

Abstract
Epidemiological and experimental data strongly suggest that cardiovascular diseases can originate from an aberrant environment during fetal development, a phenomenon referred to as perinatal programming. This review will focus on the role of the kidneys in determining blood pressure, and how (re)programming the renal development can persistently ameliorate hereditary hypertension. By combining physiologic and genomic studies we have discovered some candidate pathways suited for (re)programming the development of hypertension. This sets the stage for mechanistic analysis in future studies.
Key words: perinatal development, spontaneously hypertensive rat, citrulline, nitric oxide, hypertension
Hypertension and the Kidney
The hereditary nature of hypertension is well established,1,2 and many genes can contribute to the development of high blood pressure. However, there are many non-genetic factors, like high sodium intake or obesity, which are closely associated with hypertension.3,4 With the exception of distinct monogenetic forms of hypertension, it is hard to separate genetic and environmental components because of their complex interaction. However, it is well known that the contribution of the kidneys is paramount. For instance, transplantation studies have demonstrated that blood pressure “follows” the kidney5 and all monogenetic (Mendelian) forms of hypertension affect blood pressure by changing net renal sodium balance.2 In addition Guyton et al. inextricably linked long-term blood pressure regulation to renal excretory function,6 i.e., hypertension can only persist when renal water and sodium handling is reset. Together this underlines the dominant role of the kidney in long-term blood pressure regulation. Studying the development of hypertension from the very beginning, even before its onset, might provide new insights. We propose that four pivotal components can be identified in the development and maintenance of elevated blood pressure (Fig. 1A).
Figure 1.

The proposed four components of development of hereditary hypertension (A) and how blood pressure regulation could be programmed during perinatal development (B).
(1) The interaction between environmental factors and hereditary traits resets the transcriptome.
(2) This reset transcriptome persistently changes protein expression.
(3) This impairs renal hemodynamics and/or alters renal structure.
(4) Because of these alterations the kidneys will shift the regulatory set point of natriuresis to a higher perfusion pressure i.e., hypertension.
The renal vascular constrictor-dilator balance is believed to be vital for blood pressure regulation and development of renal damage. A shift in this balance can impair renal function, i.e., too much dilation can cause renal damage due to an increase of transmitted pressures, while too much constriction impairs pressure natriuresis. Natriuresis driven by pressure provides a primary and powerful means of stabilizing total body sodium and blood pressure over a wide range of sodium intakes, a hypothesis first proposed by Guyton.6 The corollary of this hypothesis is that hypertension cannot persist when there is no shift in the pressure-natriuresis curve to higher pressures. However, when renal hemodynamic control is shifted to other set points, e.g., increased resistance or diminished RBF autoregulation, this can affect the renal structure. Irrespective of the primary cause, when the transmission of pressure to the glomerulus increases, capillary ballooning starts to occur, finally resulting in adhesions and sclerosis.7 Thus, hypertension per se can lead to glomerular, and downstream peritubular microvascular changes in the kidney and finally to overt renal failure.8 Hence, once hypertension has developed, it can further affect the renal transcriptome and potentially contribute to further deterioration of blood pressure regulating mechanisms.
Programming the Development of the Kidney
Because fetal development is exceptionally sensitive to environmental factors it is possible that adult renal function is programmed due to gene-environment interactions in this critical period. Dr. David Barker first formulated this so-called perinatal programming hypothesis. He described an association between low birth weight and cardiovascular disease later in life, and proposed that this was due to aberrant prenatal development.9 Subsequently, a large body of experimental and epidemiological studies have supported this concept10–18 and the hypothesis of developmental plasticity was developed.19,20 Developmental plasticity is defined as the phenomenon by which one genotype can give rise to a range of different physiological or morphological states in response to different environmental conditions during development. Developmental plasticity includes a critical period when a system is sensitive to the environment, followed by a period when the system becomes fixed with respect to its functional capacity.19 For most organs and systems in humans this occurs before birth. It may be advantageous in evolutionary terms for the organism to retain plasticity during development, and allow adaptation of the phenotype to the environment.
