

Abstract
Cellular entry of human immunodeficiency virus type 1 (HIV-1) involves fusion of viral and cellular membranes and is mediated by structural transitions in viral glycoprotein gp41. The antiviral C-peptide T20 targets the gp41 N-terminal heptad repeat region (N-HR), blocking gp41 conformational changes essential for the entry process. To probe the T20 structure-activity relationship, we engineered a molecular mimic of the entire gp41 N-HR coiled coil using the 5-Helix design strategy. T20 bound this artificial protein (denoted 5H-ex) with nanomolar affinity (KD = 30 nm), close to its IC50 concentration (∼3 nm) but much weaker than the affinity of a related inhibitory C-peptide C37 (KD = 0.0007 nm). T20/C37 competitive binding assays confirmed that T20 interacts with the hydrophobic groove on the surface of the N-HR coiled coil outside of a deep pocket region crucial for C37 binding. We used 5H-ex to investigate how the T20 N and C termini contributed to the inhibitor binding activity. Mutating three aromatic residues at the T20 C terminus (WNWF → ANAA) had no effect on affinity, suggesting that these amino acids do not participate in T20 binding to the gp41 N-HR. The results support recent evidence pointing to a different role for these residues in T20 inhibition (Peisajovich, S. G., Gallo, S. A., Blumenthal, R., and Shai, Y. (2003) J. Biol. Chem. 278, 21012–21017; Liu, S., Jing, W., Cheung, B., Lu, H., Sun, J., Yan, X., Niu, J., Farmar, J., Wu, S., and Jiang, S. (2007) J. Biol. Chem. 282, 9612–9620). By contrast, mutations near the T20 N terminus substantially influenced inhibitor binding strength. When Ile was substituted for Thr in the second T20 position, a 40-fold increase in binding affinity was measured (KD = 0.75 nm). The effect of this affinity enhancement on T20 inhibitory potency varied among different viral strains. The original T20 and the higher affinity T20 variant had similar potency against wild type HIV-1. However, the higher affinity T20 variant was significantly more potent against T20-resistant virus. The findings suggest that other factors in addition to binding affinity play a role in limiting T20 potency. As a mimetic of the complete gp41 N-HR coiled coil region, 5H-ex will be a useful tool to further elucidate mechanistic profiles of C-peptide inhibitors.
The HIV-12 surface glycoprotein Env promotes viral entry through the fusion of viral and cellular membranes (3). Env consists of three gp120 surface subunits and three gp41 transmembrane subunits arranged as a trimer-of-heterodimers on the virion surface. In the current model of HIV-1 entry, cellular receptor binding to gp120 initiates a series of coordinated structural transformations that stimulate gp41 to extend and insert its N-terminal fusion peptide into target cell membranes (see Fig. 1A) (4, 5). This high energy extended intermediate structure ultimately collapses into a trimer-of-hairpins conformation that juxtaposes the gp41 fusion peptide and transmembrane domain. Because the fusion peptide and transmembrane domain are inserted in target cell and viral membranes, formation of the trimer-of-hairpins is proposed to bring these membranes into the close proximity required for efficient fusion.
FIGURE 1.

HIV-1 gp41 and its role in viral membrane fusion. A, a model of HIV-1 entry (46). In native Env prior to receptor activation, gp41 is held in a metastable conformation by a canopy of gp120 proteins (green). Receptor binding to gp120 stimulates gp41 to extend and insert its fusion peptide segment (red) into the target cell membrane. The N-HR (gray) and C-HR (blue) regions of the gp41 ectodomain are transiently exposed in this prehairpin state. Subsequently, gp41 collapses into the trimer-of-hairpins conformation that brings the gp41 fusion peptides, transmembrane regions (purple), and their associated membranes into the close proximity for membrane fusion. The actual disposition of gp120 in both the prehairpin and trimer-of-hairpins states is uncertain; for clarity, the protein is omitted in the schematic of the trimer-of-hairpins conformation. B, a diagram of HIV-1 gp41 identifying its fusion peptide (FP), N-HR, C-HR, MPER (MP), transmembrane (TM), and cytoplasmic (cyto) domains. Amino acid sequences above and below the diagram are derived from the N-HR and C-HR/MPER regions of EnvHXB2; all but the MPER sequence WNWF (magenta) were used in the design of 5H-ex. The N-HR and C-HR segments found in the original 5-Helix are boxed in gray and blue, respectively, whereas the sequences of C37 and T20 are denoted by lines. The side chains of the C-HR amino acids marked with + pack into the hydrophobic pocket at the C terminus of the N-HR coiled coil.
The core of the trimer-of-hairpins is a bundle of six α-helices formed by two hydrophobic heptad repeat sequences in the N- and C-terminal regions of the gp41 ectodomain (N-HR and C-HR, respectively) (6, 7). In the trimer-of-hairpins, the N-HR segments from three gp41 ectodomains form a central trimeric coiled coil, around which the three C-HR segments pack as antiparallel helices into hydrophobic grooves (8–11). In the prehairpin extended conformation, the N-HR and C-HR segments are unassociated and transiently accessible to inhibitors of HIV-1 entry (5, 12). Several such inhibitors are formed from the peptide sequence of the C-HR and adjacent gp41 regions (4, 6, 13, 14). Denoted C-peptides, they work in a dominant negative fashion by binding to the exposed N-HR coiled coil, thereby blocking trimer-of-hairpins formation and inhibiting viral membrane fusion (4, 15–21). One C-peptide, T20 (also called enfuvirtide), has shown antiviral activity in vivo and has been approved for use in the treatment of HIV-1 infection (22, 23).
