

Summary
The F-spondin family of extracellular matrix proteins has been implicated in axon outgrowth, fasciculation, and neuronal cell migration, as well as differentiation and proliferation of non-neuronal cells. In screens for mutants defective in C. elegans embryonic morphogenesis we identified SPON-1, the only C. elegans member of the spondin family. SPON-1 is synthesized in body muscles and localizes to integrin-containing structures on body muscles and to other basement membranes. SPON-1 maintains strong attachments of muscles to epidermis; in the absence of SPON-1, muscles progressively detach from the epidermis, causing defective epidermal elongation. In animals with reduced integrin function SPON-1 becomes dose-dependent, suggesting SPON-1 and integrins function in concert to promote attachment of muscles to the basement membrane. Although spon-1 mutants display largely normal neurite outgrowth, spon-1 synergizes with outgrowth defective mutants, revealing a cryptic role for SPON-1 in axon extension. In motor neurons SPON-1 acts in axon guidance and fasciculation, whereas in interneurons SPON-1 maintains process position. Our results show that a spondin maintains cell-matrix adhesion in multiple tissues.
Keywords: spondin, extracellular matrix, cell adhesion, morphogenesis, axon guidance
Introduction
The morphogenesis of epithelial tissues and organs is profoundly dependent on the extracellular matrix (ECM), especially the specialized forms of ECM known as basement membranes (BM) (Miner and Yurchenco, 2004). Active remodeling of BM by tissues is essential for many developmental events, and aberrant cell-ECM interactions underlie tumorigenesis and metastasis of epithelial tissues (Larsen et al., 2006). Developing axons often grow over BM substrates, and BM components play central roles both in tissue morphogenesis and in axon guidance (Hinck, 2004). Yet, rather than a passive scaffold for tissue morphogenesis, ECM can be regarded as an active participant in tissue morphogenesis and cell signaling (Nelson and Bissell, 2006).
C. elegans embryonic epidermal morphogenesis is an example of an organogenesis process that involves multiple interactions between an epithelial sheet, underlying muscle, and an intervening BM (Chisholm and Hardin, 2005). In late embryogenesis epidermal cells undergo coordinated shape changes that lead to embryo elongation, converting the ovoid embryo into a worm-shaped larva. Forces for elongation are generated within the epidermis by actomyosin-based contraction of circumferential actin bundles (Priess and Hirsh, 1986). Nevertheless epidermal elongation is also critically dependent on BM, indicating the importance of cell-cell interactions in coordinating the development of embryonic tissues.
Epidermal elongation beyond the two-fold stage requires underlying muscle (Williams and Waterston, 1994). Mutants defective in body muscle function are paralyzed and arrest at the two-fold stage of elongation, the Pat phenotype. The need for body muscle function in epidermal elongation may reflect a role for muscle in organizing the epidermal cytoskeleton. Body muscles lie underneath dorsal and ventral epidermis and adhere to adjacent epidermis via BM, which allows force transmission from muscle to epidermis. The muscle-epidermal BM itself also promotes organization of muscle and of the overlying epidermis. Importantly, different ECM components contribute to distinct aspects of epidermal morphogenesis. The earliest BM component to be deposited, laminin, is required for assembly of the myofilament lattice and localization of dense bodies. Animals completely lacking laminin arrest in early embryonic elongation with defective muscle morphogenesis (Huang et al., 2003a; Kao et al., 2006). The BM proteoglycan Perlecan/UNC-52 is essential for assembly of the myofilament lattice (Rogalski et al., 1993), whereas type IV collagen has a later role in muscle-epidermal attachment (Guo et al., 1991).
Other BM components such as nidogen/NID-1 or type XVIII collagen/CLE-1 are not required for embryonic morphogenesis, but play critical roles in axon outgrowth, guidance, and synaptogenesis (Ackley et al., 2001; Kang and Kramer, 2000). Similarly the laminin receptor dystroglycan/DGN-1 is not required in embryonic morphogenesis, but functions in axon guidance (Johnson et al., 2006). Integrin signaling is required for multiple aspects of neuronal development, including cell migration, axon fasciculation (Baum and Garriga, 1997) and guidance (Poinat et al., 2002). These findings underscore the role of BM as a central scaffold for the developing nervous system.
Spondins are a conserved family of ECM proteins originally identified as axon guidance factors in the vertebrate spinal cord (Klar et al., 1992). In vertebrates, spondins have context dependent effects on axon outgrowth and cell migration, and can promote neuronal differentiation (Schubert et al., 2006). Despite intensive analysis, the mechanisms by which spondins affect cell behavior as well as their in vivo roles remain poorly understood. Here we report that SPON-1, the sole C. elegans member of the spondin family, is essential for embryonic morphogenesis. We show that SPON-1 promotes muscle-epidermal adhesion and is required for completion of epidermal elongation. In the nervous system, SPON-1 promotes axon fasciculation and guidance, and continuously maintains neural architecture. These results are the first demonstration of an essential role for spondins in development.
Materials and Methods
Isolation of spon-1 mutations and molecular cloning
All strains were generated from Bristol N2, and grown under standard conditions (Brenner, 1974) unless stated. We used the mutations emb-9(g23ts cg46), pat-3(st564), ina-1(gm144), dig-1(ky188), unc-71(ju156), zig-4(gk34), and egl-15(n484). We used the GFP-expressing balancer mIn1 mIs14 (Edgley and Riddle, 2001) to balance spon-1 lethal alleles.
We isolated the mutations ju348, ju402, and ju430ts in a screen for EMS-induced mutants displaying defective elongation and lumpy epidermal morphology (Mei Ding, W.-M.W., and A.D.C, unpublished). Two F1 progeny of mutagenized parents were picked and placed onto a plate and their F2 broods scored for malformed L1 larvae. e2623 was isolated by Jonathan Hodgkin (personal communication). nc30 was isolated in screens for mispositioned ventral nerve cords (Shioi et al., 2001).
Genetic mapping placed spon-1 between dpy-10 and unc-4 on chromosome II. SNP mapping placed ju430 between pkP2148 and pkP2150, an interval of 420 kb. A 5.6 kb DNA fragment that contains ∼1.4 kb 5′ and ∼ 0.3 kb 3′ to the F10E7.4 coding sequence (pCZ695, Fig. 2A) rescued ju402 mutants to wild-type levels (data not shown). We sequenced a spon-1 full-length cDNA yk260h10 and confirmed the gene structure in Wormbase. The spon-1 coding sequence is spread over 12 exons and encodes a primary polypeptide of 819 amino acid residues. In strain constructions spon-1 mutations were followed using allele specific SNPs: e2623 creates a Taq I site and ju430 eliminates a BsrD I site.
Figure 2. spon-1 encodes a member of the F-spondin family.

(A) Genetic map position, gene organization, and minimal spon-1 rescuing DNA fragment (pCZ695). The nc30 mutation affects the last base pair of exon 3 (CAGgtaa to CAAgtaaa; codon 104), likely forcing use of an alternative cryptic splice site, resulting in a frameshift and a premature stop in the reelin domain. (B) Domain organization of SPON-1, and percent identities to Drosophila fat-spondin and rat F-spondin. (C) Phylogenetic tree of spondin domains of C. elegans SPON-1, Drosophila fat-spondin (CG6953), CG17739, CG30203, and CG30046, rat (Rn) F-spondin, zebrafish (Br) spondin 1a and 1b (F-spondin 1 and 2), Xenopus (Xl) spon-1A, and Amphioxus Amphi-F-spondin (Bf, Branchiostoma floridae), made with ClustalW and displayed using njplot (Perrière and Gouy, 1996). (D) Alignment of the thrombospondin type I repeats from SPON-1, showing missense alterations in red.
Analyses of embryonic morphogenesis
4-D microscopy was performed as described (Chin-Sang et al., 1999). To record the embryogenesis of ju430 at 25°C, the objective was enclosed in a copper solenoid infused with temperature controlled water to keep embryos at the desired temperature. At least ten embryos were recorded for each genotype. Statistical tests used GraphPad Prism (Carlsbad, CA).
To determine the temperature sensitive period of ju430, we cut open gravid adult hermaphrodites grown at 15°C and transferred 2- and 4- cell stage embryos to plates pre-equilibriated to 15°C. We shifted plates to 25°C at appropriate times and scored lethality after 24 and 48 hours. 10-15 embryos were recorded for each time point.
Analysis of axon guidance and muscle attachment
We analyzed D type motorneurons using the Punc-25-GFP marker juIs76 (Huang et al., 2002) or the DD marker Pflp-13-GFP ynIs37 (Kim and Li, 2004), PVQs using the Psra-6-GFP marker oyIs14 (Sarafi-Reinach et al., 2001), and the muscle marker in trIs10 (Dixon and Roy, 2005).
Electron microscopy
EM was performed as described (Woo et al., 2004). For analysis of muscle attachment we analyzed four ju430 animals (raised at 22.5°) and four e2623 animals; we cut serial sections from the posterior of the pharynx to the anterior end of the gonad. For analysis of PVQ crossing we sectioned two ju430 oyIs14 animals.
