

Abstract
The transfer of phenotypes from one individual to another is a fundamental aspect of biology. In addition to traditional nucleic acid-based genetic determinants, unique proteins known as prions can also act as elements of inheritance, infectivity, and disease. Nucleic acids and proteins encode genetic information in distinct ways, either in the sequence of bases in DNA or RNA or in the three dimensional structure of the polypeptide chain. Given these differences in the nature of the genetic repository, the mechanisms underlying the transmission of nucleic acid-based and protein-based phenotypes are necessarily distinct. While the appearance, persistence and transfer of nucleic acid determinants require the synthesis of new polymers, recent studies indicate that prions are propagated through dynamic transitions in the structure of existing protein.
Key Words: prion, PrP, [PSI+], [URE3], [Het-s], Sup35, Ure2, Het-s, Hsp104
The Protein-Only Mechanism
The prion hypothesis was originally proposed to explain the atypical etiology of the transmissible spongiform encephalopathies (TSEs), a group of progressive and fatal neuro-degenerative diseases including scrapie in sheep, bovine spongiform encephalopathy (or mad cow disease) in cattle, and Creutzfeldt-Jacob Disease (CJD) and kuru in humans.1 Strikingly, these infectious diseases may also develop spontaneously or through genetic predisposition, suggesting that the genetic determinant is actually host encoded.2
Historically, elements of infection and inheritance are thought to rely on a nucleic acid core to encode genetic information. The TSE agent proved to be enigmatic, however, resisting exposure to manipulations known to destroy nucleic acids, such as nucleases and radiation, while succumbing to treatments that disrupt protein structure, including proteases,1 detergents,3 denaturants,4 chaotropic salts5 and organic solvents.6 Consistent with these observations, a 27–30 kDa host encoded protein was found to be the major constituent of highly purified brain homogenate preparations that retained infectivity,7–10 leading Prusiner to postulate that the scrapie agent was an infectious protein (prion) termed PrP.10 This once heretical model also appears to be applicable to a number of unrelated phenotypes particularly in fungi, where protein-only models accurately describe the inheritance of a wide range of previously inexplicable phenotypes including the use of alternate nitrogen sources,11 the regulation of translation termination efficiency,11 the formation of heterokaryons,12 the appearance of other prions13 and organismal dependence on quality control pathways.14
While appealing, the idea of protein-directed inheritance is at odds with our classical view of genetics. In the simplest case, the transmission of phenotypes from one individual to another either through heredity or infectivity relies on two key events. First, the genetic determinant of the trait must be able to replicate itself to produce identical copies. Second, a mechanism must exist to ensure that these copies are efficiently partitioned between donor and recipient. As their molecular architecture is particularly well suited to self-templated replication, nucleic acids, specifically the sequence of bases found in these polymers, form an effective repository for genetic information (Fig. 1A). The idea of an infectious protein, however, immediately poses a mechanistic dilemma in the absence of a protein-directed protein synthetic pathway. Thus, protein-only genetic information must be fundamentally distinct from the sequence-based genetic information carried by nucleic acids (Fig. 1B).
Figure 1.
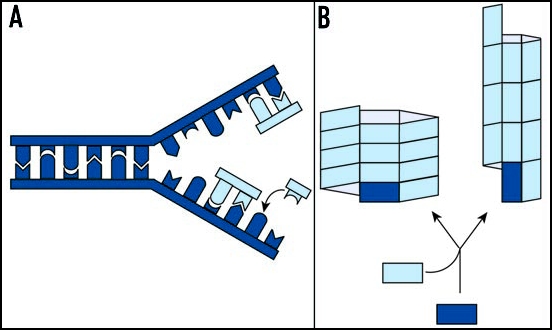
Genetic information can be replicated in two ways. (A) For nucleic acid-based determinants, replication occurs through the polymerization of free nucleotides (light blue), using the sequence of bases in an existing polymer (dark blue) as a template. (B) For protein-based determinants, genetic information is encoded in quaternary structure. For the example shown here, a simple rectangle can be assembled with the subunits aligned along either their short or their long faces, leading to two distinct self-replicating polymers. Adapted from Caughey.142
In 1967, Griffith proposed a mechanism whereby a protein could catalyze its own replication.15 Based on this proposal, self-replicating proteins, such as prions, must adopt at least two distinct conformations, a normal form and a disease-associated form, with the latter serving as a template for the conversion of the former. In line with this prediction, the PrPC (cellular) and PrPSc (scrapie)16 conformational isomers (conformers) as well as the two forms of the yeast prion proteins are readily distinguished by their sensitivity to proteases,17–22 their state of oligomerization,18,19,22–25 and their secondary structure.26
The idea of a self-replicating protein conformation gained additional support when this principle was directly demonstrated in cell-free systems. In these assays, the abnormal form of a prion protein directs the conversion of soluble prion protein to an oligomerized amyloid state sharing the biochemical characteristics of ex vivo prion-state protein.24,27–32 In many cases, material generated by in vitro conversion reactions has been shown to promote or accelerate the appearance of prion-associated phenotypes when exogenously supplied to susceptible hosts.33–39 These observations support the idea that protein-based genetic elements, unlike their nucleic acid-based counterparts, encode information in their quaternary rather than their primary structures (Fig. 1B).