T20 is a 36-amino acid peptide extending from Tyr638 in the middle of the C-HR to Phe673 in the Trp-rich membrane proximal external region (MPER) that precedes the gp41 transmembrane domain (residue numbering is according to the EnvHXB2 sequence; see Fig. 1B) (13). In T20, these C-terminal MPER-derived residues are critical for inhibitory activity, although their structure and function in the gp41-bound state are currently unknown (1, 24, 25). A second class of similarly potent C-peptides includes C34 (residues 628–661) and the slightly larger C37 (residues 625–661) (4, 6, 26, 27). These peptides are derived entirely from the C-HR sequence and thus are shifted in the N-terminal direction compared with T20 (Fig. 1B). The interactions of C34 and C37 with gp41 are greatly stabilized by residues Trp628, Trp631, and Ile635 near the C-HR N terminus (4). Their bulky hydrophobic side chains pack into a deep hydrophobic pocket on the surface of the N-HR coiled coil. T20 lacks these pocket binding residues and their stabilizing effect. However, T20 does contain bulky hydrophobic residues (Trp670, Trp672, and Phe673) at its C terminus that might pack into a similar pocket at the other end of the N-HR coiled coil.
High resolution structures of the gp41 trimer-of-hairpins have aided our understanding of the mechanism of C-peptide inhibition. These structures have enabled the design of polypeptides that mimic the gp41 N-HR coiled coil and bind C34/C37, thereby providing a tool to probe the structure-activity relationships of the inhibitors (26, 28–30). No similar tool is available for investigating T20 inhibition in detail. The structures of the gp41 trimer-of-hairpins do not include the T20 C terminus (9 residues) nor the gp41 N-terminal segments that putatively interact with it. Furthermore, gp41 N-HR-derived peptides predicted to interact with T20 are poorly soluble and difficult to use in solution phase interaction assays (6). Here we describe the design of a soluble protein (denoted 5H-ex) that mimics the putative T20-binding site on the N-HR coiled coil. 5H-ex interacts with T20 with an equilibrium dissociation constant (KD) of 30 nm, close to the T20 50% inhibitory concentration (IC50) of 3 nm. Using this protein, we explored the extent to which the N and C termini of T20 contribute to its binding activity. First, we showed that the MPER-derived residues at the peptide C terminus do not stabilize the 5H-ex/T20 interaction. Second, we identified an N-terminal substitution that significantly enhanced T20 binding affinity and improved peptide inhibitory activity against T20-resistant HIV-1. The results suggest that T20 binding to the N-HR coiled coil is stabilized primarily by residues derived from the C-HR and not the MPER. 5H-ex is likely to be a useful tool in probing the structure-activity relationship of T20.
EXPERIMENTAL PROCEDURES
Design and Purification of Extended 5-Helix Variants—The T20-binding protein 5H-ex was designed as an extended variant of the engineered HIV-1 entry inhibitor 5-Helix (26). The original 5-Helix contained three N-HR segments of 40 residues (HIV-1HXB2 residues Gln543 to Ala582) and two C-HR segments of 38 residues (His625 to Glu662) alternately connected using 5-amino acid Gly/Ser linkers. In 5H-ex, the N-HR segments were extended at their N terminus by 13 amino acids (530MGAASMTLTVQAR542), and the C-HR segments were extended at their C terminus by 7 amino acids (663LDK-WASL669). To accommodate the disparity in segment lengths, the linkers connecting each C-HR segment to the following N-HR segment were lengthened to 9 amino acids (GSSGGSGSG). In the slightly truncated 5H-ex variant (denoted 5H-exΔ5), each N-HR segment was shortened by 5 amino acids at its N terminus. In the mutant 5H-ex variant with altered C37 affinity, W571R and G572D substitutions were introduced into the third N-HR segment to disrupt the hydrophobic pocket at the C terminus of the N-HR coiled coil. All of the proteins contained a C-terminal hexahistidine tag to facilitate purification. Genes encoding each of these proteins were assembled by a combination of overlap extension PCR and QuikChange site-directed mutagenesis (Stratagene). The constructs were cloned into the pEAD4 vector (31) and confirmed by sequencing the entire open reading frame.