Generation of spon-1 reporter genes
We used duplex PCR to generate Pspon-1-GFP reporters, using 3.3 kb of 5′ DNA sequence (primer sequences available on request). This DNA was injected into wild-type animals at ∼20 ng/μ1 with pRF4, generating juEx592 and juEx593. To make the SPON-1∷GFP C-terminal fusion pCZ697, a 5 kb BamH I-Stu I fragment of pCZ695 was subcloned into pPD95.75 (Fire lab vector kit), such that GFP was fused in-frame at residue 756 of SPON-1, truncating the protein after TSR4. pCZ697 was injected into spon-1(ju402)/mIn1 mIs14 animals at 1 ng/μl with pRF4 as marker. All 17 transgenic lines failed to rescue ju402. An outcrossed version of transgenic line juEx734 was used in immunostaining experiments.
To tag SPON-1 at the N-terminus with Venus YFP we used a modular strategy (Hudson et al., 2006). We injected SPON-1∷GFP at 10 ng/μl with Pttx-3-RFP (50 ng/μl) into spon-1(ju402)/mIn1mIs14 hermaphrodites. Rescued ju402 homozygotes were selected based on absence of mIs14, yielding transgenic lines juEx1111 and juEx1112. To express SPON-1 in pharyngeal muscles we used the myo-2 promoter from pPD118.3. Pmyo-2-GFP∷SPON-1 was injected into spon-1(ju402)/mIn1mIs14 animals at 50 ng/μl with Pttx-3-RFP. Viable ju402 homozygous transformants were selected, yielding lines juEx1302-1304.
Antibody generation and immunostaining
We raised antibodies against peptides corresponding to residues 499-547 in TSR2 and purified the antisera according to standard procedures. The antisera recognize recombinant SPON-1 C-terminal proteins in bacterial lysates (not shown). Whole-mount immunofluorescence was as described (Finney and Ruvkun, 1990), except that fixation was extended to 3.5-4 hours, an optimization for ECM antigens (J.R. Crew and J.M. Kramer, personal communication). Monoclonal supernatants MH3 (anti-Perlecan), and MH25 (anti-αPAT-2 integrin) (Developmental Studies Hybridoma Bank, University of Iowa) were used at 1:200 dilution; DM5-6 (anti-MHC A) (Miller et al., 1983) was diluted 1:200. Phalloidin staining was as described (Ding et al., 2003). To detect GFP we used monoclonal 3E6 (Invitrogen, Carlsbad, CA) or a rabbit polyclonal (A11122) at 1:500 to 1:1000 dilution. Raw or purified anti-SPON-1 antisera were diluted at 1:100 in embryo staining and 1:800 in mixed stage staining, unless stated. For anti-SPON-1 immunostaining we used pCZ695 arrays juEx696 and juEx698, both in ju402 background. We acquired images on Zeiss Axioplan 2 or LSM 510 confocal microscopes.
Results
Mutations in spon-1 disrupt morphogenesis and affect a C. elegans spondin
To identify genes required for muscle-epidermal interactions we screened for mutations causing late arrest in elongation and muscle detachment. From a screen of approximately 8,000 haploid genomes we identified three mutations (ju348, ju402, ju430ts) that cause highly penetrant defects in epidermal elongation and muscle attachment. We found that these mutations were allelic to vab-13(e2623), found in screens for morphogenetic mutants (J.A. Hodgkin, personal communication), and ven-3(nc30), isolated in a screen for mutations defective in fasciculation of the ventral nerve cord (Shioi et al., 2001). All five mutations affect the same gene, here renamed spon-1. e2623 mutants are slightly dumpy, egg-laying defective (Egl) and display variably abnormal (Vab) epidermal morphology that is more pronounced in early larvae (Figure 1B, C). Other spon-1 alleles cause fully penetrant embryonic or early larval lethality with aberrant epidermal morphology and detached body muscles (Table 1; Fig. 1E, F). ju348 and ju402 are nonconditional lethals, and ju430 is a temperature sensitive allele that resembles e2623 at 15°C and is inviable at 25°C. In penetrance the five alleles rank from strongest to weakest: ju402 > nc30 > ju348 > ju430ts > e2623. In addition to the epidermal elongation, muscle attachment, and axon guidance defects described here, spon-1 mutations also disrupt development of the excretory canal and of epidermal seam cells (Shioi et al., 2001).
Figure 1. spon-1 mutants are defective in epidermal morphogenesis and muscle attachment.
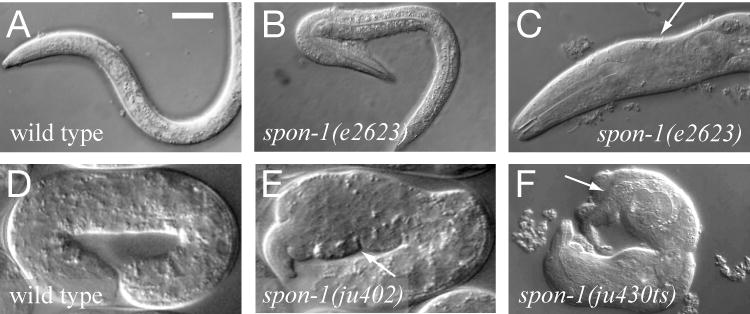
(A) The wild-type L1 larva has a smooth epidermal shape. (B, C) spon-1(e2623) L1 larvae and adults display variable defects in epidermal morphology. The bent body phenotype is more obvious during early larval stages (B) while some adults display ‘pinched’ head morphology (arrow, C); other e2623 phenotypes include egg laying defects and slightly shorter body length. (D) A wild-type embryo at three-fold stage. (E) spon-1(ju402) mutant embryo, displaying elongation arrest and uneven epidermal morphology (arrow). (F) spon-1(ju430) mutant hatchling showing elongation arrest with lumpy two-fold morphology and detached body wall muscles (F, arrow). Scale, 10 μm.
Table 1. Penetrance of spon-1 morphogenetic phenotypes.
| Allele, temperature | n | Embryonic lethality | Larval lethality | Total lethality | Vab adults | non-Vab adults |
|---|---|---|---|---|---|---|
| e2623, 15°C | 1116 a | 3.3% | 1.1% | 4.4% | 10.8% | 84.9% |
| ju430, 15°C | 721 a | 18.6% | 8.5% | 27.1% | 72.5% | 0.4% |
| ju430, 25°C | 148 b | 51.3% | 39.9% | 91.2% | 8.8% | 0% |
| nc30, 22.5°C | 635 c | 44% | 66% | 100% | (na) | (na) |
| ju348, 20°C | 155 c | 21.2% | 75.4% | 96.6% | 3.4% | 0% |
| ju402, 15°C | 285 c | 66.3% | 33.7% | 100% | (na) | (na) |
| Pmyo-2-vGFP∷SPON-1d, 20°C | 789 a | 0.4% | 0.2% | 0.6% | 0% | 99.4% |
| spon-1(ju402); Pmyo-2-vGFP∷SPON-1, 20°C | 454 a | 1.7% | 6.6% | 8.3% | 91.7% | 0% |
determined from complete broods of homozygous strains.
progeny of ju430 homozygotes grown at 15°C and shifted to 25°C as young adults.
progeny of heterozygous balanced strains. spon-1 mutants were identified as non-GFP-expressing animals segregating from spon-1/mIn1 mIs14. na, not applicable.
Transgene juEx1302.
By mapping and transformation rescue we identified the gene affected by these mutations as F10E7.4 (Fig. 2A), which encodes a member of the spondin family of ECM proteins. Spondins are defined by the spondin domain, of unknown function, and are classified into two subfamilies, the F-spondins and Mindins (Feinstein and Klar, 2004). F-spondins also contain an N-terminal reelin domain and up to six thrombospondin type I repeats (TSRs), whereas Mindins lack a reelin domain. C. elegans SPON-1 most resembles vertebrate F-spondins (Fig. 2B). Like some Drosophila and Anopheles F-spondins SPON-1 contains a Kunitz serine protease inhibitor domain between TSRs 3 and 4. In domain organization SPON-1 most resembles the Drosophila gene CG17739; however the SPON-1 spondin domain is more similar to that of Drosophila fat-spondin (49% identity, Fig. 2C).
We identified the DNA lesions of spon-1 alleles. The ju402 mutation results in a premature stop codon in the spondin domain and is a candidate null mutation. nc30 is alters the last base of exon 3 (Fig. 2A); this does not affect coding potential and may cause a strong loss of function by altering usage of the intron 3 splice donor. The ju348, e2623, and ju430 lesions cause missense alterations in TSRs 2, 3, and 4 respectively (Fig. 2D), suggesting that these TSRs are critical for SPON-1 function.