Since the appearance, spread and reversal of prion-associated phenotypes involve changes in protein state rather than the synthesis of new protein components, a clear mechanistic understanding of the process requires insight into prion protein dynamics in vivo. With this idea in mind, the prion mechanism can be broken down into three discrete fundamental steps. First, non-prion state protein must adopt the prion conformation, a process that may be either spontaneous or templated by existing prion-state protein. Second, the prion template must be continually regenerated to provide new surfaces for conversion. Finally, prion-state protein must be transmitted to other cells either by extracellular secretion and uptake for non-dividing cells or via partitioning to daughter cells in actively dividing cultures. Each step of this in vivo prion cycle must be undertaken with high precision to maintain a strong link between protein state and phenotype in order for the prion mechanism to serve as an effective alternate route for the inheritance of traits. In this review, we focus on insights into the mechanisms underlying prion propagation in vivo gleaned from studies of prion protein dynamics.
Prion Conversion In Vivo
The biosynthesis and maturation of proteins in a eukaryotic cell is an intricate process that is regulated by the action of chaperones and proteases and that is influenced by subcellular compartmentalization. 40,41 While the prion hypothesis predicts that the non-prion conformer can be directly remodeled to the prion form in the presence of a pre-existing prion template,15,16 the range of quality control pathways regulating protein biogenesis must be considered when extending the predictions of the prion hypothesis to a living cell. For example, protein maturation begins as soon as the polypeptide emerges from the ribosome exit tunnel, as co-translationally acting chaperones41 and proteolytic mechanisms42,43 engage the nascent peptide. Indeed, productive folding pathways often lead proteins to their mature folded state by the time synthesis is complete.44 With these observations in mind, a key consideration in developing a mechanistic understanding of any in vivo prion cycle is the point at which the alternate biogenesis pathway is initiated, whether it be nascent, non-native or mature prion protein (Fig. 2A).
Figure 2.
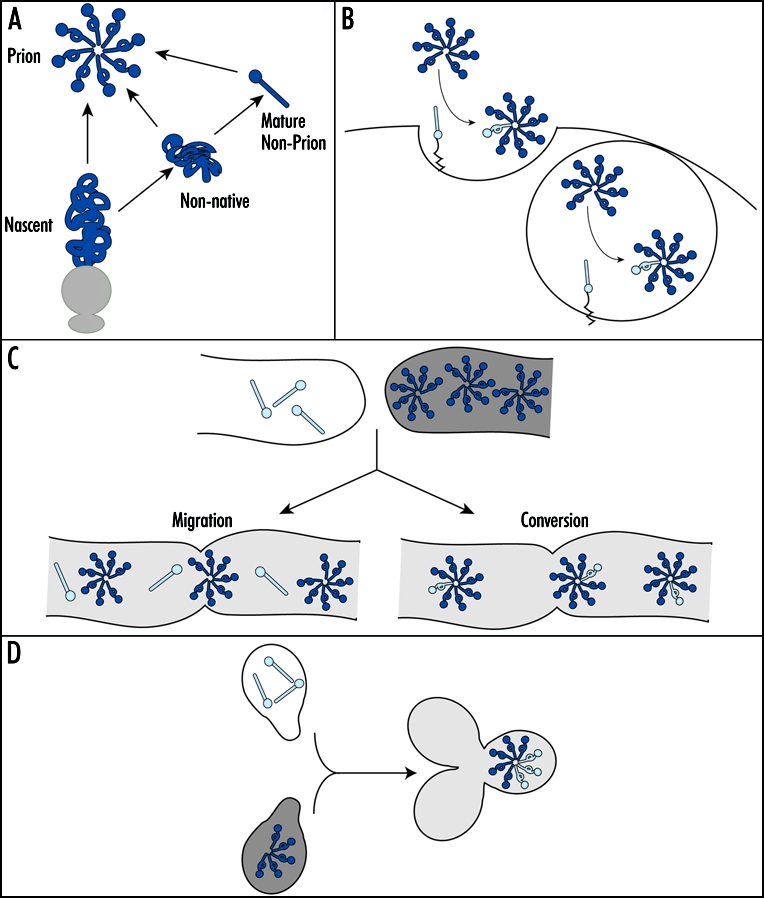
In vivo prion conversion. (A) In vivo, a prion protein (dark blue) may convert to the prion form at multiple points during its biogenesis pathway (see text for details). (B) PrPC (light blue ball and stick) likely converts to the prion form (dark blue ball and loop) at the cell surface (left) or in endocytic vesicles (right). (C) The prion phenotype is transmitted from a [Het-s] prion strain (dark gray) to [Het-s*] non-prion strain (white) through cytoplasmic mixing following hyphal fusion (light gray). This transmission may occur either through the migration of existing [Het-s] complexes (dark blue) into the [Het-s*] recipient and/or via the conversion of existing non-prion state protein (light blue ball and stick) to the prion form (light blue ball and loop). (D) Upon mating, a [psi-] cell (white) to a [PSI+] cell (dark gray), the prion phenotype establishes dominance via incorporation of existing non-prion state protein (light blue) into the introduced prion complexes (dark blue).