5-Helix was produced as previously described (26). 5H-ex proteins were recombinantly expressed in Escherichia coli (strain RP3098) grown in 2× YT broth at 37 °C and induced with isopropyl-β-d-thiogalactopyranoside (0.4 mm). After 3 h, bacterial pellets were collected by low speed centrifugation and resuspended in TBS (50 mm Tris, pH 8, 100 mm NaCl) supplemented with 4 m guanidine hydrochloride. The bacterial resuspensions were lysed by sonication and clarified by high speed centrifugation. Protein was purified by metal affinity chromatography (nickel-nitrilotriacetic acid-agarose; Qiagen) in solutions of TBS, 4 m guanidine hydrochloride and serially dialyzed, first into TBS with 6 m urea and then into TBS with 500 mm arginine. The final dialyzed sample was further purified on a Superdex 75 column (GE) running TBS, 500 mm Arg. 5H-ex protein eluted at the monomeric molecular weight was assessed to be >95% pure by SDS-PAGE. The samples were stored diluted (∼5 μm) in TBS, 500 mm Arg at 4 °C and concentrated up to 60 μm using a Centricon 10 (Millipore) immediately prior to use. Concentration was determined by absorbance at 280 nm using the method of Edelhoch (32).
C-peptide Production—T20 and its variants (T20YI, T20NYT, T20NYI, and f-T20) were synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry by the peptide synthesis facility at the Kimmel Cancer Center, Thomas Jefferson University. Cleaved, desalted peptides were purified to homogeneity by reverse phase high pressure liquid chromatography (HPLC) using a Vydac C-18 column and a linear gradient of acetonitrile in water containing 0.1% trifluoroacetic acid. Absorbance at 280 nm was used to ascertain the concentration of all T20 variants except f-T20. The f-T20 peptide contained a fluorescein label (Molecular Probes) at its N terminus; its concentration was determined in 50 mm potassium phosphate (pH 9) using an extinction coefficient of 87,500 m-1 cm-1 at 490 nm (Molecular Probes).
Rhodamine-labeled C37 (R-C37) and its variants (R-C37N656D, R-C37N656S, and R-C37T639I) were produced as previously described (33). Briefly, a C37 peptide containing an N-terminal Cys residue (Cys-C37-H6) was generated by proteolysis of a recombinantly expressed trimer-of-hairpins construct (CGG-NC1). The peptide was fluorescently labeled using rhodamine-5-maleimide (Molecular Probes) and purified to homogeneity using reverse phase HPLC. R-C37 concentration was determined in methanol using an extinction coefficient of 95,000 m-1 cm-1 at 520 nm (Molecular Probes). The identities of all T20 and C37 peptides were confirmed using surface-enhanced laser desorption/ionization-time-of-flight mass spectrometry (Bio-Rad).
Production of MBP-T20 and MBP-C37 Fusion Proteins—Genes encoding MBP-T20 and MBP-C37 (27) in the pET-14b vector were kindly provided by Dr. Michael S. Kay (University of Utah). In these fusion proteins, the C-peptide sequence is attached to the C terminus of MBP (maltose-binding protein) through a variable linker (SSSENLYFQGS for MBP-T20 and SSSSSGG for MBP-C37). The proteins also contain a C-terminal hexahistidine tag. Constructs encoding MBP-T20 mutant variants described in the text were generated using QuikChange site-directed mutagenesis (Stratagene) and confirmed by sequencing. These fusion proteins were recombinantly expressed in E. coli (strain RP3098) and purified by metal affinity chromatography and gel filtration (Sephacryl S200 HR, GE). Purified protein (>95% pure by SDS-PAGE analysis) was dialyzed into water and stored lyophilized at -20 °C. The protein was resuspended in TBS, and its concentration was determined using absorbance at 280 nm.
Interaction Assays—The reported KD values were measured for the interaction of C-peptides and their binding proteins in solution. The binding parameter was determined by sampling the free (unbound) concentration of R-C37 or f-T20 in reaction mixtures using a Kinexa 3000 flow fluorimeter (Sapidyne Instruments). As reaction mixtures were passed through the instrument's optical flow cell, a minute fraction of the total unbound C-peptide was captured by a small amount (∼5 μl) of chromatography beads coated with 5-Helix or 5H-ex. The resulting increase in bead fluorescence (Δf) was directly proportional to the concentration of unbound C-peptide in the reaction mixture.
The beads were prepared as previously described (33). Briefly, 5-Helix or 5H-ex (100 μg) was incubated with activated azlactone (50 mg; Pierce) in 50 mm sodium carbonate (pH 9.5, 1 ml) at 4 °C overnight. Nonspecific binding sites were subsequently blocked using bovine serum albumin (10 mg) in 1 m Tris (pH 8.0, 1 ml) at 20 °C for 1 h. Before use, the bead mixture was diluted into 35 ml of 10TBS (10 mm Tris, pH 8, 100 mm NaCl) containing 0.02% NaN3. For each sample, ∼0.5 ml of the diluted bead mixture was passed through the flow cell capillary, and a small bead pack was retained on a 20-μm screen. All of the bead preparations were tested for linear responses to both sample volume and fluorescent C-peptide concentration to ensure that the fluorescent measurement accurately reflected the free C-peptide concentration in reaction mixtures.
Interaction experiments were carried out at 25 °C in solutions of 10TBS containing 100 μg/ml bovine serum albumin, 0.02% NaN3, and 1 mm phenylmethylsulfonyl fluoride. For direct binding measurements, 5-Helix or 5H-ex was titrated into a fixed concentration of R-C37 or f-T20 and incubated for 4–72 h (empirically determined to ensure that equilibrium had been achieved). Each sample was run through the Kinexa 3000 in duplicate, and concentration dependence to Δf was fit to a general bimolecular equilibrium binding model using the manufacturer's software (Sapidyne Instruments).