The complex interactions among genes and environmental factors brought about by alterations in nutrition, maternal behavior and conditions (e.g., temperature, oxygenation), utero-placental circulation and exogenous noxious agents such as nicotine will affect gene transcription patterns in the developing organism. Different environmental stresses likely give rise to unique gene activity patterns, which in turn determine organogenesis and embryonic, fetal and placental growth (Fig. 1B). Specific to the kidney, this will affect systems regulating extracellular fluid homeostasis and thus determine blood pressure in adult life.12 Most studies focus on how aberrant perinatal factors induce pathology later in life. However, perinatal factors can also have beneficial long-lasting effects.
During its development the kidney is very susceptible to environmental stimuli determining renal function and structure.21 It has been proposed that a reduced number of nephrons can lead to glomerular hypertension.22 Could improvement of perinatal conditions increase the number of nephrons? An interesting model is protein restriction during lactation. Rat pups with a normal birth weight born to dams fed a 20% protein diet were cross fostered to 8% protein, but isocalorically-fed, mothers until weaning.23 This resulted in slower growth during lactation and decreased body weight throughout their lifespan as compared to controls (offspring cross fostered to mothers fed a 20% diet during pregnancy and lactation). In concordance with studies relating growth rates to aging, they had a longer lifespan23 and aging-related development of albuminuria was delayed.24 At 12 months of age the rats with postnatal low-protein (PLP) had higher antioxidant enzyme levels, and did not show the age-related reduction of telomere length in their renal cortex as compared to the control offspring.25 They also tended to have less glomerular injury. In a follow-up analysis we measured glomerular diameter and density in these control and PLP rats (Table 1).25a There was a very significant reduction in glomerular diameter in the PLP rats, suggesting that these aging male Wistar rats were protected from the glomerular injury that is common to aging male laboratory rats.26,27 Unexpectedly, glomerular density was increased in the PLP rats by about 30%. Of course, this increase may well be partly the consequence of less nephron loss in the PLP group in comparison to the aging control group. Nevertheless, these data suggest that reducing protein intake very early in life may have long-term renal protective effects. Whether this protection runs via pathways analogous to the well-known protection afforded by reducing protein intake in adult life28 is not clear. This model is an example of how manipulations early in life may have long-term beneficial effects in the kidney.
Table 1.
Glomerular diameter glomerular density, and kidney weight (mean left and right) of male control and postnatal low-protein (PLP) rats at 12 months of age)
| n | Glomerular diameter (µm) | Glomerular density (n/mm3) | 12 m absolute kidney wt (mg) | |
| Control | 12 | 118 ± 2 | 53 ± 2 | 2182 ± 45 |
| PLP | 13 | 109 ± 1# | 69 ± 3# | 1686 ± 57# |
p < 0.001 vs. control, mean ± SEM.25a
(Re)programming Development of Hypertension
As outlined above hypertension is associated with a resetting of the renal transcriptome and renal hemodynamics, and primary or secondary renal damage. It is plausible that manipulations during development that correct these alterations could result in beneficial long-lasting effects. To be able to correct one or more of these issues during early development one needs specific targets that can be manipulated by exogenous agents. Because the balance between nitric oxide (NO) and reactive oxygen species (ROS) plays a role in many blood pressure regulating systems,29,30 manipulating the NO/ROS balance in early development might be a useful strategy for reprogramming developmental renal gene expression programs and organogenesis.