SPON-1 is synthesized in body wall muscles and localizes to specific basement membranes
To understand how SPON-1 functions we first assayed the activity of the spon-1 promoter. Pspon-1-GFP reporters were expressed exclusively in body wall muscles from early elongation onwards (Fig. 3A-C). We constructed translational GFP fusions in which SPON-1 was tagged with GFP either at the N-terminus following the signal sequence, or after the 4th TSR (see Materials and Methods). N-terminal SPON-1∷GFP transgenes rescued all phenotypes of spon-1(ju402); C-terminal GFP fusions showed similar localization but did not rescue spon-1 mutants, suggesting that C-terminal TSRs are required for SPON-1's function in morphogenesis. Rescuing SPON-1∷GFP was expressed by muscles from the comma stage onwards and showed localization to embryonic BM (Fig. 3D). In larvae and adults SPON-1∷GFP localized to dense bodies and M lines on muscle surfaces (Fig. 3E,F), as determined by colocalization with the dense body component vinculin (Barstead and Waterston, 1989). Dense bodies and M lines are sites of integrin based adhesions (Francis and Waterston, 1991) and are also enriched for the BM components UNC-52/Perlecan (Rogalski et al., 1993) and EPI-1/αB laminin (Huang et al., 2003a). We consistently detected SPON-1∷GFP on BM surrounding the pharynx (not shown), and within coelomocytes (Fig 3H). As coelomocytes endocytose extracellular proteins (Fares and Greenwald, 2001) we infer that SPON-1∷GFP fusions are secreted. As the spon-1 promoter is not active in the pharynx, SPON-1 can move from sites of synthesis to sites of localization, like type IV collagen (Graham et al., 1997). Unlike type IV collagen we did not detect SPON-1∷GFP at intestinal or gonadal BM. We also generated antibodies that detect SPON-1 in whole mount immunostaining of animals overexpressing SPON-1, but not in wild-type animals. The pattern of anti-SPON-1 staining in overexpressing animals was similar to that of SPON-1∷GFP, including expression in embryonic and larval muscles (Fig. 3J,K), the excretory canal (Fig. 3K, arrowhead), pharyngeal BM (Fig. 3G) and coelomocytes (not shown). We conclude that SPON-1 is secreted from muscle and incorporates into some but not all BM, and that it is enriched at integrin-based adhesion sites.
Figure 3. SPON-1 is synthesized in muscle and localizes to basement membranes.
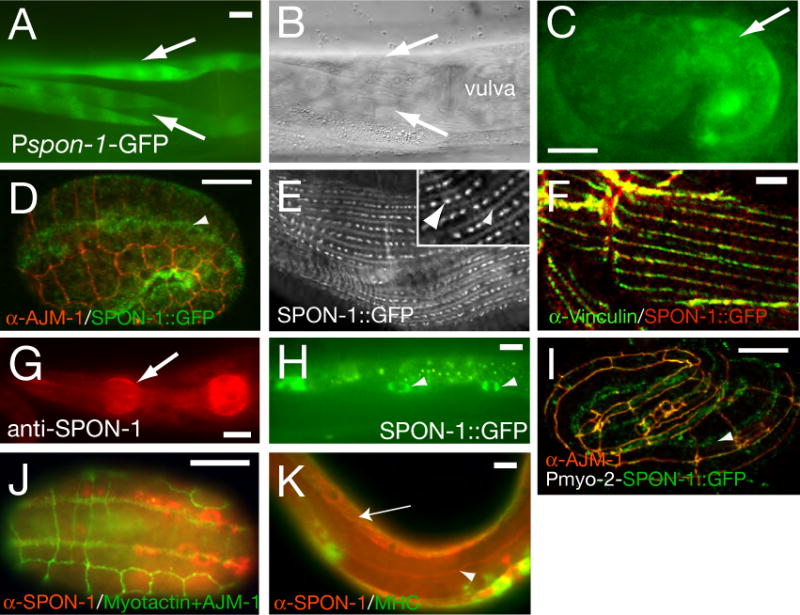
(A) Pspon-1-GFP (juEx593) is expressed in body wall muscles (arrows, two muscle quadrants), ventral view of adult midbody; DIC optics (B) showing the muscle quadrants (arrows) on ventral side near the vulva. (C) Embryonic expression of Pspon-1-GFP from the comma stage onwards. (D-F,H) SPON-1∷GFP (genotype ju402; juEx1111 (D,E) and juEx734 (F-H) localization, anti-GFP immunostaining. (D) In 1.5-fold stage embryos SPON-1∷GFP is localized to muscle quadrants (E) SPON-1∷GFP localizes at muscle dense bodies (inset, large arrowhead) and at M lines (inset, small arrowhead). (F) Colocalization of SPON-1∷GFP (anti-GFP) and the dense body component vinculin (MH24 immunostaining) in adult muscles. SPON-1 was detected on the pharynx surface (anti-SPON-1 staining of overexpressing lines, arrow, G) and within coelomocytes (SPON-1∷GFP, arrowheads, H). (I) Functional SPON-1∷GFP expressed under the control of the pharynx-specific myo-2 gene (juEx1302) localizes to muscle basement membranes (arrowheads) in 3-fold stage embryos (anti-GFP immunostaining). (J,K) Immunostaining with anti-SPON-1 antibodies of animals of genotype spon-1(ju402); juEx698[spon-1(+)]. (J) Dorsal view of a 1.5-fold stage embryo stained with anti-SPON-1 (red), and anti-Myotactin (MH46) and anti-AJM-1 (MH27) (both green) mark muscle-epidermal attachments and epidermal adherens junctions respectively. SPON-1 is in muscle cells, beneath Myotactin staining. (K) Larva stained with anti-SPON-1 (red) and anti-MHC (green) to show body muscles. SPON-1 expression was detected in or adjacent to body muscles (arrowhead) and the excretory canal (arrow). Scales, 10 μm.
The accumulation of SPON-1 on pharyngeal basement membrane suggested that SPON-1 is recruited to BM distant from the cells that synthesize it. To test whether SPON-1 can function distant from its sites of synthesis we expressed SPON-1∷GFP in pharyngeal muscles using the myo-2 promoter (Okkema et al., 1993). Such transgenes rescued the epidermal elongation and lethality of spon-1(ju402) null mutants (Table 1). Pharyngeally expressed SPON-1∷GFP specifically accumulated at muscle-epidermal basement membranes during late embryogenesis (Fig. 3I). These results show that SPON-1 can be produced by cells that do not normally express it, and that it translocates within the ECM to function in specific basement membranes.
spon-1 mutants display late-onset defects in epidermal elongation and muscle attachment
To determine when spon-1 functions in epidermal morphogenesis we performed timelapse microscopy on spon-1 embryos. Most (18/19) spon-1(ju402) mutants developed normally past the two-fold stage of elongation, then either elongated to the three-fold stage and retracted or did not reach a three-fold stage. spon-1 embryos displayed muscle movements before arrest (Fig. 4E-H; see Movie 1 in supplementary material): they twitched at 1.75-fold (∼430 minutes) and kept twitching to three-fold stage. However only 3/18 embryos displayed vigorous rolling movements similar to those of the wild type. Muscle movements slowed down between the two- and three-fold stages, and then stopped (paralysis). During this period of slower muscle twitching the epidermis retracted and became uneven in shape (Fig. 4G, H). ju348 and ju430 mutations caused similar or slightly weaker elongation phenotypes than did ju402. Thus, although many spon-1 mutants had a body length comparable to that of a two- or 2.5-fold stage embryo, they usually elongate beyond this stage and then retract. Compared to other extracellular matrix mutants, spon-1 most resembles mutants such as emb-9, which lack type IV collagen.
Figure 4. SPON-1 is required for late elongation and interacts with INA-1 α-integrin.