For PrP, the conversion process has been extensively studied within the context of a number of scrapie infected cell culture systems, which support conversion of newly made PrP to the prion state at a low frequency (∼10% of total PrP).20,45,46 Using these systems, several lines of evidence suggest that PrP converts to the prion state after synthesis (Fig. 2B). First, pulse-labeled PrP transits from a protease sensitive to a protease resistant state on the time scale of hours.45,47 Second, PrP localization appears to impact the conversion process. PrP is a cell-surface glycoprotein,48 and retention of PrP in the endoplasmic reticulum by treatment with either brefeldin A49 or intracellular antibodies50 or removal of PrP from the cell surface by phospholipase treatment45,51 inhibits the accumulation of protease resistant PrP. Third, exposure of infected tissue culture cells or animals to anti-PrP antibodies inhibits accumulation of protease resistant PrP and infectious prions by inducing PrP degradation or by retaining it at the cell surface, suggesting that conversion may normally occur in endocytic vesicles.52–55 These observations clearly indicate that the final hallmarks of conversion, protease resistance and infectivity, appear late in the maturation of PrP. A future challenge is to determine if this alternate biogenesis pathway is initiated at the time of synthesis or if mature PrPC is a direct substrate for this transition.45,47,49,51
The in vivo conversion process has also been studied in the propagation of two fungal prions, [Het-s], a regulator of heterokaryon compatibility in Podospora anserina,12 and [PSI+], a regulator of translation termination efficiency in Saccharomyces cerevisiae.56 In each case, conversion was studied by introducing the non-prion and prion forms of the corresponding proteins into the same cell by fusion. For [Het-s], heterokaryons formed between non-prion ([Het-s*]) and prion ([Het-s]) cells display the prion phenotype: incompatibility with strains harboring the het-S allele.57 Upon fusion of [Het-s*] and [Het-s] strains, the prion phenotype can be tracked as it migrates into the non-prion recipient from the site of fusion by dissecting and challenging fragments of the mycelium with a het-S strain.12 In these experiments, spread of the prion phenotype occurs in the presence of cycloheximide, indicating that the process does not require new protein synthesis.12 This observation can be explained either by direct conversion of non-prion state Het-s protein to the prion form12 or alternately by the migration of existing prion state protein from the donor into the recipient cell, as the [Het-s] phenotype is a gain-of-function (Fig. 2C).25 In either case, a dynamic transition in the physical state and/or localization of the Het-s protein is a key contributor to the infectious spread of the [Het-s] prion.
The [PSI+] prion, a conformer of the Sup35 protein, is perhaps the most extensively studied of the fungal prions. Conversion of Sup35 from its non-prion [psi-] state to its prion [PSI+] state is accompanied by a partial inhibition of normal Sup35 function in translation termination.18,19,58 This easily scored link between the physical and functional states of the protein has been a useful tool in dissecting the mechanism of prion conversion in vivo. By mating a [PSI+] cell expressing unmarked Sup35 to a [psi-] cell expressing a Sup35 fusion to the green fluorescent protein (Sup35-GFP), conversion of soluble pre-existing Sup35 to an aggregated state can be directly observed in live cells, and this process occurs on the time scale of minutes (Fig. 2D).59 This transition in Sup35 physical state is accompanied by loss of Sup35 activity, as measured by stop codon read-through.59 These studies indicate that the prion state does not need to be specified at the time of synthesis but instead can be transmitted to existing, mature non-prion state protein.
Regeneration of the Prion Template In Vivo
Although the original formulations of the prion hypothesis suggested that the template for prion conversion could be a monomer,15,16 mathematical models of prion infectivity are inconsistent with this idea.60,61 Rather, such modeling studies suggest that small oligomers would function as the most infectious units.62,63 Consistent with these predictions, ionizing radiation,64–66 fractionation,67,68 and denaturation69 studies support a minimum infectious complex of ∼5 PrP subunits, with peak infectivity in the range of 14–28 monomers.68 Moreover, the threshold behavior of scrapie appearance, in which the sporadic rate of disease is exceedingly low, can only be explained by a mechanism in which multiple, spontaneously arising PrPSc monomers must come together to form a stable complex that functions as an active template.60,70 In the alternate scenario, every individual in the population would eventually develop a TSE, as each spontaneously arising PrPSc monomer would be sufficient to establish disease.60 The concept of an oligomeric template is also supported by fungal experiments in which external delivery of protein to live cells and structure-based mutagenesis studies have linked heritable and infectious [Het-s], [PSI+], [URE3], and [PIN+] prions to ordered amyloid fibers of the Het-s,39,71 Sup35,35,37 Ure2,33 and Rnq138 proteins, respectively.
Assuming a linear polymer model, the number of free ends (i.e., the templating surfaces) is a limiting factor for the rate of conversion and therefore disease progression.60,72 Since the number of complexes spontaneously arising or those existing in an infectious inoculum are likely to be insufficient to establish an infection,62 early theoretical models of disease predicted a second vital step in the process: the continual generation of additional catalytic surfaces.60,72 Various mechanistic scenarios are possible to generate these secondary sites of nucleation,60 but the continual fragmentation of existing linear templates is the most widely favored pathway.60,70,72,73