Oxidative stress is often the net result of NO deficiency and increased ROS production.29,30 Oxidative stress is invariably present in animal models of induced and hereditary hypertension, e.g., angiotensin (ANG) II induced hypertension, the Spontaneously Hypertensive Rat (SHR) and the Dahl salt-sensitive rat. Antioxidant treatment in all these models can lower blood pressure.31–33 In concordance, inducing oxidative stress can cause hypertension.34 Moreover, a positive feedback loop seems to exist because a high blood pressure per se can cause oxidative stress.35 Taken together, it is conceivable that a shift in the (renal) redox balance towards ROS formation can induce hypertension. Correction of this imbalance in early development, by a perinatal intervention, might lead to normalization of blood pressure in adult life with persistent correction of at least some renal characteristics including preserved nephrogeneisis, sodium excretion and blood flow autoregulation.
The spontaneously hypertensive rat (SHR) is a model of essential hypertension with increased angiotensin II sensitivity and a high preglomerular resistance resulting in a high blood pressure with almost no renal injury. Racasan et al. perinatally manipulated the NO/ROS balance in SHR by perinatally supplementing pregnant and lactating SHR and offspring up to 4 weeks of age with a combination of L-arginine and antioxidants (taurine, vitamins C and E). This resulted in SHR offspring that had persistently lower blood pressure until 48 weeks of age. Interestingly, no effect on nephron number was observed.36 Perinatal treatment of SHR with the NO donor molsidomine also persistently lowered blood pressure in the offspring, suggesting that in SHR manoeuvres aimed either at changing the overall NO/ROS balance or aiming at solely increasing NO availability, can persistently reduce blood pressure.37 Nevertheless, many issues still need to be addressed. At this moment, it is still unclear which specific component(s) of the L-arginine and antioxidants treatment is/are responsible for the observed effects. Moreover, at a more mechanistic level, it is as yet unresolved how perinatal L-arginine, antioxidant or molsidomine supplements affect blood pressure control mechanisms.
Using microarray technology we can investigate the transcriptional response to manipulated NO/ROS balance. For instance Chon et al. found, using microarrays, that both NOS inhibition and glutathione depletion-induced hypertension resulted in partially overlapping adaptations in gene expression of energy and protein metabolism in the heart prior to cardiac hypertrophy.38 In addition, Wesseling et al. observed that NO depletion affected renal expression of genes involved in synthesis of the antioxidants glutathione and bilirubin but had no direct effects on pro-oxidant systems.39 Braam et al. found that gene expression patterns of fibroblasts obtained from aging human donors in culture were strongly affected by anti-oxidant treatment.40 In endothelial cells Braam et al. observed NO-dependent gene expressions and identified shear-sensitive and NO-dependent transcriptional regulators.41,42 To track down genes that are responsible for the proposed shift in NO/ROS balance, we performed microarray studies on renal cortex during renal development in SHR and WKY.
L-Arginine, Citrulline and Nitric Oxide in the SHR
Arginine availability is a potential candidate for perinatal intervention. NO has vasodilator and natriuretic properties and induced NO deficiency by inhibition of NOS has been shown to cause marked and persistent hypertension. Because NO is synthesized by NOS by converting arginine to citrulline, it is likely that reduced arginine availability may cause NO deficiency. In kidney proximal tubular cells, arginine is synthesized by the sequential action of argininosuccinate synthetase and argininosuccinate lyase (ASS and ASL) that convert citrulline to argininosuccinate and argininosuccinate to arginine, respectively. Citrulline delivery to proximal tubule cells is rate-limiting for renal arginine synthesis.43,44 A second source of arginine in the kidney is tubular uptake by the cationic amino acid Y-transporter, so called Y+LAT1, located on the basolateral side of renal epithelial cells, which exchanges neutral for cationic amino acids. As arginine is a cationic amino acid, the Y-transporters may regulate the rate of NO synthesis by controlling the uptake of arginine. A defect in this citrulline-arginine pathway could reduce arginine availability and hence cause a NO deficiency in the pre-hypertensive SHR kidney. Our laboratory and others have observed increased NOS gene and protein expression in young SHR vs. age-matched control WKY rats.45,46 However, Jones et al. observed a decreased arginine pool in both plasma and skeletal muscle in 3.5-week-old SHR47 and we observed a decreased urinary excretion of stable NO metabolites in 4-week-old SHR (Fig. 2). These data are suggestive for an NO deficiency due to a defect citrulline-arginine pathway in SHR, well before the onset of hypertension. Using microarray methodology we indeed observed decreased ASS, ASL and Y+LAT1 transporter expression in kidneys of young SHR, and this was confirmed by quantitative PCR.46 We propose that reduced cationic amino acid transporter disable the young SHR kidney to use arginine reabsorption to compensate for reduced arginine synthesis.