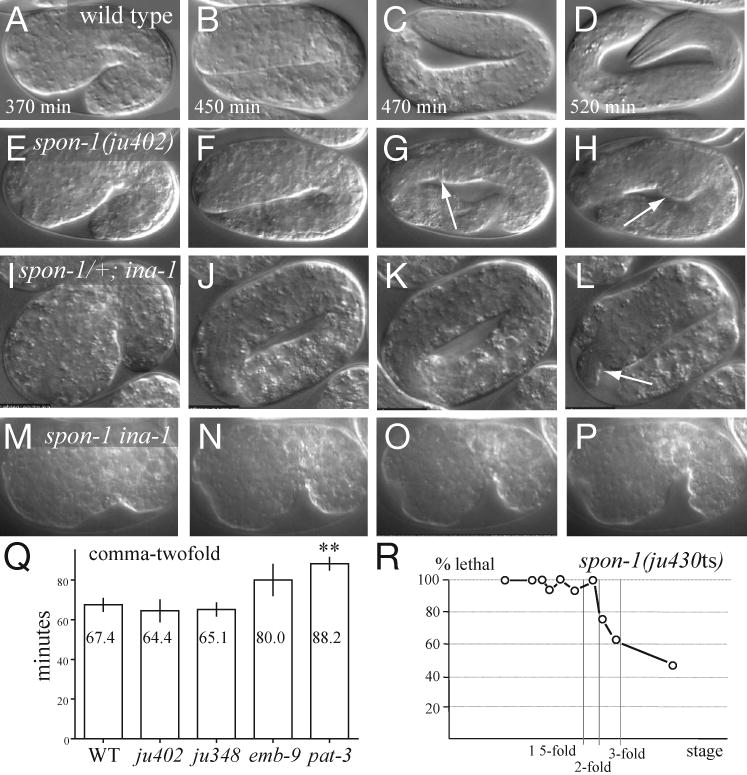
(A-P) Frames from time-lapse analysis of wild type (A-D), spon-1(ju402) mutants (E-H), putative ina-1(gm144); spon-1(ju430)/+ mutants (I-L) and spon-1 ina-1 double mutants (M-P). Most spon-1(ju402) embryos developed normally up to either two- or three-fold stage (F, G), but then retracted to 2.5-fold (H) by the time the wild-type embryo has developed to the three-fold stage. spon-1(ju402) embryos displayed normal muscle twitching movements from 1.75- to three-fold stage before elongation arrest (see Movie 1 in supplementary material). Body constrictions became apparent when muscle movements slowed down and stopped later. At the restrictive temperature spon-1(ju430ts) mutants resemble ju402 in phenotype. More spon-1(ju402) embryos retracted to a two-fold stage (12/19, 63%) than did spon-1(ju430ts) embryos (7/33, 21%). 63% of spon-1(ju402) and 79% of spon-1(ju430ts) embryos reached the three-fold stage before retraction. spon-1(ju348) embryos were indistinguishable from ju402 in embryonic phenotype. ina-1(gm144) mutants displayed normal elongation (not shown). From strains of genotype ina-1(gm144); spon-1(ju430)/mIn1 mIs14 mutants, 41% (18/44) GFP-positive embryos (either ina-1; spon-1/mIn1 mIs14 or ina-1; mIn1 mIs14) displayed late-onset elongation defects. 0/11 ina-1 mIn1 mIs14 embryos showed elongation defects. We infer that ∼60% of spon-1/+ heterozygotes have elongation defects in the gm144 background. 6/7 ju430; gm144 homozygotes (GFP-negative embryos) displayed 1.5-fold arrest (panels M-P). (Q) Early elongation rates (mean ± s.e.m.). spon-1 mutants elongate normally from comma to two-fold stage; emb-9 mutants were slightly slower; pat-3 mutants are significantly slower (P < 0.05, ANOVA). (R) The spon-1(ju430ts) temperature sensitive period begins at the two-fold stage.
To address when spon-1 acts in epidermal elongation, we determined the temperature sensitive period for spon-1(ju430) (Fig. 4R). When spon-1(ju430) embryos grown at the permissive temperature (15°C) were shifted to the restrictive temperature (25°C) at the 1.5-fold stage, 92-100% arrested as embryos or early larvae. When these embryos were shifted up after the 2-fold stage, lethality progressively dropped to levels observed in animals raised at the permissive temperature. We infer that SPON-1 acts directly in elongation after the two-fold stage.
Although SPON-1 does not appear to be essential for the early integrin-dependent assembly of the myofilament lattice, the localization of SPON-1 to integrin attachment sites suggested SPON-1 might have a subtle or redundant role in integrin-mediated adhesion. We therefore used double mutant analysis to test whether spon-1 interacts with integrin signaling. As animals lacking PAT-2 or PAT-3 arrest at the two-fold stage, we focused on the α-integrin INA-1, which is not essential for embryonic elongation yet colocalizes with PAT-3 in embryonic basement membranes (Baum and Garriga, 1997). Animals carrying the hypomorphic mutation ina-1(gm144) display normal epidermal elongation (not shown). spon-1(ju430) ina-1(gm144) double mutants were completely inviable (in contrast to either single mutant at 20°C) and arrested in early elongation (Fig 4M-P), suggesting SPON-1 and INA-1 may act redundantly in early elongation. Strikingly, spon-1/+ ina-1 animals resembled spon-1 homozygotes in that they showed a late elongation arrest (Fig 4I-L). These results suggest that when integrin function is reduced, SPON-1 becomes dose-dependent for embryonic elongation.
SPON-1 maintains muscle attachment during later elongation and larval development
The phenotype of spon-1 mutants suggests that, unlike integrins or perlecan/UNC-52, SPON-1 was not essential for initial assembly of the myofilament lattice. Similarly, the late onset of epidermal elongation defects in spon-1 implies that it is not essential for assembly of trans-epidermal attachments, as these are required for early elongation (Bosher et al., 2003; Woo et al., 2004). Instead, the late block in elongation appears to be a result of muscle detachment, which in turn leads to failure to maintain epidermal cell shape. We examined epidermal and muscle morphology using the adherens junction protein AJM-1 to mark epidermal cell boundaries and body muscle myosin heavy chain (MHC) to mark muscle cells (Miller et al., 1983). MHC staining was normal in early elongation (Fig. 5A,B), but as elongation progressed we observed frequent gaps along muscle quadrants (Fig. 5C,D) due to muscle detachment. MHC organization in the remaining muscle was essentially normal. Muscle-associated PAT-3/β-integrin and UNC-52/perlecan likewise became fragmented after the twofold stage due to muscle detachment (Fig. 5E-H). Epidermal actin bundles became misoriented following elongation arrest in regions where muscles had detached (Fig. 5I,J). Epidermal attachment structure components, including intermediate filaments and Myotactin, appeared normal before elongation arrest, but became discontinuous in arrested embryos (Fig. K, L). These results confirm that SPON-1 is not essential for early muscle or epidermal organization, but becomes essential in later muscle-BM-epidermal adhesion.
Figure 5. SPON-1 maintains muscle-epidermal attachment and muscle organization.
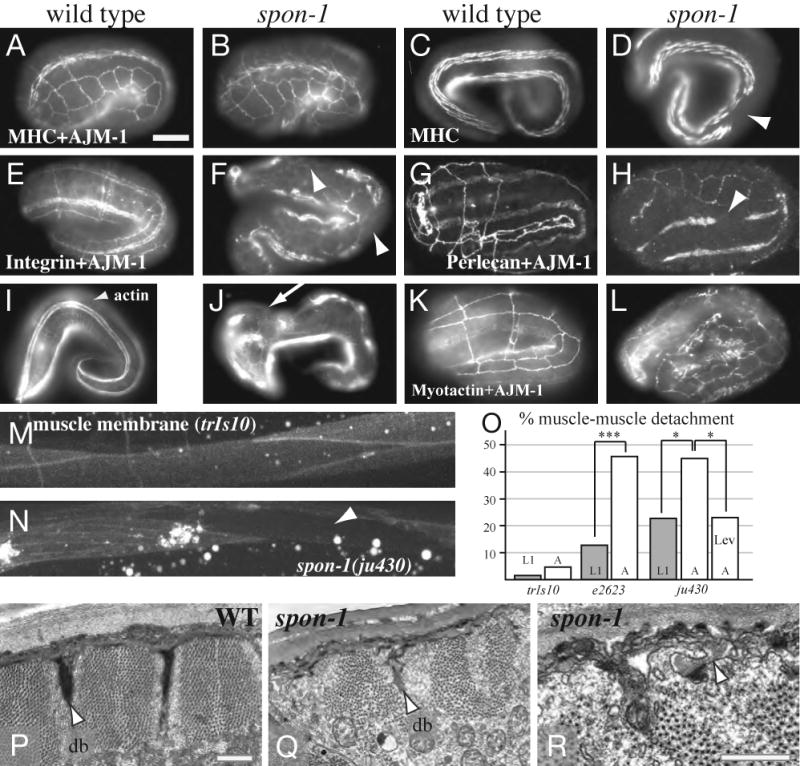
(A-D) Muscle quadrants in two-fold (A, B) and three-fold (C, D) embryos; anti-MHC-A staining counterstained with anti-AJM-1 in A and B. 3-fold stage spon-1(ju402) mutants display gaps in MHC staining due to muscle detachment (D). (E, F) Anti-PAT-3/β-integrin (MH25) staining in wild-type and in arrested spon-1(ju402) embryos. (G,H) Perlecan (MH3) in wild-type and spon-1(ju402) embryos. Epidermal cell outlines are shown by anti-AJM-1 staining. Similar to MHC-A, both β-integrin and perlecan staining were discontinuous due to muscle detachment (arrowhead in F,H), but were normal in other regions; the gaps were variable in size and position from embryo to embryo. (I, J) Epidermal circumferential F-actin bundles (CFBs), detected by phalloidin labeling of in wild-type and arrested spon-1(ju402) embryos, respectively. In the wild type, CFBs in lateral epidermal cells were circumferentially oriented (I, arrowhead). In arrested spon-1(ju402) embryos, CFBs in lateral epidermal cells were misoriented (J, arrow), becoming perpendicular to the circumference. (K, L) Myotactin patterns (MH46 antibody staining) in the wild type (K) and in arrested spon-1(ju402) embryos (L). Epidermal cell boundaries are outlined by anti-AJM-1 staining for adherens junctions. Myotactin patterns are discontinuous along the body; however, normal circumferential double band patterns were observed in the remaining Myotactin. Scale (A-L), 10 μm. (M, N) Post-embryonic muscle surfaces visualized using trIs10; note muscle detachment in ju430 (arrowhead, N). (O) Percent animals displaying muscle-muscle detachment; n > 40 for each condition; Fisher exact test. (P-R) Ultrastructure of larval body wall muscle and basement membrane (P) wild-type body muscle, showing dense bodies (db) and regular myofilament lattice; cuticle is at top. (Q) muscle in spon-1(ju430) larvae grown at 22.5°C; note absence of dense bodies (db), disorganized myofilament lattice and invagination of basal lamina (arrowhead). (R) higher magnification of basal lamina invagination into muscle (arrowhead). Scale (P-R), 500 nm.