For C37 displacement assays, MBP-T20 was titrated into solutions containing 1 nm R-C37N656D and 10 nm 5H-ex (or 5-Helix). Each experiment also included the following two control solutions: 1) 1 nm R-C37N656D alone and 2) 1 nm R-C37N656D and 1 μm 5-Helix (enough 5-Helix to chelate 99.9% of the R-C37N656D). The unbound R-C37N656D in each reaction mixture was sampled twice in the Kinexa 3000 flow cell using 5-Helix-coated beads. The fraction of unbound R-C37N656D was determined by normalizing the measured Δf by the maximum and minimum Δf values obtained from the two control solutions. The dependence of free R-C37N656D on MBP-T20 concentration was fit to an exact expression for three-state competitive binding (34) using a least squares algorithm implemented in Visual Basic. The output of the algorithm (MBP-T20 KD value) required input of the R-C37N656D KD value, determined through direct binding experiments to be 1.6 nm. This value is consistent with the ∼15% free R-C37N656D observed in the absence of MBP-T20 in each experiment (see Fig. 3). Importantly, the presence of MBP-T20 at high concentrations did not perturb the efficiency of R-C37N656D capture by 5-Helix-coated beads. This observation was made for each C37 displacement assay by showing that a control solution containing 1 nm R-C37N656D and MBP-T20 at its highest concentration generated a fluorescence signal within 10% of the maximum Δf (data not shown).
FIGURE 3.

Properties of the 5H-ex/T20 interaction. A, overview of the C37 displacement assay. T20 and R-C37 compete for the same binding site on 5H-ex, and therefore titration of T20 (or MBP-T20) leads to an increase in the free (unbound) concentration of R-C37. The circled R denotes the rhodamine label linked to C37. B, Kinexa 3000 fluorescence response as solutions of 5H-ex (10 nm), R-C37N656D (1 nm), and MBP-T20 (indicated concentration) were passed over beads that specifically captured unbound R-C37N656D. The dashed and dotted lines were generated by control samples in which R-C37N656D is fully bound or fully unbound, respectively (see “Experimental Procedures”). The fraction of unbound R-C37N656D in each sample was calculated by normalizing the difference fluorescence signal Δf to the minimum and maximum Δf obtained from the control solutions. C, C37N656D displacement from 5H-ex (triangles) or 5-Helix (circles) as T20 (open symbols) or MBP-T20 (filled symbols) was titrated into reaction mixtures. The data points represent the means ± S.E. of at least three separate measurements and have been fit to a three-state competitive binding model (solid lines). D, tabulated KD values for the interaction of T20 with either 5-Helix or 5H-ex as determined using the direct binding assay (f-T20) or the C37 displacement assay (T20, MBP-T20). The values represent the means and S.E. of three or more separate measurements. E, effect of the W571R/G572D substitution in the N-HR hydrophobic pocket on C-peptide binding affinities. The data are represented as fold change in KD value relative to the wild type 5H-ex/C-peptide KD. The KD values were determined using the direct binding assay for C37 values or the C37 displacement assays for MBP-T20 values.
For C37 displacement assays using the 5H-ex double mutant variant W572R-G572D, the higher affinity C-peptide R-C37T639I was substituted for R-C37N656D. The R-C37T639I/mutant 5H-ex KD value was determined to be 1.2 nm by direct binding measurements. For C37 displacement assays using MBP-T20NYI, the higher affinity R-C37N656S was likewise substituted for R-C37N656D. The R-C37N656S/5H-ex KD value was determined to be 0.024 nm. Finally, for C37 displacement assays using T20 peptide, the maximum Δf values were determined for each T20 concentration using control solutions containing R-C37N656D and T20 peptides. This protocol change was implemented because T20 peptide in excess of 125 nm was observed to reduce the maximum fluorescent signal through a nonspecific mechanism.
Inhibition Experiments—Antiviral activities of peptides and proteins were measured in cell-cell fusion and viral infectivity assays that have been described in detail previously (4, 33). For cell-cell fusion assays, EnvHXB2 and TAT-expressing Chinese hamster ovary cells (clone 7d2, kindly provided by M. Krieger, Massachusetts Institute of Technology) were cocultured with HeLa-CD4-LTR-β-Gal cells (M. Emerman, obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health) in the presence of varying concentrations of inhibitor for 15 h at 37 °C. Multinucleated syncytia were identified by staining with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal; Sigma) and manually scored. For the infectivity assay, virions pseudotyped with EnvHXB2 were generated by cotransfection of the Env-deficient HIV-1NL4-3 genome (pNL4-3E-V-Luc+) and an Env-expressing plasmid (pEBB_HXB2) into 293T cells. HIV-1 harvested 48 h post-transfection was used to infect HOS-LES cells (N. Landau, obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health) in the presence of varying inhibitor concentrations. The level of viral infectivity was measured 48 h later by assaying for luciferase production in infected cells (luciferase assay system; Promega).