Figure 2.
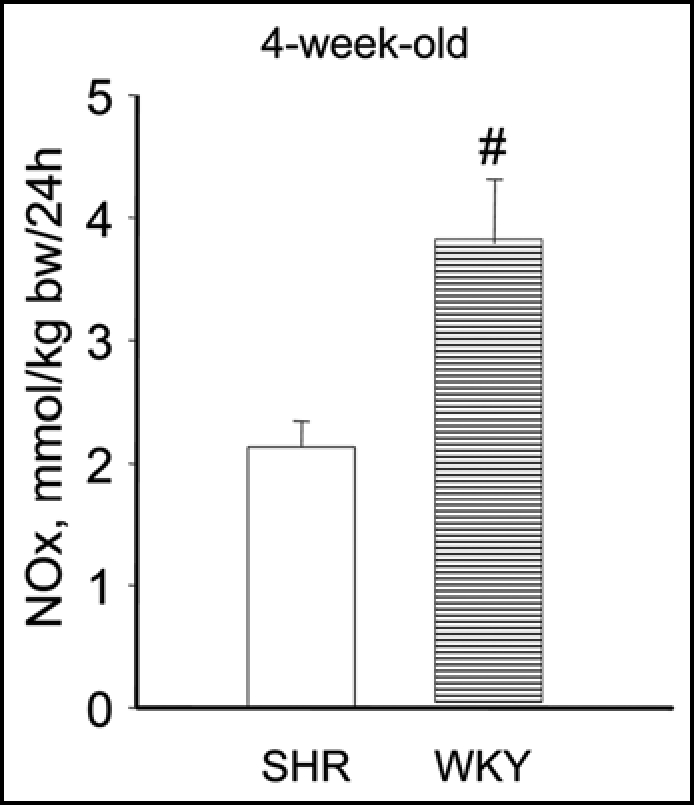
Urinary excretions of stable NO metabolites in 4 week-old SHR (open bar) and WKY (horizontal striped bar). #p < 0.001 vs. SHR.
This led us to investigate the challenging issue whether renal NO deficiency precedes hypertension in SHR due to reduced renal arginine availability. We speculated that sufficient arginine availability to produce NO in the kidney depends on both endogenous arginine synthesis and arginine reabsorption. Thus we hypothesized that NO deficiency in the developing SHR kidney, is caused by a defect in the citrulline-arginine pathway, arginine reabsorption, or both. Hence, we measured renal and cardiac amino acid pools and NO in 2-week-old pre-hypertensive SHR and perinatally supplemented SHR with citrulline. Quantification of NO is particularly difficult because NO is a highly reactive substance with a very short half-life. To compare the in vivo renal NO content we developed a new method to trap NO in vivo with which we could quantify NO with electron paramagnetic resonance in tissues of very young rats. Indeed, using this novel measurement we could show that in 2-week-old SHR NO was reduced in the kidneys but not in the heart, pointing to an organ specific deficiency at a very early age (Fig. 3). Perinatal citrulline supplements partially corrected this renal NO deficiency and persistently ameliorated hypertension in SHR (Fig. 4). Moreover, the decrease in renal vascular resistance during acute Tempol infusion observed in 28 week-old control SHR, was no longer significant at the same age after perinatal citrulline supplements. The latter is compatible with the idea that there has been a shift in renal constrictor-dilator balance towards relaxation that has persisted for 5 months after stopping citrulline supplements. Whatever the precise mechanism, it is very promising that brief dietary supplements of a single amino acid (arginine, citrulline or taurine) during development can cause long-lasting improvement of blood pressure regulation.