Indirect evidence points to the importance of fragmentation of PrP complexes in vivo. For example, PrPSc accumulation is exponential following infection,74–76 and only mathematical models that consider fragmentation can accurately describe these kinetics.63,77 Moreover, prion infectivity greatly increases upon partial disruption of ex vivo preparations of PrP by denaturation,69 liposome dispersion,67,78 homogenization79 and sonication,68,80 highlighting the potent effect of fragmentation on prion titre. Finally, the yield of in vitro conversion reactions is greatly improved by cyclic rounds of polymerization and sonication,31 a process that increases titres to a level sufficient to establish an infection in vivo.34 Thus, although an endogenous fragmentation activity has not been directly observed in the mammalian prion system, such a process, whether stochastic or catalyzed, has the potential to greatly impact the establishment and progression of disease.62
In S. cerevisiae, template fragmentation is an active process, catalyzed by the molecular disaggregase Hsp104. Hsp104 is a member of the AAA+ ATPase family81 and is required for survival of yeast at high temperatures, provided the organism is first exposed to a more modest heat stress.82 At these elevated temperatures, Hsp104's essential function is the resolution of thermally induced aggregates.83,84 Hsp104 was first identified as a prion modulator in a screen to identify factors stimulating loss of [PSI+] when overexpressed.85 In this and subsequent studies, an essential role for Hsp104 in the propagation of all fungal prions was uncovered, as inactivation of Hsp104's ATPase activity by deletion, expression of a dominant mutant85 or treatment with guanidine hydrochloride (GdnHCl)86–90 leads to prion loss in all cases.13,82,85,91,92
Insight into the mechanism of Hsp104 action arose from early studies on the kinetics of prion loss or curing by GdnHCl. When [PSI+] cultures are grown in the presence of GdnHCl, [psi-] cells begin to appear after a 4–5 generation lag, and mathematical models of the kinetics of this curing event suggest a two-step process: (1) a failure to replicate or fragment existing prion templates and (2) subsequent dilution of these complexes during cell division.93,94 Consistent with these ideas, Sup35 complexes increase in size upon Hsp104 inactivation,95–97 and [PSI+] curing only occurs in actively dividing cultures upon Hsp104 inhibition.93 Hsp104 appears to provide similar fragmentation activity in the propagation of [PIN+], as Rnq1 complexes also increase in size upon Hsp104 inhibition.98 Intriguingly, loss of Sis1, an Hsp40 family member, similarly affects Rnq1 complex size, suggesting that Sis1 and Hsp104 cooperate to generate additional Rnq1 templates in vivo.98
Complementing in vitro observations of Hsp104-dependent fragmentation of Sup35 amyloid fibers99,100 (see also refs. 101 and 102), direct proof of Hsp104-dependent fragmentation in vivo was gleaned from observations of prion protein dynamics in live cells. When new protein synthesis is inhibited in cells with wildtype Hsp104 activity, existing prion complexes marked with Sup35-GFP become undetectable by microscopy within hours;97,103 however, upon Hsp104 inhibition, the same complexes persist.97 Since Hsp104 does not alter the metabolic stability of Sup35,97 the observed loss of fluorescence in wildtype cells provides an assay for fragmentation. Through the repeated fragmentation of complexes by Hsp104 and the subsequent incorporation of constitutively expressed untagged Sup35, the original pool of Sup35-GFP monomers is redistributed among a greater number of prion complexes, leading to their decreased intensity. Consistent with this idea, quantitative imaging techniques indicate that Sup35-GFP complexes remain the same size despite the progressive loss of fluorescence in wildtype cells.97,103
Secondary Effects of Fragmentation
Fragmentation is predicted to impact two events during prion propagation: conversion efficiency and prion transmission (Fig. 3).62,72,73,77 Experimental proof of these predictions has been provided by studies of the Sup35/[PSI+] prion in which dynamic transitions in aggregation state, as assayed by fluorescence microscopy and biochemical analyses, were correlated with changes in functional state, as assayed by the efficiency of translational termination. Using these powerful approaches, efficient inactivation of newly synthesized Sup35 by incorporation into existing prion complexes is evident in wildtype cells; however, nascent Sup35 accumulates in a soluble and functional pool upon Hsp104 inhibition, suggesting a defect in the kinetics of conversion likely due to a limitation in the number of prion templates.18,97,104,105 This defect is immediately apparent within the first generation of Hsp104 inhibition and can be reversed within the same time frame by reactivation of Hsp104.97,104–106 Thus, the [PSI+] prion cycle is finely tuned in vivo, with the continual generation of new prion templates by fragmentation making a key contribution to this razor's edge balance.
Figure 3.
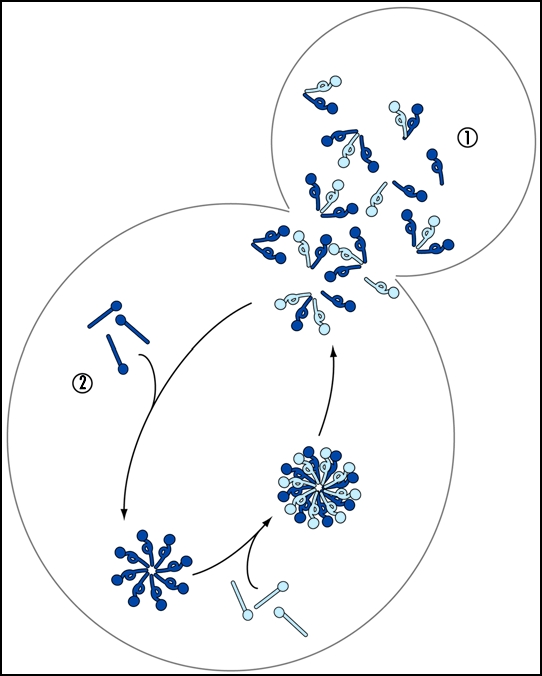
Fragmentation has multiple consequences. Non-prion state protein (light blue ball and stick) converts to the prion form (light blue ball and loop) upon incorporation into existing prion complexes (dark blue ball and loop), which are subsequently fragmented, either stochastically or catalytically, to generate smaller complexes. This fragmentation facilitates spread of the prion templates, shown here as partitioning to daughter cells in a dividing culture—pathway 1, and increases the efficiency of incorporation of additional non-prion subunits (dark blue ball and stick)—pathway 2.
Despite the importance of fragmentation for conversion efficiency, newly synthesized Sup35 does continue to join the static prion complexes that persist upon Hsp104 inhibition.97,104 This incorporation gradually decreases the mobility of complexes in the yeast cytosol, as revealed by fluorescence recovery after photobleaching (FRAP) experiments.97,105 The resulting, largely immobile complexes are inefficiently transferred to daughter cells, leading to a partitioning defect and ultimately loss of [PSI+].97,106
Modulating Prion Dynamics
The role of Hsp104 in fungal prion propagation is a dramatic example of the importance of dynamic transitions in prion protein physical state for the propagation of prion-associated phenotypes. In addition to chaperone effects, homotypic and heterotypic interactions between prion proteins themselves have emerged as potent modulators of in vivo prion cycles.