To test whether SPON-1 was required in later muscle adhesion we analyzed post-embryonic muscle anatomy using GFP markers and electron microscopy. In addition to defective muscle-epidermal attachment, spon-1 mutants also displayed defects in muscle-muscle adhesion within a muscle quadrant (Fig. 5M, N). Muscle-muscle adhesion became progressively more defective during larval development and was suppressed by levamisole paralysis, indicating that spon-1 mutant muscles are pulled apart by the force of muscle contraction (Fig. 5O). Muscle ultrastructure in spon-1 mutant larvae was disorganized; in particular, BM between muscle and epidermis was thickened and invaginated into the muscle (Fig. 5P-R). We conclude that SPON-1 acts continuously throughout development to promote strong adhesion both at muscle-muscle and at muscle-BM interfaces.
SPON-1 promotes axon fasciculation and guidance, and acts in parallel to ADAM UNC-71 in outgrowth
F-spondin has context-dependent roles in axonal outgrowth and fasciculation in vertebrates (Burstyn-Cohen et al., 1998; Tzarfati-Majar et al., 2001). We therefore examined specific axon morphologies to determine the roles of SPON-1 in C. elegans nervous system architecture. As spon-1 null mutants arrest in embryogenesis we examined animals mutant for the weak allele e2623 or the conditional allele ju430. spon-1 mutants displayed widespread defects in axon guidance and fasciculation of motor neurons and PVQ interneurons, described below, as well as defects in migration, fasciculation, and guidance of mechanosensory neurons (not shown).
spon-1 mutants displayed extensive defasciculation of motor neuron processes within the ventral cord (Fig. 6A,B; Table 2). Motor neuron commissures also displayed defects in left-right choice of outgrowth (Table 2) and often deflected laterally upon reaching the subdorsal muscle quadrant, before eventually reaching the dorsal cord (Table 2A; Fig. 6C-E). To determine if such guidance defects might be a secondary effect of muscle detachment we examined motor commissure and muscle morphology simultaneously using the trIs10 marker. Of 20 commissures with dorsoventral guidance defects, only 5 were in regions of muscle detachment (Fig. 6F); conversely in regions of muscle detachment we frequently saw normal commissural guidance (Fig. 6G). We conclude that dorsoventral guidance defects appear to arise independently of muscle detachment.
Figure 6. SPON-1 is required for motor neuron fasciculation and dorsoventral guidance.
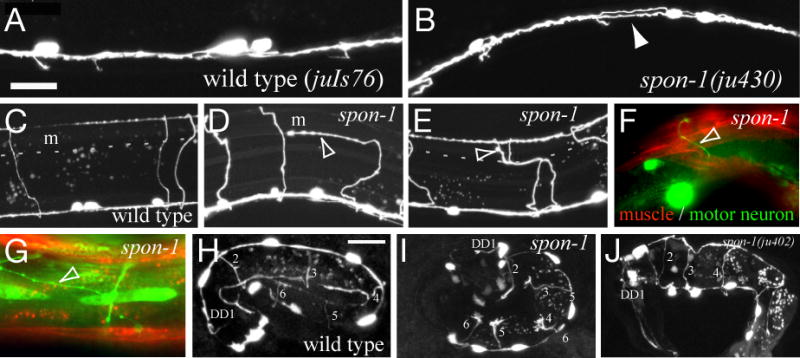
(A-D) Motor neuron processes (juIs76) in wild type (A,C) and spon-1(ju430) mutant L4s (B,D). (B) defasciculation of D neuron ventral processes in ju430 mutants (arrowhead). (C) motor neuron commissures in the wild type extend from ventral to dorsal on the right side. (D,E) Motor commissures in spon-1(ju430) mutants turn laterally (arrowhead) upon contacting the dorsal muscle quadrant (m, dotted line) and either extend and terminate subdorsally (D) or turn back to the dorsal midline (arrowhead, E). (F,G) Commissural guidance defects occur independently of muscle detachment (trIs10); commissure guidance can be normal (arrow) in regions of body wall muscle detachment (F) and can be aberrant in regions of normal attachment (G). (H-J) D neuron outgrowth in wild type (juIs76) (H), in ju430 juIs76 embryo (I, progeny of animal shifted to 25°C as L4) and in spon-1(ju402) arrested hatchling (J, ynIs37 marker); all six D neuron commissures (DD1-6) extend dorsally. Scale, 10 μm.
Table 2. Motor neuron fasciculation and guidance defects in spon-1 mutants.
| (A) spon-1 single mutant defects | ||||
|---|---|---|---|---|
| Genotype, temp | n | Ventral cord defasciculationa | Commissure handedness defect | Dorsoventral guidance defect |
| N2 20° | 43 | 0% | 2.7% (0.2%) | 0% |
| ju430ts 15° | 42 | 40.5% (6.5%) ** | 30.9% (2.9%) ** | 9.5% (0.5%) ns |
| ju430ts 20° | 41 | 31.7% (4.9%) ** | 56.1% (8.7%) ** | 29.3% (2.4%) ** |
| ju430ts 22.5° | 40 | 17.5% (2.5%) * | 87.5% (15%) ** | 40% (4.1%) ** |
| ju430ts 15°-25° | 37 | 16.2% (4.1%) ** | 43.2% (4.7%) ** | 2.7% (0.2%) ns |
| upshift | ||||
| (B) Interaction between spon-1 and unc-71 | ||||
|
| ||||
| Genotype | n | Missing 1 or more commissures | Commissure handedness defect | Dorsoventral guidance defect |
|
| ||||
| N2 | 61 | 0% | 0% (0.2%) | 0% |
| unc-71(ju156) | 45 | 73% | 24% (5%) | 0% (0%) |
| ju430ts | 45 | 0% | 11% (3%) | 0% (0%) |
| ju430ts; ju156 | 48 | 100% *** | 44% (10%) | 44% (9%) ** |
D morphology was scored using the Punc-25-GFP (juIs76) marker in young adults (A), or in L1 larvae (B); only commissures of DD2-6 were scored in L1s. All defects are scored per animal (n) and per neuron/commissure (or per ventral cord segment for defasciculation). Data in part B are all at 20°C. Tests of significance use the Fisher exact test;
P < 0.05,
P < 0.01,
P < 0.001.
‘Ventral cord defasciculation’: The ventral nerve cord was divided into 10 sections between D neuron cell bodies. An animal is scored as defasciculated if 2 or more segments are defasciculated.
‘Commissure handedness defect’ is outgrowth of a commissure on the left side. The two anterior commissures (DD1/VD2) of the left side and most anterior commissure on the right side (VD3) were not scored.
‘Dorsoventral guidance defect’ is % animals with >1 commissure that fails to reach dorsal cord. About 8% of D neurons did not extend commissures from the ventral cord. We observed similar defects in A and B type motor neurons (not shown).
spon-1 mutants raised at semipermissive temperatures had a normal number of D neuron commissures, suggesting SPON-1 is not essential for outgrowth of commissures (Table 2). To test if further reduction in SPON-1 function could affect outgrowth, we examined the progeny of ju430ts animals shifted to the restrictive temperature as L4s, and found that 100% (60/60) of DD commissures reached the dorsal midline, as in the wild type (Fig. 6I-J). spon-1(ju402) embryos also displayed a normal number of DD commissures (Fig. 6H). Although motor neuron outgrowth appeared normal in spon-1 single mutants, spon-1 mutations significantly enhanced outgrowth defects of unc-71 ADAM mutants (Huang et al., 2003b) (Table 2B). We infer that SPON-1 has a minor role in outgrowth that is masked by redundancy with other pathways such as UNC-71.