Figure 3.

NO yields in kidney (A) and heart (B) determined by electron paramagnetic resonance in SHR (open bars), SHR perinatally supplemented with citrulline (SHRcitr; diagonally striped bars) and WKY (horizontal striped bar).46 †p < 0.05, ‡p < 0.01 and #p < 0.001 vs. SHR.
Figure 4.

Systolic blood pressure (A) in control SHR (open circles; n = 24) and SHRcitr (closed circles; n = 16) and WKY (triangles; n = 9).46 WKY was lower than SHR from 8 weeks onwards (p < 0.001). Mean arterial pressure (MAP) at 50 weeks of age (B) in SHR (open bars), SHRcitr (diagonally striped bars) and WKY (striped bars). †p < 0.05, #p < 0.001 vs. SHR.
Potential Mechanism(s)
It is striking that, given the polygenic and diverse nature of hypertension, all Mendelian forms of hypertension (and hypotension) affect blood pressure by changing sodium balance.2 Therefore the capacity of the kidney to maintain precise sodium balance seems to be crucial. Hypertension induced by a maternal low protein diet during pregnancy in Sprague Dawley rats upregulated two critical sodium transporters at 4 weeks of age, the bumetanide-sensitive Na-K-2Cl cotransporter and, to a much lesser extent, the thiazidesensitive Na-Cl cotransporter.48 This suggests that increased sodium reabsorption at an early age is amenable to environmental influences. We need more studies that assess the pressure-natriuresis curve in models of programmed hypertension to support that different perinatal maneuvers can reset sodium balance during early development and thereby persistently alter blood pressure regulation.
Brenner et al. proposed that hypertension, including programming due to restricted nutrition, is a consequence of reduced nephron number, leading to a shift in the pressure-natriuresis curve22 and hypertension. The observation by Keller et al. that the number of glomeruli is lower in the kidneys of patients with hypertension than in matched normotensive controls49 supports this hypothesis. However, using a F2 generation, where there was random mixing of SHR/WKY genes, Black et al. demonstrated50 that there was no cosegregation of reduced numbers of nephrons in the kidney with increased levels of blood pressure, or of reduced total renal filtration surface area with increased levels of adult blood pressure. In concordance we did not find significant differences in nephron number after perinatal treatment.36 Conversely, hypertension was found in the absence of changed nephron numbers in pups born from pregnancies with uteroplacental insufficiency (induced by arterial ligation) in combination with a reduced litter size during lactation,51 and even in the presence of increased nephron numbers after postnatal overfeeding by reducing litter size after normal pregnancy.52 Thus more subtle changes in sodium transport are more likely to play a role in some blood pressure programming models than a change in nephron number.
Perspectives
Low levels of NO in the kidney precede hypertension in SHR, an accepted model of essential hypertension. Studies from our laboratory indicate that perinatal interventions that shift the NO/ROS balance towards an increase in NO availability during renal development in SHR have persistent antihypertensive effects. Obviously more research needs to be done, including determination of actual NO and ROS in different renal compartments. These compartments could be defined at the level of gross anatomy e.g., cortex vs. medulla, cell type e.g., endothelial vs. epithelial or tissue compartments i.e., intracellular vs. extracellular. Localization of endogenous antioxidant pathways, e.g., SOD, could be responsible for such localized fine tuning of ambient ROS levels.53 In addition it is of great interest whether perinatal treatment changes renal function in such a way that homeostasis is facilitated in the face of a sodium load. This will give an indication of the components that may be altered during early development.
Acknowledgements
The Dutch Kidney Foundation (C03.2039) and the European Union Sixth Framework Programme for Research and Technical Development of the European Community—The Early Nutrition Programming Project (FOOD-CT-2005-007036) supported our studies.