IC50 values were determined by fitting the normalized syncytia number or luciferase activity (F) to inhibitor concentration ([Inh]) using the Langmuir equation: F = (1 + [Inh]/IC50)-1. For viral infectivity experiments performed in the presence of T20 and either 5-helix or 5H-ex, combination indices (CI) were determined to asses synergistic, additive, or antagonistic behavior of inhibitor pairs (35, 36). Using mixtures of T20 and 5-Helix as an example, the CI is defined for inhibitors that act in a mutually exclusive manner as shown in the following equation,
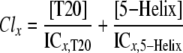 |
(Eq. 1) |
Here x is the percentage of inhibition (0 < x < 100) observed in a mixture of T20 at concentration [T20] and 5-Helix at concentration [5-Helix]. ICx, T20 is the T20 concentration required to achieve x percent inhibition in the absence of 5-Helix, and ICx, 5-Helix is the 5-Helix concentration required to achieve x percent inhibition in the absence of T20. Synergistic activity leads to CI values less than 1, whereas antagonistic behavior leads to CI values greater than 1. A CI value equal to 1 indicates that the activities are additive. The average CI values shown in Fig. 6D were calculated from various levels of inhibition observed using different mixtures of T20 and 5-Helix or 5H-ex.
FIGURE 6.

Inhibitory properties of 5H-ex and 5-Helix. A and B, Titration of HIV-1 infectivity by 5H-ex (A) or 5-Helix (B) in the absence (open circles) or presence of 100 nm MBP-T20 (filled circles) or 100 nm MBP-C37 (squares). The data represent the mean ± S.E. from three or more separate experiments. The dotted line in both panels is a fit of the 5H-ex data to the Langmuir equation (see “Experimental Procedures”). C, titration of HIV-1 infectivity by 5H-ex (circles) or 5-Helix (squares) in the presence of 3 nm T20. The solid line depicts the expected infection level if the mixed inhibitors bound gp41 in a mutually exclusive manner and their antiviral activities were additive: Fraction infection = (1 + ([T20]/IC50, T20) + ([5H]/IC50, 5H))-1. D, tabulated inhibitory parameters for 5H-ex and 5-Helix. IC50 values are reported for cell-cell fusion (Cell) and HIV-1 infectivity (Virus) assays and represent the means and S.E. of three or more independent experiments. The CI determined for multiple mixtures of T20 and 5-Helix or 5H-ex was calculated as described under “Experimental Procedures.”
RESULTS
Design of an Extended 5-Helix Variant—The design of the T20-binding protein 5H-ex followed the strategy utilized to develop the HIV-1 entry inhibitor 5-Helix (26). 5-Helix contains three peptide segments derived from the N-HR and two peptide segments derived from the C-HR alternately connected (N-C-N-C-N) through short Gly-Ser linkers to form a single polypeptide. The alternating connectivity enables the N-HR and C-HR segments to pack in the antiparallel fashion observed in the trimer-of-hairpins structure. 5-Helix adopts a conformation that resembles the helical core of the gp41 trimer-of-hairpins except that one C-HR segment is absent (5, 37). This vacancy creates a single C-peptide-binding site that interacts fully with the C37 sequence but only partially with the shifted T20 sequence (Fig. 2A). Consequently, 5-Helix binds strongly to C37 (KD = 0.00065 nm) but relatively poorly to T20 (KD ∼ 850 nm; Fig. 2, B and C). T20 lacks the 13 N-terminal residues of C37 that include the amino acids Trp628, Trp631, and Ile635, the side chains of which pack into the deep hydrophobic pocket at the C terminus of the N-HR coiled coil. The data suggest that this sequence accounts for approximately half of the free energy that stabilizes C37 binding.3
FIGURE 2.

C-peptide binding properties of 5-Helix and 5H-ex. A and D, schematic diagrams of 5-Helix (A) and 5H-ex (D). The N-HR (dark gray) and C-HR (light gray) segments are shown as cylinders, whereas the Gly/Ser linkers are represented by black lines. The black oval in the C-peptide binding site indicates the location of the deep hydrophobic pocket at the N-HR C terminus. The cylinders labeled C37 and T20 show the extent of the binding site of each C-peptide. The N and C designate the N and C termini for each polypeptide. B and E, fluorescence response as equilibrated mixtures of f-T20 (20 nm), and either 5-Helix (B) or 5H-ex (E) were washed through the Kinexa 3000 flow fluorimeter. The instrument flow cell contained beads that specifically captured free (unbound) f-T20. The difference in fluorescence signals (Δf) measured before sample load (0 ml) and after the sample washout (2 ml) was proportional to the free f-T20 concentration in reaction mixtures (see “Experimental Procedures”). C and F, titrations of fluorescent C37 (2.5 pm) and T20 (20 pm) with either 5-Helix or 5H-ex. In C, R-C37 (squares) and f-T20 (circles) were titrated by 5-Helix, whereas in F, f-T20 was titrated by either 5-Helix (circles) or 5H-ex (triangles). The fraction of unbound C-peptide was calculated by normalizing each Δf to a minimum and maximum Δf obtained from samples in which the C-peptide is fully bound or fully unbound. The KD values were determined by fitting these data to a bimolecular equilibrium binding model (solid lines).