The prion hypothesis originally predicted that prion proteins can physiologically access two physical states, but studies in both mammals and lower eukaryotes now indicate that this conformational flexibility is much more complex than the model first proposed, with a range of physical and phenotypic states (strains or variants) possible.56,107–109 These variants have important ramifications for prion biology in vivo. For example, PrP variants differ in both their incubation times and in their patterns of neurodegeneration,110 and variants of the [PSI+], [PIN+] and [URE3] yeast prions can be distinguished by the severity of their phenotypes.107,109,111 Intriguingly, recent studies of both PrP and Sup35 variants revealed a difference in the stability of prion complexes, assessed by denaturation either with GdnHCl or SDS,95,112–114 and this range of stabilities is likely due to conformational differences between the assembled subunits.35,37,115–122 In these studies, an increase in the stability of prion complexes, and presumably a decrease in the rate of endogenous fragmentation,62 diminished the severity of the prion phenotype (i.e., incubation time for PrP and degree of functional inactivation for Sup35). Thus, conformational differences alone can have profound effects on the physiological consequences of prion propagation.
In addition to sequence-independent effects, the efficiency of prion propagation by wildtype protein can be dramatically altered by co-expression of sequence variants. For example, co-expression of PrP proteins derived from different species interferes with templated conversion of PrP to a protease-resistant form both in vivo123 and in vitro,124 and in some cases, inhibition can be linked to single amino acid changes.125,126 Similar findings have been reported in the yeast system in which fragments of Ure223 or point mutations within Sup35 disrupt or diminish prion propagation by wildtype protein.127–129 Three mechanisms have been proposed to explain these dominant effects. First, sequence differences could alter prion protein interaction with an essential trans regulator.123,127,130 Second, mutant proteins could interact with and cap existing aggregates, thereby decreasing their templating ability.23,127,131,132 Finally, mixed complexes, containing wildtype and mutant proteins, could adopt conformations that are not efficiently inherited.127,131 Future studies of prion dynamics in the presence of these dominant inhibitors will likely be instructive in revealing their mechanisms of inhibition in vivo.
Inter-prion interactions can also profoundly alter an in vivo prion cycle. A case in point is the de novo appearance of a new prion. In both yeast and mammals, such events are rare, but the frequency of de novo prion induction increases greatly if the prion protein is overexpressed, perhaps by increasing the frequency of a stochastic misfolding event.11,12,111,133,134 Whether spontaneous misfolding occurs during or after synthesis is unclear; however, studies in yeast suggest that this initiating event is itself nucleated. In the case of the [PSI+] prion, de novo appearance by overexpression of Sup35 depends on the presence of a pre-existing protein template, most frequently the [PIN+] prion, encoded by Rnq1.13,92,133 Recent studies in vitro and in vivo suggest that Rnq1 complexes are likely to effect Sup35 dynamics: Rnq1 complexes heterogeneously nucleate the formation of Sup35 complexes135,136 and are required for conversion of these nascent Sup35 oligomers to a heritable form.137
In addition to positive interactions between prions, co-existence of multiple prion forms, whether they are different sequences or different variants of the same sequence, is often disfavored. For example, co-inoculation of mice with distinct PrP variants extends incubation timing;138,139 de novo induction of [URE3] by overexpression of Ure2 is diminished in [PSI+] strains,107 and despite their positive interactions under other conditions, some variants of [PIN+] destabilize variants of [PSI+].140 These observations suggest a competition for some component common to the propagation pathways. One example of this idea is the competition between different variants of the same prion protein. When two different variants are introduced into the same cytoplasm by mating yeast, only one form persists when the resulting diploids are allowed to grow into colonies, and the protein state is subsequently analyzed.114,141 This dominance likely results from differences in fragmentation rate, allowing one variant to out compete another for the incorporation of the same pool of nascent prion protein.112
Concluding Remarks
The prion hypothesis has expanded our view of genetics to include proteins as potential determinants of phenotypic traits. Several examples of this alternate route of inheritance and infectivity now exist, in which protein conformations impart unique phenotypes to an organism. The distinctions between prion and non-prion conformers and phenotypes are, however, extreme points on a continuum. This inherent metastability of protein-based genetics underlies its fascinating biology; that is, the potential for new phenotypes to arise, persist, spread and be lost within the lifetime of an organism. The multi-step prion cycle in vivo provides many exquisitely sensitive points of regulation and potential intervention into this process. Remarkably, slight variations in prion protein dynamics, mediated by either cis or trans effectors, have the capacity to profoundly and swiftly impact the biological consequences of protein-based genetic elements.
Acknowledgements
We would like to thank E. Craig for sharing unpublished observations, R. Lesiak for assistance with the illustrations, and Martina Strbuncelj, Sara Langseth and Susanne DiSalvo for comments on the manuscript. This work is supported by grants from the Pew Scholars Program in the Biomedical Sciences (3274sc) and NIGMS (GM069802) to T.R.S.