Footnotes
Previously published online as an Organogenesis E-publication: http://www.landesbioscience.com/journals/organogenesis/article/6504
References
- 1.Robinson RF, Batisky DL, Hayes JR, Nahata MC, Mahan JD. Significance of heritability in primary and secondary pediatric hypertension. Am J Hypertens. 2005;18:917–921. doi: 10.1016/j.amjhyper.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 3.Hall JE. The kidney, hypertension and obesity. Hypertension. 2003;41:625–633. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 4.Law M. Salt, blood pressure and cardiovascular diseases. J Cardiovasc Risk. 2000;7:5–8. doi: 10.1177/204748730000700102. [DOI] [PubMed] [Google Scholar]
- 5.Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res. 1975;36:692–696. doi: 10.1161/01.res.36.6.692. [DOI] [PubMed] [Google Scholar]
- 6.Guyton AC. Physiologic regulation of arterial pressure. Am J Cardiol. 1961;8:401–407. doi: 10.1016/0002-9149(61)90159-x. [DOI] [PubMed] [Google Scholar]
- 7.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 8.Johnson RJ, Rodriguez-Iturbe B, Kang DH, Feig DI, Herrera-Acosta J. A unifying pathway for essential hypertension. Am J Hypertens. 2005;18:431–440. doi: 10.1016/j.amjhyper.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 9.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:700–707. doi: 10.1038/ncpneph0344. [DOI] [PubMed] [Google Scholar]
- 11.Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol. 2007;19:1–19. doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- 12.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 13.Gennser G, Rymark P, Isberg PE. Low birth weight and risk of high blood pressure in adulthood. Br Med J (Clin Res Ed) 1988;296:1498–1500. doi: 10.1136/bmj.296.6635.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, Willett WC, Hennekens CH. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ. 1997;315:396–400. doi: 10.1136/bmj.315.7105.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet. 1996;348:1269–1273. doi: 10.1016/s0140-6736(96)04547-3. [DOI] [PubMed] [Google Scholar]
- 16.Leon DA, Koupilova I, Lithell HO, Berglund L, Mohsen R, Vagero D, Lithell UB, McKeigue PM. Failure to realise growth potential in utero and adult obesity in relation to blood pressure in 50 year old Swedish men. BMJ. 1996;312:401–406. doi: 10.1136/bmj.312.7028.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsen T, Eriksson JG, Tuomilehto J, Teramo K, Osmond C, Barker DJ. Mother's weight in pregnancy and coronary heart disease in a cohort of Finnish men: Follow up study. BMJ. 1997;315:837–840. doi: 10.1136/bmj.315.7112.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunnarsdottir I, Birgisdottir BE, Benediktsson R, Gudnason V, Thorsdottir I. Relationship between size at birth and hypertension in a genetically homogeneous population of high birth weight. J Hypertens. 2002;20:623–628. doi: 10.1097/00004872-200204000-00018. [DOI] [PubMed] [Google Scholar]
- 19.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 20.Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, et al. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- 21.Bagby SP. Maternal nutrition, low nephron number, and hypertension in later life: pathways of nutritional programming. J Nutr. 2007;137:1066–1072. doi: 10.1093/jn/137.4.1066. [DOI] [PubMed] [Google Scholar]
- 22.Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens. 1988;1:335–347. doi: 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 23.Jennings BJ, Ozanne SE, Dorling MW, Hales CN. Early growth determines longevity in male rats and may be related to telomere shortening in the kidney. FEBS Lett. 1999;448:4–8. doi: 10.1016/s0014-5793(99)00336-1. [DOI] [PubMed] [Google Scholar]