Three properties of the original 5-Helix protein made it an attractive scaffold for further engineering. First, 5-Helix is extremely stable to thermal and chemical denaturation (TM > 100 °C, C½ > 7 m guanidine hydrochloride at 20 °C) (26). We hypothesized that such rigidity would assist in stabilizing extensions of N-HR and C-HR segments that otherwise may be highly flexible in the context of unlinked peptides. Second, 5-Helix is much more soluble than N-HR peptides of similar length because of the presence of two attached C-HR segments (6). We reasoned that this enhanced solubility would reduce the tendency of the hydrophobic extensions to promote aggregation. Third, C-peptide binding to 5-Helix is a simple bimolecular interaction that is much less complicated to analyze than the assembly of a hexameric complex formed by N-HR and C-HR peptides.
In engineering a 5-Helix-like T20-binding protein, we assumed that the helical structures of N-HR and C-HR segments extend through the T20-binding site, although no structural data exists to support this conjecture. In 5H-ex, each N-HR segment was lengthened by 13 amino acid residues at its N terminus (Fig. 2D). Initially, we attempted to stabilize this N-HR extension by adding 11 amino acid residues to the C terminus of each C-HR segment so that its sequence would extend as far as the T20 sequence. However, this engineered protein was insoluble in aqueous solutions. The aggregation problem was linked to the last four residues of each extended C-HR segment: Trp670, Asn671, Trp672, and Phe673, collectively denoted WNWF. Substituting the WNWF sequence with Gly-Ser-Gly-Ser produced a soluble, well behaved protein with the stable helical structure expected from the design (supplemental Fig. S1). In the final version of 5H-ex, each N-HR segment extended from Met530 to Ala581, and each C-HR segment extended from His625 to Leu669.
Binding Properties of 5H-ex—When correctly folded, 5H-ex should contain an intact C37-binding site as well as the putative T20 interaction sequence. 5H-ex had the same binding affinities for C37 and two C37 variants (N656S (KD = 0.024 nm) and N656D (KD = 1.6 nm)) as the original 5-Helix (not shown), confirming the proper fold of the extended molecule. In a solution phase interaction assay, 5H-ex bound fluorescein-tagged T20 (f-T20) with a KD value of 32 nm, an ∼30-fold improvement over the 5-Helix/T20 affinity (Fig. 2, E and F). The KD value is significantly closer to the observed T20 IC50 (∼3 nm), but the T20 affinity is still more than 4 orders of magnitude weaker than the C37 affinity (see “Discussion”).
T20 binding 5H-ex is sufficient to displace the lower affinity C37 variant N656D from its binding site (Fig. 3, A–C). Titrating either free T20 peptide or a maltose-binding protein-T20 fusion (MBP-T20, kindly provided by Dr. Michael Kay, University of Utah) into a solution of prebound 5H-ex/C37N656D complexes led to increasing amounts of free (unbound) C37N656D. By comparison, MBP-T20 did not significantly displace C37N656D bound to 5-Helix, consistent with the much lower affinity of T20 for this original construct (Fig. 3C). Quantitative characterization of C37N656D displacement from 5H-ex using a three state competitive binding model yielded KD values for T20 and MBP-T20 of ∼30 nm, in close agreement with the affinity obtained using f-T20 in the direct-binding assay (Fig. 3D).
To confirm the predicted binding site for T20, we generated a 5H-ex variant with the double mutation W571R/G572D. Because Trp571 and Gly572 line the hydrophobic pocket at the C terminus of the N-HR coiled coil, the dual substitution should significantly destabilize C37 interaction but have only a minor effect on T20 binding. Consistent with this prediction, C37 affinity for the mutant 5H-ex variant was reduced by 4 orders of magnitude (KD = 7 nm). By contrast, the dual substitution did not significantly alter the KD value for T20 binding to 5H-ex (KD = 49 nm) (Fig. 3E). Together, the binding and displacement data imply that T20 adopts the predicted mode of interaction with the N-HR coiled coil, packing into the same hydrophobic groove as C37 but outside of the pocket region.