Abbreviations
- GdnHCl
guanidine hydrochloride
- Prp
prion protein
- TSEs
Transmissible Spongiform Encephalopathies
- CJD
Creutzfeldt-Jacob Disease
Footnotes
Previously published online as a Prion E-publication: http://www.landesbioscience.com/journals/Prion/abstract.php?id=3992
References
- 1.Prusiner SB, McKinley MP, Groth DF, Bowman KA, Mock NI, Cochran SP, Masiarz FR. Scrapie agent contains a hydrophobic protein. Proc Natl Acad Sci USA. 1981;78:6675–6679. doi: 10.1073/pnas.78.11.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2:861–871. doi: 10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB, Groth DF, Cochran SP, Masiarz FR, McKinley MP, Martinez HM. Molecular properties, partial purification, and assay by incubation period measurements of the hamster scrapie agent. Biochemistry. 1980;19:4883–4891. doi: 10.1021/bi00562a028. [DOI] [PubMed] [Google Scholar]
- 4.Hunter GD, Gibbons RA, Kimberlin RH, Millson GC. Further studies of the infectivity and stability of extracts and homogenates derived from scrapie affected mouse brains. J Comp Pathol. 1969;79:101–108. doi: 10.1016/0021-9975(69)90033-4. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner SB, Groth DF, McKinley MP, Cochran SP, Bowman KA, Kasper KC. Thiocyanate and hydroxyl ions inactivate the scrapie agent. Proc Natl Acad Sci USA. 1981;78:4606–4610. doi: 10.1073/pnas.78.7.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hunter GD, Millson GC. Attempts to release the scrapie agent from tissue debris. J Comp Pathol. 1967;77:301–307. doi: 10.1016/0021-9975(67)90039-4. [DOI] [PubMed] [Google Scholar]
- 7.Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46:417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 8.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 9.McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. doi: 10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- 10.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 11.Wickner RB. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 12.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507–519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collin P, Beauregard PB, Elagoz A, Rokeach LA. A non-chromosomal factor allows viability of Schizosaccharomyces pombe lacking the essential chaperone calnexin. J Cell Sci. 2004;117:907–918. doi: 10.1242/jcs.00943. [DOI] [PubMed] [Google Scholar]
- 15.Griffith J. Self-replication and Scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 16.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 17.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 18.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 19.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 20.Butler DA, Scott MR, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol. 1988;62:1558–1564. doi: 10.1128/jvi.62.5.1558-1564.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caughey B, Neary K, Buller R, Ernst D, Perry LL, Chesebro B, Race RE. Normal and scrapie-associated forms of prion protein differ in their sensitivities to phospholipase and proteases in intact neuroblastoma cells. J Virol. 1990;64:1093–1101. doi: 10.1128/jvi.64.3.1093-1101.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci USA. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA. 1999;96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sondheimer N, Lindquist S. Rnq1: An epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 25.Coustou-Linares V, Maddelein ML, Begueret J, Saupe SJ. In vivo aggregation of the HET-s prion protein of the fungus Podospora anserina. Mol Microbiol. 2001;42:1325–1335. doi: 10.1046/j.1365-2958.2001.02707.x. [DOI] [PubMed] [Google Scholar]
- 26.Pan KM, Baldwin MA, Nguyen J, Gasset M, Serban A, Grothe D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of alpha-helices into beta-sheet features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 28.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 29.King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 31.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 32.Dos Reis S, Coulary-Salin B, Forge V, Lascu I, Begueret J, Saupe SJ. The HET-s prion protein of the filamentous fungus Podospora anserina aggregates in vitro into amyloid-like fibrils. J Biol Chem. 2002;277:5703–5706. doi: 10.1074/jbc.M110183200. [DOI] [PubMed] [Google Scholar]
- 33.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO J. 2005;24:3082–3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 35.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 36.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 38.Patel BK, Liebman SW. “Prion-proof” for [PIN(+)]: Infection with In vitro-made amyloid aggregates of Rnq1p-(132–405) induces [PIN(+)] J Mol Biol. 2007;365:773–782. doi: 10.1016/j.jmb.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci USA. 2002;99:7402–7407. doi: 10.1073/pnas.072199199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 41.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 42.Turner GC, Varshavsky A. Detecting and measuring co-translational protein degradation in vivo. Science. 2000;289:2117–2120. doi: 10.1126/science.289.5487.2117. [DOI] [PubMed] [Google Scholar]
- 43.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 44.Netzer WJ, Hartl FU. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 45.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- 46.Taraboulos A, Serban D, Prusiner SB. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J Cell Biol. 1990;110:2117–2132. doi: 10.1083/jcb.110.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol. 1990;110:743–752. doi: 10.1083/jcb.110.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caughey B, Race RE, Ernst D, Buchmeier MJ, Chesebro B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J Virol. 1989;63:175–181. doi: 10.1128/jvi.63.1.175-181.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell. 1992;3:851–863. doi: 10.1091/mbc.3.8.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardinale A, Filesi I, Vetrugno V, Pocchiari M, Sy MS, Biocca S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J Biol Chem. 2005;280:685–694. doi: 10.1074/jbc.M407360200. [DOI] [PubMed] [Google Scholar]
- 51.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem. 1992;267:16188–16199. [PubMed] [Google Scholar]