- 24.Petry CJ, Jennings BJ, James LA, Hales CN, Ozanne SE. Suckling a protein-restricted rat dam leads to diminished albuminuria in her male offspring in adult life: A longitudinal study. BMC Nephrol. 2006;7:14. doi: 10.1186/1471-2369-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarry-Adkins JL, Joles JA, Chen JH, Martin-Gronert MS, van der Giezen DM, Goldschmeding R, Hales CN, Ozanne SE. Protein restriction in lactation confers nephroprotective effects in the male rat and is associated with increased antioxidant expression. Am J Physiol Regul Integr Comp Physiol. 2007;293:1259–1266. doi: 10.1152/ajpregu.00231.2007. [DOI] [PubMed] [Google Scholar]
- 25a.Joles JA, Tarry-Adkins JL, Snoeijs S, van der Giezen DM, Goldschmeding R, Ozanne SE. Reply to: Schreuder. Am J Physiol Regul Integr Comp Physiol. 2007;294:277–278. [Google Scholar]
- 26.Baylis C. Age-dependent glomerular damage in the rat. Dissociation between glomerular injury and both glomerular hypertension and hypertrophy. Male gender as a primary risk factor. J Clin Invest. 1994;94:1823–1829. doi: 10.1172/JCI117531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldstein RS, Tarloff JB, Hook JB. Age-related nephropathy in laboratory rats. Faseb J. 1988;2:2241–2251. doi: 10.1096/fasebj.2.7.3280378. [DOI] [PubMed] [Google Scholar]
- 28.Brenner BM, Meyer TW, Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med. 1982;307:652–659. doi: 10.1056/NEJM198209093071104. [DOI] [PubMed] [Google Scholar]
- 29.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol. 2005;289:913–935. doi: 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]
- 30.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582–593. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- 31.Manning RD, Jr, Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol. 2005;25:311–317. doi: 10.1159/000086411. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension. 2000;36:957–964. doi: 10.1161/01.hyp.36.6.957. [DOI] [PubMed] [Google Scholar]
- 33.Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension. 2001;38:655–659. doi: 10.1161/01.hyp.38.3.655. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri ND, Wang XQ, Oveisi F, Rad B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension. 2000;36:142–146. doi: 10.1161/01.hyp.36.1.142. [DOI] [PubMed] [Google Scholar]
- 35.Sindhu RK, Roberts CK, Ehdaie A, Zhan CD, Vaziri ND. Effects of aortic coarctation on aortic antioxidant enzymes and NADPH oxidase protein expression. Life Sci. 2005;76:945–953. doi: 10.1016/j.lfs.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Racasan S, Braam B, van der Giezen DM, Goldschmeding R, Boer P, Koomans HA, Joles JA. Perinatal L-arginine and antioxidant supplements reduce adult blood pressure in spontaneously hypertensive rats. Hypertension. 2004;44:83–88. doi: 10.1161/01.HYP.0000133251.40322.20. [DOI] [PubMed] [Google Scholar]
- 37.Racasan S, Braam B, Koomans HA, Joles JA. Programming blood pressure in adult SHR by shifting perinatal balance of NO and reactive oxygen species toward NO: the inverted Barker phenomenon. Am J Physiol Renal Physiol. 2005;288:626–636. doi: 10.1152/ajprenal.00314.2004. [DOI] [PubMed] [Google Scholar]
- 38.Chon H, Bluyssen HA, Holstege FC, Koomans HA, Joles JA, Braam B. Gene expression of energy and protein metabolism in hearts of hypertensive nitric oxide- or GSH-depleted mice. Eur J Pharmacol. 2005;513:21–33. doi: 10.1016/j.ejphar.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 39.Wesseling S, Joles JA, van Goor H, Bluyssen HA, Kemmeren P, Holstege FC, Koomans HA, Braam B. Transcriptome-based identification of pro- and antioxidative gene expression in kidney cortex of nitric oxide-depleted rats. Physiol Genomics. 2007;28:158–167. doi: 10.1152/physiolgenomics.00077.2006. [DOI] [PubMed] [Google Scholar]