Probing the T20 Structure-Activity Relationship: Contributions of the C-terminal CHR/MPER Sequence to T20 Binding—The increased stability of the T20/5H-ex complex (compared with T20/5-Helix) must be due to additional interactions of the C-peptide C terminus with the N-HR N terminus. We initially focused on the last four residues of T20: the WNWF sequence known to play a critical role in HIV-1 inhibition. Substituting Ala residues for the three aromatic amino acids (WNWF → ANAA) functionally ablates the potent antiviral activity of the peptide (1, 24, 25). However, recent studies suggest that the WNWF sequence may not directly bind the gp41 N-HR (38). To test this possibility, we investigated how the WNWF sequence contributes to the interaction between T20 and 5H-ex in two ways. First, we generated the mutant MBP-T20ANAA variant with Ala residues in place of the aromatic amino acids. Second, we produced a slightly truncated 5H-ex variant (5H-exΔ5) that lacked the residues modeled to interact with the WNWF sequence (Met530, Gly531, Ala532, Ala533, and Ser534 in each of the three N-HR segments; Fig. 4A). C37N656D displacement assays were used to measure the binding affinities of the following two interactions: 1) MBP-T20ANAA and full-length 5H-ex and 2) MBP-T20 and the truncated 5H-exΔ5. In both cases, the C37N656D displacement curves were indistinguishable from results obtained using MBP-T20 and the full-length 5H-ex (Fig. 4B). The data indicated that the three interacting molecular pairs had the same KD value, implying that the WNWF sequence does not contribute to the stability of the T20/5H-ex complex. The results corroborated recent findings that demonstrated minimal destabilization of T20/N-HR peptide complexes when amino acids near the T20 C terminus were replaced with their enantiomers (38). These observations suggest that the WNWF sequence of T20 is involved in interactions outside of the gp41 N-HR region during inhibition of HIV-1 entry (see “Discussion”).
FIGURE 4.

Role of the C-terminal C-HR/MPER sequence in stabilizing T20 binding to 5H-ex. A, schematic representations of 5-Helix, 5H-ex, and the slightly truncated 5H-exΔ5. The extent of the C-peptide binding site of each protein is indicated relative to T20 and T20ANAA peptides. B, C37N656D displacement measured using 5H-ex and MBP-T20ANAA (open circles), 5H-ex and MBP-T20 (filled circles), 5H-exΔ5 and MBP-T20 (open squares), and 5-Helix and MBP-T20 (filled diamonds). The data represent the means and S.E. of three or more independent experiments and were analyzed as described in the legend to Fig. 3. C, effect of C-peptide point mutations on the stability of the T20/5H-ex complex. Binding free energies were calculated using the formula ΔGo = -RTln(KD), where the KD values were obtained using the C37N656D displacement assay with MBP-T20 constructs as titrants. The horizontal lines indicate the binding energies of wild type MBP-T20 with either 5H-ex (solid) or 5-Helix (dotted).
Because the WNWF segment did not appear to interact with the N-HR, we refocused our attention on the adjacent eight residues at the C-HR/MPER junction (662ELDKWASL). Using scanning mutagenesis, we confirmed a role for this segment in stabilizing the T20/5H-ex interaction. Alanine substitution for Leu663, Asp664, and Trp666 and glutamine substitution for Ala667 significantly decreased free energy of binding (Fig. 4C). The greatest effect was seen for the mutation at Leu663, a residue previously predicted to lie within the hydrophobic N-HR/C-HR interface (8). Three and four residues down from Leu663, Trp666 and Ala667 would be expected to make contact with the N-HR if the C-HR helical register were maintained. Interestingly, glycine substitution at Ala667 had almost no impact on T20 binding affinity (KD = 35 nm). Taken together, the consequences of mutations at Ala667 suggest that the residue packs within the N-HR/C-HR interface in a manner that cannot accommodate the added bulk of a glutamine side chain. Finally, a putative helical structure would also position Asp664 in close proximity to Arg542 of the N-HR; the small destabilizing effect of the D664A mutation might reflect disruption of this potential electrostatic interaction. Together, the pattern of destabilization strongly suggests that the C-HR α-helix extends into this region at least through Ala667.
Probing the T20 Structure-Activity Relationship: Affinity Enhancement through the T639I Substitution—We have previously shown that C37 binding to 5-Helix is exquisitely sensitive to mutations in the C-HR locus formed by amino acids Asn637, Tyr638, and Thr639.4 Specifically, a C37 peptide variant with the T639I substitution was found to bind 5-Helix with ∼9-fold higher affinity (KD ∼ 70 fM). The T20 sequence begins at Tyr638 and thus does not include Asn637 at the N terminus of the tripeptide segment. To investigate the effect of this C-HR locus on T20 affinity, we first elongated the peptide sequence to include Asn637. This new MBP fusion variant, denoted MBP-T20NYT, possessed the same activity in the C37 displacement assay as the original MBP-T20, indicating that the additional Asn residue did not significantly change binding affinity (Fig. 5A). However, substituting Ile for Thr639 created an MBP construct (denoted MBP-T20NYI) that was able to displace C37N656D fully from 5H-ex at extremely low concentrations (Fig. 5A). The T639I variant also showed significantly greater activity when the higher affinity C37N656S peptide (KD = 0.024 nm) was used in place of C37N656D in the displacement assay (Fig. 5B). The KD value of T20NYI is 0.75 nm, indicating that the T639I substitution increased T20 peptide affinity 40-fold.
FIGURE 5.

