems biology of the human immune system. Cell Immunol. 2006;244:87–89. doi: 10.1016/j.cellimm.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Ortutay C, Siermala M, Vihinen M. Molecular characterization of the immune system: emergence of proteins, processes, and domains. Immunogenetics. 2007;59:333–348. doi: 10.1007/s00251-007-0191-0. [DOI] [PubMed] [Google Scholar]
- 22.Ortutay C, Vihinen M. Efficiency of the immunome protein interaction network increases during evolution. Immunome Res. 2008;4:4. doi: 10.1186/1745-7580-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortutay C, Siermala M, Vihinen M. ImmTree: database of evolutionary relationships of genes and proteins in the human immune system. Immunome Res. 2007;3:4. doi: 10.1186/1745-7580-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beissbarth T. Interpreting experimental results using gene ontologies. Methods Enzymol. 2006;411:340–352. doi: 10.1016/S0076-6879(06)11018-6. [DOI] [PubMed] [Google Scholar]
- 26.Ochs MF, Peterson AJ, Kossenkov A, Bidaut G. Incorporation of gene ontology annotations to enhance microarray data analysis. Methods Mol. Biol. 2007;377:243–254. doi: 10.1007/978-1-59745-390-5_15. [DOI] [PubMed] [Google Scholar]
- 27.Gaj S, van Erk A, van Haaften RI, Evelo CT. Linking microarray reporters with protein functions. BMC Bioinformatics. 2007;8:360. doi: 10.1186/1471-2105-8-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 29.Middendorf M, Ziv E, Wiggins CH. Inferring network mechanisms: the Drosophila melanogaster protein interaction network. Proc. Natl Acad. Sci. USA. 2005;102:3192–3197. doi: 10.1073/pnas.0409515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhodes DR, Tomlins SA, Varambally S, Mahavisno V, Barrette T, Kalyana-Sundaram S, Ghosh D, Pandey A, Chinnaiyan AM. Probabilistic model of the human protein-protein interaction network. Nat. Biotechnol. 2005;23:951–959. doi: 10.1038/nbt1103. [DOI] [PubMed] [Google Scholar]
- 31.Estrada E. Virtual identification of essential proteins within the protein interaction network of yeast. Proteomics. 2006;6:35–40. doi: 10.1002/pmic.200500209. [DOI] [PubMed] [Google Scholar]
- 32.Wu X, Zhu L, Guo J, Zhang DY, Lin K. Prediction of yeast protein-protein interaction network: insights from the gene ontology and annotations. Nucleic Acids Res. 2006;34:2137–2150. doi: 10.1093/nar/gkl219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amaral LA, Scala A, Barthelemy M, Stanley HE. Classes of small-world networks. Proc. Natl Acad. Sci. USA. 2000;97:11149–11152. doi: 10.1073/pnas.200327197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doherty IA, Padian NS, Marlow C, Aral SO. Determinants and consequences of sexual networks as they affect the spread of sexually transmitted infections. J. Infect. Dis. 2005;191(Suppl 1):S42–S54. doi: 10.1086/425277. [DOI] [PubMed] [Google Scholar]
- 35.Huber W, Carey VJ, Long L, Falcon S, Gentleman R. Graphs in molecular biology. BMC Bioinformatics. 2007;8(Suppl 6):S8. doi: 10.1186/1471-2105-8-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pieroni E, de la Fuente van Bentem S, Mancosu G, Capobianco E, Hirt H, de la Fuente A. Protein networking: insights into global functional organization of proteomes. Proteomics. 2008;8:799–816. doi: 10.1002/pmic.200700767. [DOI] [PubMed] [Google Scholar]
- 37.Jeger MJ, Pautasso M, Holdenrieder O, Shaw MW. Modelling disease spread and control in networks: implications for plant sciences. New Phytol. 2007;174:279–297. doi: 10.1111/j.1469-8137.2007.02028.x. [DOI] [PubMed] [Google Scholar]
- 38.Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–382. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 39.Xulvi-Brunet R, Sokolov IM. Growing networks under geographical constraints. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2007;75:046117. doi: 10.1103/PhysRevE.75.046117. [DOI] [PubMed] [Google Scholar]
- 40.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 41.Mathivanan S, Periaswamy B, Gandhi TK, Kandasamy K, Suresh S, Mohmood R, Ramachandra YL, Pandey A. An evaluation of human protein-protein interaction data in the public domain. BMC Bioinformatics. 2006;7(Suppl 5):S19. doi: 10.1186/1471-2105-7-S5-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Csardi G, Nepusz T. The igraph software package for complex network research. Int. J. Complex Syst. 2006:1695. [Google Scholar]
- 43.Latora V, Marchiori M. Efficient behavior of small-world networks. Phys. Rev. Lett. 2001;87:198701. doi: 10.1103/PhysRevLett.87.198701. [DOI] [PubMed] [Google Scholar]
- 44.Gol’dshtein V, Koganov G, Surdutovich G. Vulnerability and hierarchy of complex networks. Arxiv. Prepr. Cond.-Mater. 2004:0409298. [Google Scholar]
- 45.Freeman L. Centrality in social networks: conceptual clarification. Social Networks. 1979;1:215–239. [Google Scholar]
- 46.Smith CL, Goldsmith CA, Eppig JT. The Mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6:R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc. Natl Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE. The mouse genome database (MGD): new features facilitating a model system. Nucleic Acids Res. 2007;35:D630–D637. doi: 10.1093/nar/gkl940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 50.Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc. Natl Acad. Sci. USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ideker T, Sharan R. Protein networks in disease. Genome Res. 2008;18:644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oti M, Snel B, Huynen MA, Brunner HG. Predicting disease genes using protein-protein interactions. J. Med. Genet. 2006;43:691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 54.George RA, Liu JY, Feng LL, Bryson-Richardson RJ, Fatkin D, Wouters MA. Analysis of protein sequence and interaction data for candidate disease gene prediction. Nucleic Acids Res. 2006;34:e130. doi: 10.1093/nar/gkl707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Hughes-Oliver JM, Menius JA., Jr Domain-enhanced analysis of microarray data using GO annotations. Bioinformatics. 2007;23:1225–1234. doi: 10.1093/bioinformatics/btm092. [DOI] [PubMed] [Google Scholar]
- 57.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, Takada H, Hara T, Kawamura N, Ariga T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 58.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 59.Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, Casanova JL. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr. Opin. Immunol. 2008;20:39–48. doi: 10.1016/j.coi.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 60.Xavier RJ, Rioux JD. Genome-wide association studies: a new window into immune-mediated diseases. Nat. Rev. Immunol. 2008;8:631–643. doi: 10.1038/nri2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis —a genomewide study. N. Engl. J. Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.