- 52.White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 53.Kim CL, Karino A, Ishiguro N, Shinagawa M, Sato M, Horiuchi M. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J Gen Virol. 2004;85:3473–3482. doi: 10.1099/vir.0.80113-0. [DOI] [PubMed] [Google Scholar]
- 54.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA. 2001;98:9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 56.Cox B. [PSI], a cytoplasmic suppressor of super-suppression in yeast. Heredity. 1965;20:505–521. [Google Scholar]
- 57.Rizet G. Les phenomenes de Barrage chez Podspora anserina I.—Analyse genetique des barrages entre souches S ET s. Rev Cytol Biol Veg. 1952;13:51–92. [Google Scholar]
- 58.Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature. 2005;437:262–265. doi: 10.1038/nature03981. [DOI] [PubMed] [Google Scholar]
- 60.Eigen M. Prionics or the kinetic basis of prion diseases. Biophysical Chemistry. 1996;63:A1–A18. doi: 10.1016/s0301-4622(96)02250-8. [DOI] [PubMed] [Google Scholar]
- 61.Masel J, Genoud N, Aguzzi A. Efficient inhibition of prion replication by PrP-Fc(2) suggests that the prion is a PrP(Sc) oligomer. J Mol Biol. 2005;345:1243–1251. doi: 10.1016/j.jmb.2004.10.088. [DOI] [PubMed] [Google Scholar]
- 62.Hall D, Edskes H. Silent prions lying in wait: A two-hit model of prion/amyloid formation and infection. J Mol Biol. 2004;336:775–786. doi: 10.1016/j.jmb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 63.Masel J, Jansen VA. The measured level of prion infectivity varies in a predictable way according to the aggregation state of the infectious agent. Biochim Biophys Acta. 2001;1535:164–173. doi: 10.1016/s0925-4439(00)00095-8. [DOI] [PubMed] [Google Scholar]
- 64.Alper T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochemical and Biophysical Research Communications. 1966;22:278–284. doi: 10.1016/0006-291x(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 65.Gibbs CJ, Jr, Gajdusek DC, Latarjet R. Unusual resistance to ionizing radiation of the viruses of kuru, Creutzfeldt-Jakob disease, and scrapie. Proc Natl Acad Sci USA. 1978;75:6268–6270. doi: 10.1073/pnas.75.12.6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellinger-Kawahara CG, Kempner E, Groth D, Gabizon R, Prusiner SB. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology. 1988;164:537–541. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- 67.Gabizon R, McKinley MP, Prusiner SB. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc Natl Acad Sci USA. 1987;84:4017–4021. doi: 10.1073/pnas.84.12.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caughey B, Raymond GJ, Kocisko DA, Lansbury PT., Jr Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J Virol. 1997;71:4107–4110. doi: 10.1128/jvi.71.5.4107-4110.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 71.Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orgel LE. Prion replication and secondary nucleation. Chemistry and Biology. 1996;3:413–414. doi: 10.1016/s1074-5521(96)90087-3. [DOI] [PubMed] [Google Scholar]
- 73.Nowak MA, Krakauer DC, Klug A, May RM. Prion infection dynamics. Integrative Biology. 1998;1:3–15. [Google Scholar]
- 74.Bolton DC, Rudelli RD, Currie JR, Bendheim PE. Copurification of Sp33–37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease. J Gen Virol. 1991;72:2905–2913. doi: 10.1099/0022-1317-72-12-2905. [DOI] [PubMed] [Google Scholar]
- 75.Jendroska K, Heinzel FP, Torchia M, Stowring L, Kretzschmar HA, Kon A, Stern A, Prusiner SB, DeArmond SJ. Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity. Neurology. 1991;41:1482–1490. doi: 10.1212/wnl.41.9.1482. [DOI] [PubMed] [Google Scholar]
- 76.Beekes M, Baldauf E, Diringer H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J Gen Virol. 1996;77(Pt 8):1925–1934. doi: 10.1099/0022-1317-77-8-1925. [DOI] [PubMed] [Google Scholar]
- 77.Masel J, Jansen VA, Nowak MA. Quantifying the kinetic parameters of prion replication. Biophysical Chemistry. 1999;77:139–152. doi: 10.1016/s0301-4622(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 78.Gabizon R, McKinley MP, Groth DF, Kenaga L, Prusiner SB. Properties of scrapie prion protein liposomes. J Biol Chem. 1988;263:4950–4955. [PubMed] [Google Scholar]
- 79.Malone TG, Marsh RF, Hanson RP, Semancik JS. Membrane-free scrapie activity. J Virol. 1978;25:933–935. doi: 10.1128/jvi.25.3.933-935.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rohwer R, Gajdusek DC. Scrapie—Virus or viroid the case for a virus. In: Boese A, editor. Search for the Cause of Multiple Sclerosis and Other Chronic Diseases of the Central Nervous System. Weinheim: Verlag Chemie; 1980. pp. 333–355. [Google Scholar]
- 81.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 82.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 83.Glover JR, Lindquist S. Hsp104 Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 84.Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 85.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 86.Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem. 2004;279:7378–7383. doi: 10.1074/jbc.M312403200. [DOI] [PubMed] [Google Scholar]
- 87.Jung G, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 2002;99:9936–9941. doi: 10.1073/pnas.152333299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr Microbiol. 2001;43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 89.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 2001;40:1357–1369. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 90.Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics. 1981;98:691–711. doi: 10.1093/genetics/98.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell. 2001;106:183–194. doi: 10.1016/s0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 93.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kushnirov VV, Ter-Avanesyan MD. Structure and replication of yeast prions. Cell. 1998;94:13–16. doi: 10.1016/s0092-8674(00)81216-7. [DOI] [PubMed] [Google Scholar]
- 95.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 96.Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol. 2001;21:4656–4669. doi: 10.1128/MCB.21.14.4656-4669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Satpute-Krishnan P, Langseth SX, Serio TR. Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance. PLoS Biol. 2007;5:e24. doi: 10.1371/journal.pbio.0050024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Craig EA. Personal communication.