SPON-1 continuously maintains axon positions at the ventral midline
F-spondin regulates crossing of midline axons at the floor plate of the vertebrate spinal cord (Burstyn-Cohen et al., 1999). To learn whether SPON-1 is involved in axon behavior at the C. elegans ventral midline, we examined the PVQ neurons, which undergo regulated midline crossing in the wild type. PVQ neurons in spon-1 mutants frequently displayed inappropriate crossing of the ventral midline (Fig. 7A,B). These defects became more severe during larval development, suggesting SPON-1 actively maintains process positions. PVQ midline crossing errors were increased in ju430ts animals shifted from 15°C to 25°C in the L1 stage compared to unshifted controls (Fig. 7C), indicating that SPON-1 functions in larval development to prevent midline crossing. This post-embryonic increase in midline crossing errors was also suppressed by levamisole, indicating that crossing errors arise due to movement of the animal. Such post-embryonic ‘flip-overs’ have been distinguished from crossing over during initial axon outgrowth, and imply a defect in maintenance of process positions (Hobert and Bulow, 2003). For PVQ axons to ‘flip-over’ they must traverse the ventral hypodermal ridge. To determine if PVQ flip-overs might be secondary to an epidermal defect we performed correlative light and electron microscopy of PVQ flip-over regions, and found that the overall structure of the ventral nerve cords and hypodermal ridge was normal (Fig. 7F-J). We infer that PVQ flip-overs are a direct result of reduced SPON-1 function in the microenvironment of the ventral cord.
Figure 7. SPON-1 maintains process position at the ventral midline.
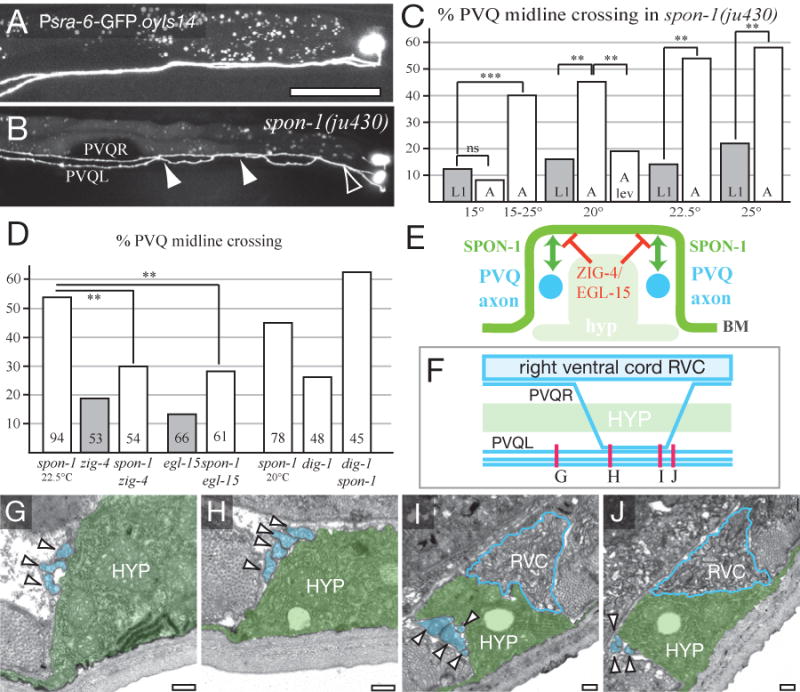
(A) PVQ axons in wild type (oyIs14). (B) In spon-1(ju430) PVQL crosses over to the right-hand VNC (open arrowhead) as in the wild type, and undergoes two ectopic crossovers (filled arrowheads). (C) Midline crossing is more penetrant in spon-1 adults than in L1s, is enhanced by shifting L1s from 15 to 25°C, and suppressed by growth in 1 mM levamisole (see Table S1). (D) Suppression of spon-1(ju430) midline crossing by zig-4 and egl-15 (at 22.5°C; number of animals shown in column) and enhancement by dig-1 (at 20°C). (E) Model for the interaction of SPON-1 and ZIG-4/EGL-15A pathways; cross-section of ventral nerve cord. SPON-1 in the BM (green) mediates adhesion of PVQ axons (blue) to their normal environment; ZIG-4 and EGL-15 (red) locally inhibit axon-BM adhesion at the midline. (F-J) spon-1(ju430ts) oyIs14 animals were shifted from 15°C to 25°C in the L1 stage and PVQ scored in young adults. The adults were sectioned in the region of PVQ crossing over posterior to the vulva; approximate levels of sections are shown in F. (G) Anterior to PVQR crossover (0 μm) the left-hand ventral nerve cord contains its normal three processes, PVQL, PVPR and AVKR (arrowheads and colored blue in G-J). (H,I) Between 90 and 150 μm a fourth process, presumably PVQR, has crossed over and fasciculated with the left hand bundle. (J) At ∼160 μm posterior the left hand bundle now again contains three processes. The ventral hypodermal ridge (HYP, green) and the right-hand ventral nerve cord (RVC, blue outline) appear normal in both crossover regions. Similar results were obtained for a second animal (not shown). Scale, 0.2 μm (G-J).
Several other pathways act to maintain PVQ axon positions, including the secreted Ig domain protein ZIG-4 (Aurelio et al., 2002), the FGF receptor EGL-15 (Bulow et al., 2004), and the giant ECM protein DIG-1 (Benard et al., 2006). To investigate how SPON-1 might interact with these midline maintenance factors, we constructed double mutants. Strikingly, zig-4 and egl-15A, significantly suppressed midline crossing defects of spon-1 mutants, whereas spon-1 and dig-1 displayed additive effects on crossing (Fig. 7D). These results suggest that the SPON-1 pathway is distinct from the DIG-1 and ZIG-4/EGL-5 pathways, and moreover suggests that although SPON-1 and ZIG-4/EGL-15 maintain PVQ axon positions, they do so using opposing mechanisms.
Discussion
The founding member of the spondin family, F-spondin, was identified by its expression in the floor plate of the rat embryonic spinal cord (Klar et al., 1992). Since the discovery of F-spondin, a family of spondin-related genes has been defined, containing two subfamilies, F-spondins and the Mindins (Higashijima et al., 1997). Most animals encode at least one member of each subfamily. Genetic analysis of the spondins in other animals has been hampered by putative redundancy between family members, which are often expressed in overlapping patterns (Higashijima et al., 1997). For example, loss of M-spondin function in Drosophila does not cause detectable phenotypes (Umemiya et al., 1997), and mindin mutant mice have a normal nervous system (He et al., 2004). In contrast, we find that the sole C. elegans spondin SPON-1 is essential for embryonic development. The essential nature of SPON-1 function suggests that it may combine the functions separately performed by F-spondins and mindins in other animals.
Roles of SPON-1 domains
F-spondins contain a reelin domain, a spondin domain and multiple thombospondin repeats (TSRs). Our genetic and transgenic analysis suggests the TSRs of SPON-1 are critical for its roles in morphogenesis and axon guidance. Three spon-1 alleles cause missense alterations in TSRs, including the strong allele ju348 and the weak allele e2623. These two mutations affect equivalent residues in the WXXW motifs of TSRs 2 and 3; as these residues are not highly conserved, TSR2 may be more critical for the embryonic morphogenesis function of SPON-1 than TSR3. The temperature sensitive allele ju430 affects a highly conserved cysteine in TSR4, and could render SPON-1 thermolabile due to disruption of one of the disulfide bridges important in TSR tertiary structure (Tan et al., 2002). Although all these mutations could cause a global disruption of SPON-1 folding, they suggest the TSRs are required for SPON-1 function in vivo. TSRs 5 and 6 of vertebrate F-spondin mediate interactions with ECM proteoglycans; plasmin-mediated cleavage within TSRs 5 and 6 can release the rest of the protein from the ECM (Tzarfaty-Majar et al., 2001). As the motifs in TSRs 5 and 6 that mediate proteoglycan interactions are not present in invertebrate spondins, SPON-1 may be less stably attached to the ECM than vertebrate spondins, consistent with our findings that SPON-1 can accumulate on basement membranes far from its site of synthesis.
SPON-1 may act in integrin mediated muscle-epidermal adhesion
Several lines of evidence suggest SPON-1 acts either in integrin mediated adhesion, or in a closely related parallel process. SPON-1 is localized to integrin-containing sites and SPON-1 function becomes dosage-sensitive when α-integrin INA-1 function is reduced, indicating SPON-1 and INA-1 have closely related functions in embryonic development. C. elegans expresses two integrin heterodimers, αPAT-2/βPAT-3 and αINA-1/βPAT-3 (Brown, 2000; Gettner et al., 1995). PAT-2 is a member of the RGD-binding subfamily of β-integrins and is likely to bind Perlecan. INA-1 is a member of the laminin-binding subfamily; it is expressed in embryonic BM but is not required for embryonic morphogenesis (Baum and Garriga, 1997; Poinat et al., 2002). As spon-1 ina-1 double homozygotes arrest earlier in elongation than either single mutant, SPON-1 at least in part acts in parallel to INA-1. We speculate that SPON-1 might function in a subset of both INA-1 and PAT-2 dependent adhesion processes. Interestingly, F-spondin has also been linked to integrin signaling (Terai et al., 2001), as has Mindin (Jia et al., 2005; Li et al., 2006). An important goal for the future is to determine whether spondins interact directly with integrins in vivo, or whether they indirectly influence integrin mediated adhesion via candidate receptors such as APP or ApoER2 (Ho and Sudhof, 2004; Hoe et al., 2005).