- 40.Braam B, Langelaar-Makkinje M, Verkleij A, Bluyssen H, Verrips T, Koomans HA, Joles JA, Post JA. Anti-oxidant sensitivity of donor age-related gene expression in cultured fibroblasts. Eur J Pharmacol. 2006;542:154–161. doi: 10.1016/j.ejphar.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 41.Braam B, de Roos R, Dijk A, Boer P, Post JA, Kemmeren PP, Holstege FC, Bluysen HA, Koomans HA. Nitric oxide donor induces temporal and dose-dependent reduction of gene expression in human endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287:1977–1986. doi: 10.1152/ajpheart.00323.2004. [DOI] [PubMed] [Google Scholar]
- 42.Braam B, de Roos R, Bluyssen H, Kemmeren P, Holstege F, Joles JA, Koomans H. Nitric oxide-dependent and nitric oxide-independent transcriptional responses to high shear stress in endothelial cells. Hypertension. 2005;45:672–680. doi: 10.1161/01.HYP.0000154683.33414.94. [DOI] [PubMed] [Google Scholar]
- 43.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur J Biochem. 2003;270:1887–1899. doi: 10.1046/j.1432-1033.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 44.Brosnan ME, Brosnan JT. Renal arginine metabolism. J Nutr. 2004;134:2791–2795. doi: 10.1093/jn/134.10.2791S. [DOI] [PubMed] [Google Scholar]
- 45.Vaziri ND, Ni Z, Oveisi F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension. 1998;31:1248–1254. doi: 10.1161/01.hyp.31.6.1248. [DOI] [PubMed] [Google Scholar]
- 46.Koeners MP, van Faassen EE, Wesseling S, de Sain-van der Velden M, Koomans HA, Braam B, Joles JA. Maternal supplementation with citrulline increases renal nitric oxide in young spontaneously hypertensive rats and has long-term antihypertensive effects. Hypertension. 2007;50:1077–1084. doi: 10.1161/HYPERTENSIONAHA.107.095794. [DOI] [PubMed] [Google Scholar]
- 47.Jones MR. Free amino acid pools in the spontaneously hypertensive rat: A longitudinal study. J Nutr. 1988;118:579–587. doi: 10.1093/jn/118.5.579. [DOI] [PubMed] [Google Scholar]
- 48.Manning J, Beutler K, Knepper MA, Vehaskari VM. Upregulation of renal BSC1 and TSC in prenatally programmed hypertension. Am J Physiol Renal Physiol. 2002;283:202–206. doi: 10.1152/ajprenal.00358.2001. [DOI] [PubMed] [Google Scholar]
- 49.Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N Engl J Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- 50.Black MJ, Briscoe TA, Constantinou M, Kett MM, Bertram JF. Is there an association between level of adult blood pressure and nephron number or renal filtration surface area? Kidney Int. 2004;65:582–588. doi: 10.1111/j.1523-1755.2004.00406.x. [DOI] [PubMed] [Google Scholar]
- 51.Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18:1688–1696. doi: 10.1681/ASN.2007010015. [DOI] [PubMed] [Google Scholar]
- 52.Boubred F, Buffat C, Feuerstein JM, Daniel L, Tsimaratos M, Oliver C, Lelièvre-Pégorier M, Simeoni U. Effects of early postnatal hypernutrition on nephron number and long-term renal function and structure in rats. Am J Physiol Renal Physiol. 2007;293:F1944–F1996. doi: 10.1152/ajprenal.00141.2007. [DOI] [PubMed] [Google Scholar]
- 53.Madamanchi NR, Moon SK, Hakim ZS, Clark S, Mehrizi A, Patterson C, Runge MS. Differential activation of mitogenic signaling pathways in aortic smooth muscle cells deficient in superoxide dismutase isoforms. Arterioscler Thromb Vasc Biol. 2005;25:950–956. doi: 10.1161/01.ATV.0000161050.77646.68. [DOI] [PubMed] [Google Scholar]