- 99.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 100.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell. 2006;23:425–438. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krzewska J, Melki R. Molecular chaperones and the assembly of the prion Sup35p, an in vitro study. EMBO J. 2006;25:822–833. doi: 10.1038/sj.emboj.7600985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Inoue Y, Taguchi H, Kishimoto A, Yoshida M. Hsp104 binds to yeast Sup35 prion fiber but needs other factor(s) to sever it. J Biol Chem. 2004;279:52319–52323. doi: 10.1074/jbc.M408159200. [DOI] [PubMed] [Google Scholar]
- 103.Kawai-Noma S, Ayano S, Pack CG, Kinjo M, Yoshida M, Yasuda K, Taguchi H. Dynamics of yeast prion aggregates in single living cells. Genes Cells. 2006;11:1085–1096. doi: 10.1111/j.1365-2443.2006.01004.x. [DOI] [PubMed] [Google Scholar]
- 104.Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol. 2002;22:5593–5605. doi: 10.1128/MCB.22.15.5593-5605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu YX, Greene LE, Masison DC, Eisenberg E. Curing of yeast [PSI+] prion by guanidine inactivation of Hsp104 does not require cell division. Proc Natl Acad Sci USA. 2005;102:12789–12794. doi: 10.1073/pnas.0506384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cox B, Ness F, Tuite M. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics. 2003;165:23–33. doi: 10.1093/genetics/165.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mabbott NA, Bruce ME. The immunobiology of TSE diseases. J Gen Virol. 2001;82:2307–2318. doi: 10.1099/0022-1317-82-10-2307. [DOI] [PubMed] [Google Scholar]
- 109.Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 2001;21:7035–7046. doi: 10.1128/MCB.21.20.7035-7046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bruce M, Fraser H, McBride P, Scott J, Dickinson A. The basis of strain variation in scrapie. In: Prusiner S, Collinge J, Powell J, Anderton B, editors. Prion Diseases of Humans and Animals. New York: Ellis Horwood; 1992. pp. 497–508. [Google Scholar]
- 111.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 113.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bagriantsev S, Liebman SW. Specificity of prion assembly in vivo: [PSI+] and [PIN+] form separate structures in yeast. J Biol Chem. 2004;279:51042–51048. doi: 10.1074/jbc.M410611200. [DOI] [PubMed] [Google Scholar]
- 115.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, DeArmond SJ, Prusiner SB, Scott MR. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34:921–932. doi: 10.1016/s0896-6273(02)00726-2. [DOI] [PubMed] [Google Scholar]
- 119.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nature Medicine. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 120.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 121.Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell. 2004;14:139–145. doi: 10.1016/s1097-2765(04)00155-8. [DOI] [PubMed] [Google Scholar]
- 122.Diaz-Avalos R, King CY, Wall J, Simon M, Caspar DL. Strain-specific morphologies of yeast prion amyloid fibrils. Proc Natl Acad Sci USA. 2005;102:10165–10170. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 124.Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci USA. 2000;97:5836–5841. doi: 10.1073/pnas.110523897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol. 1994;68:4873–4878. doi: 10.1128/jvi.68.8.4873-4878.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA. 1997;94:10069–10074. doi: 10.1073/pnas.94.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 128.Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2-mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics. 1994;137:659–670. doi: 10.1093/genetics/137.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Young C, Cox B. Extrachromosomal elements in a super-suppression system of yeast. I. A nuclear gene controlling the inheritance of the extrachromosomal elements. Heredity. 1971;26:413–422. [Google Scholar]
- 130.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol. 2004;2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Masel J, Jansen VA. Designing drugs to stop the formation of prion aggregates and other amyloids. Biophysical Chemistry. 2000;88:47–59. doi: 10.1016/s0301-4622(00)00197-6. [DOI] [PubMed] [Google Scholar]
- 132.Kochneva-Pervukhova NV, Paushkin SV, Kushnirov VV, Cox BS, Tuite MF, Ter-Avanesyan MD. Mechanism of inhibition of Psi+ prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. EMBO J. 1998;17:5805–5810. doi: 10.1093/emboj/17.19.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN(+)] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 134.Westaway D, Mirenda CA, Foster D, Zebarjadian Y, Scott M, Torchia M, Yang SL, Serban H, DeArmond SJ, Ebeling C, Prusiner SB, Carlson GA. Paradoxical shortening of scrapie incubation times by expression of prion protein transgenes derived from long incubation period mice. Neuron. 1991;7:59–68. doi: 10.1016/0896-6273(91)90074-a. [DOI] [PubMed] [Google Scholar]
- 135.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vitrenko YA, Gracheva EO, Richmond JE, Liebman SW. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–1787. doi: 10.1074/jbc.M609269200. [DOI] [PubMed] [Google Scholar]
- 137.Kimura Y, Koitabashi S, Kakizuka A, Fujita T. The role of pre-existing aggregates in Hsp104-dependent polyglutamine aggregate formation and epigenetic change of yeast prions. Genes Cells. 2004;9:685–696. doi: 10.1111/j.1356-9597.2004.00759.x. [DOI] [PubMed] [Google Scholar]
- 138.Kimberlin RH, Walker CA. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology. 1985;23:74–81. doi: 10.1159/000149588. [DOI] [PubMed] [Google Scholar]
- 139.Bartz JC, Aiken JM, Bessen RA. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J Gen Virol. 2004;85:265–273. doi: 10.1099/vir.0.19394-0. [DOI] [PubMed] [Google Scholar]
- 140.Bradley ME, Liebman SW. Destabilizing interactions among [PSI(+)] and [PIN(+)] yeast prion variants. Genetics. 2003;165:1675–1685. doi: 10.1093/genetics/165.4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.King CY. Supporting the structural basis of prion strains: Induction and identification of [PSI] variants. J Mol Biol. 2001;307:1247–1260. doi: 10.1006/jmbi.2001.4542. [DOI] [PubMed] [Google Scholar]
- 142.Caughey B. Interactions between prion protein isoforms: The kiss of death? Trends Biochem Sci. 2001;26:235–242. doi: 10.1016/s0968-0004(01)01792-3. [DOI] [PubMed] [Google Scholar]