Context-dependent roles of SPON-1 in axonal development
SPON-1 has distinct roles in axon development depending on the neuron type. Such context dependence is reminiscent of the known roles for F-spondin in the vertebrate nervous system. F-spondin promotes fasciculation of commissural axons after they cross the floor plate (Burstyn-Cohen et al., 1999), and also repels growth cones of motor neurons that do not cross the midline (Tzarfati-Majar et al., 2001). F-spondin also promotes outgrowth of sensory and hippocampal neurons in vitro (Burstyn-Cohen et al., 1998; Burstyn-Cohen et al., 1999; Feinstein et al., 1999). In C. elegans SPON-1 promotes fasciculation of motor neuron axons in the ventral nerve cord, and acts partly redundantly to promote motor neuron outgrowth. By contrast, SPON-1 appears to prevent midline crossing (fasciculation) of PVQ interneurons.
The deflections of motor commissures at the muscle boundary in spon-1 mutants suggest SPON-1 promotes the entry of growth cones into the muscle-epidermal basement membrane. Defects in entry of the commissural growth cone at the dorsal muscle quadrant are also found in integrin mutants (Poinat et al., 2002), indicating integrins act at this choice point. The similarity between spon-1 and integrin defects in motor axon development is consistent with SPON-1 promoting integrin-mediated adhesion or a closely related process in axon guidance.
Midline crossing defects could result from lack of repulsion from a midline repellent, or lack of axonal adhesion to the normal environment (Hobert and Bulow, 2003). Because we have not seen differential localization of SPON-1 at the ventral midline we favor the interpretation that the inappropriate crossing of PVQs in spon-1 mutants reflects lack of adhesion. As dig-1 and spon-1 show additive effects on crossing over, these two ECM molecules may act in distinct adhesive pathways. The ZIG-4 and FGFR(EGL-15A) pathways also maintain PVQ positions, but unexpectedly zig-4 and egl-15A mutations suppress spon-1 midline crossing defects, suggesting that spon-1 and zig-4/egl-15 maintain PVQ positions by opposing mechanisms. We speculate that PVQ maintenance involves a balance of adhesive and repulsive mechanisms. SPON-1 could promote the normal adhesion of PVQs in their respective fascicles, so that lack of SPON-1 leads to defasciculation and flip-overs. Several models have been proposed for how ZIG-4 might prevent midline crossing (Hobert and Bulow, 2003). One possibility is that ZIG-4 interferes with axon-BM adhesion at the midline, so that axons are unable to cross the midline region. As depicted in Figure 7E, SPON-1 in the adjacent basement membrane (green) may promote adhesion of PVQ axons (blue) to their appropriate locations, whereas extracellular ZIG-4/EGL-15 (red) might act locally to inhibit axon-BM adhesion at the midline. Thus, impaired axon-BM adhesion in spon-1 mutants might be compensated for by the loss of the ZIG/FGFR antiadhesive pathway. ZIG/FGFR signaling might directly inhibit SPON-1 based adhesion, or could inhibit a parallel adhesion pathway. Our results underscore the view that maintenance of process position in the nervous system involves a balance of multiple interacting forces.
In conclusion, our results demonstrate that an F-spondin-related protein promotes tissue adhesion in multiple contexts. SPON-1 functions in parallel to integrin-mediated adhesion in embryogenesis, and is antagonized by other extracellular axon maintenance factors in post-embryonic growth. These results should be useful in elucidating the in vivo cellular receptors for spondins.
Supplementary Material
Acknowledgments
We thank Mei Ding for isolating spon-1 mutations, Christie Emigh and Jian Cao for mapping, Chris Suh and members of the Chisholm and Jin laboratories for help and support. Some reagents used in this work were obtained from the Caenorhabditis Genetic Center, which is supported by the NIH NCRR. We thank Yuji Kohara for cDNA clones, Andy Fire for vectors, and Jim Kramer for advice on immunostaining. We thank Lindsay Hinck for discussions. W.-M.W. was supported by NIH training grant T32 GM08646 and by a University of California President's Dissertation-Year Fellowship. E.C.B. was supported in part by Diversity Awards from the UC Santa Cruz Center for Biomolecular Sciences. A.G. is supported by HHMI funding to Yishi Jin. This work was supported by the Whitehall Foundation and the US Public Health Service (NIH R01 GM54657).
References
- Ackley BD, Crew JR, Elamaa H, Pihlajaniemi T, Kuo CJ, Kramer JM. The NC1/endostatin domain of Caenorhabditis elegans type XVIII collagen affects cell migration and axon guidance. J Cell Biol. 2001;152:1219–32. doi: 10.1083/jcb.152.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio O, Hall DH, Hobert O. Immunoglobulin-domain proteins required for maintenance of ventral nerve cord organization. Science. 2002;295:686–90. doi: 10.1126/science.1066642. [DOI] [PubMed] [Google Scholar]
- Barstead RJ, Waterston RH. The basal component of the nematode dense-body is vinculin. J Biol Chem. 1989;264:10177–85. [PubMed] [Google Scholar]
- Baum PD, Garriga G. Neuronal migrations and axon fasciculation are disrupted in ina-1 integrin mutants. Neuron. 1997;19:51–62. doi: 10.1016/s0896-6273(00)80347-5. [DOI] [PubMed] [Google Scholar]
- Benard CY, Boyanov A, Hall DH, Hobert O. DIG-1, a novel giant protein, non-autonomously mediates maintenance of nervous system architecture. Development. 2006;133:3329–40. doi: 10.1242/dev.02507. [DOI] [PubMed] [Google Scholar]
- Bosher JM, Hahn BS, Legouis R, Sookhareea S, Weimer RM, Gansmuller A, Chisholm AD, Rose AM, Bessereau JL, Labouesse M. The Caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J Cell Biol. 2003;161:757–68. doi: 10.1083/jcb.200302151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NH. Cell-cell adhesion via the ECM: integrin genetics in fly and worm. Matrix Biol. 2000;19:191–201. doi: 10.1016/s0945-053x(00)00064-0. [DOI] [PubMed] [Google Scholar]
- Bulow HE, Boulin T, Hobert O. Differential functions of the C. elegans FGF receptor in axon outgrowth and maintenance of axon position. Neuron. 2004;42:367–74. doi: 10.1016/s0896-6273(04)00246-6. [DOI] [PubMed] [Google Scholar]
- Burstyn-Cohen T, Frumkin A, Xu YT, Scherer SS, Klar A. Accumulation of F-spondin in injured peripheral nerve promotes the outgrowth of sensory axons. J Neurosci. 1998;18:8875–85. doi: 10.1523/JNEUROSCI.18-21-08875.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstyn-Cohen T, Tzarfaty V, Frumkin A, Feinstein Y, Stoeckli E, Klar A. F-Spondin is required for accurate pathfinding of commissural axons at the floor plate. Neuron. 1999;23:233–46. doi: 10.1016/s0896-6273(00)80776-x. [DOI] [PubMed] [Google Scholar]
- Chin-Sang ID, George SE, Ding M, Moseley SL, Lynch AS, Chisholm AD. The ephrin VAB-2/EFN-1 functions in neuronal signaling to regulate epidermal morphogenesis in C. elegans. Cell. 1999;99:781–90. doi: 10.1016/s0092-8674(00)81675-x. [DOI] [PubMed] [Google Scholar]
- Chisholm AD, Hardin J. Epidermal Morphogenesis. T C e R Community, editors. Wormbook. 2005 doi: 10.1895/wormbook.1.35.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Goncharov A, Jin Y, Chisholm AD. C. elegans ankyrin repeat protein VAB-19 is a component of epidermal attachment structures and is essential for epidermal morphogenesis. Development. 2003;130:5791–5801. doi: 10.1242/dev.00791. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Roy PJ. Muscle arm development in Caenorhabditis elegans. Development. 2005;132:3079–92. doi: 10.1242/dev.01883. [DOI] [PubMed] [Google Scholar]
- Edgley ML, Riddle DL. LG II balancer chromosomes in Caenorhabditis elegans: mT1(II;III) and the mIn1 set of dominantly and recessively marked inversions. Mol Genet Genomics. 2001;266:385–95. doi: 10.1007/s004380100523. [DOI] [PubMed] [Google Scholar]
- Fares H, Greenwald I. Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics. 2001;159:133–45. doi: 10.1093/genetics/159.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein Y, Borrell V, Garcia C, Burstyn-Cohen T, Tzarfaty V, Frumkin A, Nose A, Okamoto H, Higashijima S, Soriano E, et al. F-spondin and mindin: two structurally and functionally related genes expressed in the hippocampus that promote outgrowth of embryonic hippocampal neurons. Development. 1999;126:3637–48. doi: 10.1242/dev.126.16.3637. [DOI] [PubMed] [Google Scholar]
- Feinstein Y, Klar A. The neuronal class 2 TSR proteins F-spondin and Mindin: a small family with divergent biological activities. Int J Biochem Cell Biol. 2004;36:975–80. doi: 10.1016/j.biocel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Francis R, Waterston RH. Muscle cell attachment in Caenorhabditis elegans. J Cell Biol. 1991;114:465–79. doi: 10.1083/jcb.114.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettner SN, Kenyon C, Reichardt LF. Characterization of beta pat-3 heterodimers, a family of essential integrin receptors in C. elegans. J Cell Biol. 1995;129:1127–41. doi: 10.1083/jcb.129.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham PL, Johnson JJ, Wang S, Sibley MH, Gupta MC, Kramer JM. Type IV collagen is detectable in most, but not all, basement membranes of Caenorhabditis elegans and assembles on tissues that do not express it. J Cell Biol. 1997;137:1171–83. doi: 10.1083/jcb.137.5.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo XD, Johnson JJ, Kramer JM. Embryonic lethality caused by mutations in basement membrane collagen of C. elegans. Nature. 1991;349:707–9. doi: 10.1038/349707a0. [DOI] [PubMed] [Google Scholar]
- He YW, Li H, Zhang J, Hsu CL, Lin E, Zhang N, Guo J, Forbush KA, Bevan MJ. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat Immunol. 2004;5:88–97. doi: 10.1038/ni1021. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Nose A, Eguchi G, Hotta Y, Okamoto H. MIndin/F-spondin family: novel ECM proteins expressed in the zebrafish embryonic axis. Dev Biol. 1997;192:211–227. doi: 10.1006/dbio.1997.8760. [DOI] [PubMed] [Google Scholar]
- Hinck L. The versatile roles of “axon guidance” cues in tissue morphogenesis. Dev Cell. 2004;7:783–93. doi: 10.1016/j.devcel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Ho A, Sudhof TC. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci U S A. 2004;101:2548–53. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O, Bulow H. Development and maintenance of neuronal architecture at the ventral midline of C. elegans. Curr Opin Neurobiol. 2003;13:70–8. doi: 10.1016/s0959-4388(03)00002-3. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Wessner D, Beffert U, Becker AG, Matsuoka Y, Rebeck GW. F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol Cell Biol. 2005;25:9259–68. doi: 10.1128/MCB.25.21.9259-9268.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hall DH, Hedgecock EM, Kao G, Karantza V, Vogel BE, Hutter H, Chisholm AD, Yurchenco PD, Wadsworth WG. Laminin alpha subunits and their role in C. elegans development. Development. 2003a;130:3343–58. doi: 10.1242/dev.00481. [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y. MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron. 2002;34:563–76. doi: 10.1016/s0896-6273(02)00672-4. [DOI] [PubMed] [Google Scholar]
- Huang X, Huang P, Robinson MK, Stern MJ, Jin Y. UNC-71, a disintegrin and metalloprotease (ADAM) protein, regulates motor axon guidance and sex myoblast migration in C. elegans. Development. 2003b;130:3147–61. doi: 10.1242/dev.00518. [DOI] [PubMed] [Google Scholar]
- Hudson ML, Kinnunen T, Cinar HN, Chisholm AD. C. elegans Kallmann syndrome protein KAL-1 interacts with syndecan and glypican to regulate neuronal cell migrations. Dev Biol. 2006;294:352–65. doi: 10.1016/j.ydbio.2006.02.036. [DOI] [PubMed] [Google Scholar]
- Jia W, Li H, He YW. The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood. 2005;106:3854–9. doi: 10.1182/blood-2005-04-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, Kang SH, Kramer JM. C. elegans dystroglycan DGN-1 functions in epithelia and neurons, but not muscle, and independently of dystrophin. Development. 2006;133:1911–21. doi: 10.1242/dev.02363. [DOI] [PubMed] [Google Scholar]
- Kang SH, Kramer JM. Nidogen is nonessential and not required for normal type IV collagen localization in Caenorhabditis elegans. Mol Biol Cell. 2000;11:3911–23. doi: 10.1091/mbc.11.11.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao G, Huang CC, Hedgecock EM, Hall DH, Wadsworth WG. The role of the laminin beta subunit in laminin heterotrimer assembly and basement membrane function and development in C. elegans. Dev Biol. 2006;290:211–9. doi: 10.1016/j.ydbio.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Kim K, Li C. Expression and regulation of an FMRFamide-related neuropeptide gene family in Caenorhabditis elegans. J Comp Neurol. 2004;475:540–50. doi: 10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- Klar A, Baldassare M, Jessell TM. F-spondin: a gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell. 1992;69:95–110. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–71. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Li H, Oliver T, Jia W, He YW. Efficient dendritic cell priming of T lymphocytes depends on the extracellular matrix protein mindin. Embo J. 2006;25:4097–107. doi: 10.1038/sj.emboj.7601289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DMr, Ortiz I, Berliner GC, Epstein HF. Differential localization of two myosins within nematode thick filaments. Cell. 1983;34:477–90. doi: 10.1016/0092-8674(83)90381-1. [DOI] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–84. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrière G, Gouy M. WWW-Query: An on-line retrieval system for biological sequence banks. Biochimie. 1996;78:364–9. doi: 10.1016/0300-9084(96)84768-7. [DOI] [PubMed] [Google Scholar]
- Poinat P, De Arcangelis A, Sookhareea S, Zhu X, Hedgecock EM, Labouesse M, Georges-Labouesse E. A conserved interaction between beta1 integrin/PAT-3 and Nck-interacting kinase/MIG-15 that mediates commissural axon navigation in C. elegans. Curr Biol. 2002;12:622–31. doi: 10.1016/s0960-9822(02)00764-9. [DOI] [PubMed] [Google Scholar]
- Priess JR, Hirsh DI. Caenorhabditis elegans morphogenesis: the role of the cytoskeleton in elongation of the embryo. Dev Biol. 1986;117:156–73. doi: 10.1016/0012-1606(86)90358-1. [DOI] [PubMed] [Google Scholar]
- Rogalski TM, Williams BD, Mullen GP, Moerman DG. Products of the unc-52 gene in Caenorhabditis elegans are homologous to the core protein of the mammalian basement membrane heparan sulfate proteoglycan. Genes Dev. 1993;7:1471–84. doi: 10.1101/gad.7.8.1471. [DOI] [PubMed] [Google Scholar]
- Sarafi-Reinach TR, Melkman T, Hobert O, Sengupta P. The lin-11 LIM homeobox gene specifies olfactory and chemosensory neuron fates in C. elegans. Development. 2001;128:3269–81. doi: 10.1242/dev.128.17.3269. [DOI] [PubMed] [Google Scholar]
- Schubert D, Klar A, Park M, Dargusch R, Fischer WH. F-spondin promotes nerve precursor differentiation. J Neurochem. 2006;96:444–53. doi: 10.1111/j.1471-4159.2005.03563.x. [DOI] [PubMed] [Google Scholar]
- Shioi G, Shoji M, Nakamura M, Ishihara T, Katsura I, Fujisawa H, Takagi S. Mutations affecting nerve attachment of Caenorhabditis elegans. Genetics. 2001;157:1611–22. doi: 10.1093/genetics/157.4.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, Lawler J, Wang JH. Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication. J Cell Biol. 2002;159:373–82. doi: 10.1083/jcb.200206062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai Y, Abe M, Miyamoto K, Koike M, Yamasaki M, Ueda M, Ueki M, Sata Y. Vascular smooth muscle cell growth-promoting factor/F-spondin inhibits angiogenesis via the blockade of integrin αvβ3 on vascular endothelial cells. J Cell Physiol. 2001;188:394–402. doi: 10.1002/jcp.1122. [DOI] [PubMed] [Google Scholar]
- Tzarfati-Majar V, Burstyn-Cohen T, Klar A. F-spondin is a contact-repellent molecule for embryonic motor neurons. Proc Natl Acad Sci U S A. 2001;98:4722–7. doi: 10.1073/pnas.081062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzarfaty-Majar V, Lopez-Alemany R, Feinstein Y, Gombau L, Goldshmidt O, Soriano E, Munoz-Canoves P, Klar A. Plasmin-mediated release of the guidance molecule F-spondin from the extracellular matrix. J Biol Chem. 2001;276:28233–41. doi: 10.1074/jbc.M102585200. [DOI] [PubMed] [Google Scholar]
- Umemiya T, Takeichi M, Nose A. M-spondin, a novel ECM protein highly homologous to vertebrate F-spondin, is localized at the muscle attachment sites in the Drosophila embryo. Dev Biol. 1997;186:165–76. doi: 10.1006/dbio.1997.8591. [DOI] [PubMed] [Google Scholar]
- Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J Cell Biol. 1994;124:475–90. doi: 10.1083/jcb.124.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo WM, Goncharov A, Jin Y, Chisholm AD. Intermediate filaments are required for C. elegans epidermal elongation. Dev Biol. 2004;267:216–229. doi: 10.1016/j.ydbio.2003.11.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.