Effect of the T639I substitution on T20 binding and inhibitory activities. A, C37N656D displacement from 5H-ex by MBP-T20 (squares), MBP-T20NYT (triangles), and MBP-T20NYI (circles). The data were obtained and analyzed as described in the legend to Fig. 3. B, as in A, except that the higher affinity C37N656S was used in place of C37N656D. Note that only the MBP-T20NYT (triangles) and MBP-T20NYI (circles) titrations are shown. C, titration of viral infectivity by T20 (squares) and T20YI containing the T639I substitution (circles). HIV-1 was pseudotyped with either wild type EnvHXB2 (filled symbols) or its mutant variant containing T20 escape mutations G547D/I548T (open symbols). The data represent the means ± S.E. of three to seven separate experiments and have been fit to a Langmuir function (solid lines) to determine IC50 (see “Experimental Procedures”). D, C-peptide IC50 values determined for the inhibition of HIV-1 psuedotyped with wild type Env (gray) or the G547D/I548T mutant variant (black). Inset, IC50 values for inhibition of wild type HIV-1 are plotted on an expanded scale. E, table of C-peptide IC50 and KD values. Res refers to the T20-resistant Env containing the G547D/I548T substitutions. ND, not determined.
The observed enhancement in binding affinity caused by the T639I substitution did not translate into increased potency against wild type HIV-1HXB2 (Fig. 5, C and D). The IC50 values for T20 and the slightly elongated T20NYT (with the additional N-terminal Asn residue) were ∼3 nm, indistinguishable from the IC50 values of the corresponding peptides with the T639I substitution (T20YI and T20NYI, respectively). The absence of a correlation between IC50 and KD is similar to results obtained in studies of 5-Helix inhibition (33). The finding implies that T20 inhibitory potency against wild type Env is limited by factors independent of equilibrium binding affinity (e.g. association kinetics, see “Discussion”). However, T20YI and T20NYI were significantly more potent against T20-resistant HIV-1 (Fig. 5, C–E). The dual substitution G547D/I548T in the gp41 N-HR region led to a 100-fold reduction in T20 inhibitory potency that was partially overcome by the T639I substitution (39). Similar results were also obtained with a different T20-resistant Env variant, V549E (data not shown) (40). The data suggest that T20 inhibition of these resistant Env glycoproteins is at least partially dependent on peptide binding affinity.
Inhibitory Properties of 5H-ex—Like T20, 5-Helix and 5H-ex potently inhibit gp41-mediated membrane fusion in both cell-cell fusion and HIV-1 infectivity assays (Fig. 6). 5-Helix was previously shown to target the gp41 C-HR region and disrupt trimer-of-hairpins formation in a manner complementary to C-peptide inhibition (26). The inhibitory mechanism was established, in part, by demonstrating that 5-Helix and C37 mutually antagonized each other's inhibitory activity; the residues that mediate gp41 binding for each inhibitor were sequestered in the interface of associated 5-Helix/C37 complexes, thereby reducing the concentrations of active inhibitor in the fusion assays. To confirm that the extended 5H-ex also inhibited by targeting the C-terminal region of the gp41 ectodomain, we similarly tested for antagonism using MBP-C37 and MBP-T20 constructs. Because of the bulkiness of the attached MBP, neither C-peptide fusion protein significantly blocked viral entry at 100 nm (27). However, titration of 5H-ex into assays containing 100 nm of either MBP construct resulted in a significant reduction in 5H-ex potency (Fig. 6A). By contrast, 5-Helix inhibitory activity was only reduced in the presence of MBP-C37 (Fig. 6B). The presence of 100 nm MBP-T20 led to no significant change in 5-Helix potency, consistent with the low affinity of the 5-Helix/T20 interaction. Thus, we conclude that, despite its extra bulk (∼30% increase in mass and estimated length), 5H-ex can still target the C-terminal region of the gp41 ectodomain. In fact, the similarity in IC50 values for 5-Helix and 5H-ex suggests that these inhibitors encounter little steric hindrance when binding gp41 (see “Discussion”).
We next explored the antiviral activity in mixtures of T20 peptide and either 5-Helix or 5H-ex. Because 5-Helix poorly interacts with T20, we specifically asked whether these two inhibitors might display additive or synergistic antiviral activity. In the experiments shown in Fig. 6C, viral infectivity was measured in the presence of 3 nm T20 and 8, 40, or 200 nm 5-Helix. The level of inhibition correlated well with that expected if the two inhibitors acted additively by binding gp41 in a mutually exclusive manner (solid line). By comparison, considerably less inhibitory activity was observed for the same levels of T20 and 5H-ex, which can interact over this concentration range. Synergistic, additive, and antagonistic behaviors can be discriminated quantitatively using the combination index (CI, see “Experimental Procedures” for details) (35, 36). The average CI value determined for multiple T20 and 5-Helix concentrations was approximately 1, expected for additive inhibitory activities. The CI value greater than 2 for T20 and 5H-ex mixtures indicated antagonistic inhibitory activities and was consistent with lower active inhibitor concentrations due to 5H-ex/T20 binding.
DISCUSSION
In this study, we have utilized the 5-Helix design strategy to create a molecule that mimics the gp41 N-HR coiled coil region that interacts with the HIV-1 inhibitory peptide T20. The engineered 5H-ex protein binds T20 with a KD value of 30 nm, substantially larger than the C37 KD value of 650 fm. The comparatively low T20 affinity might signify that 5H-ex does not fully recapitulate the gp41 structure that binds T20. Recent studies have suggested that the gp41 MPER and fusion peptide regions can form a stable complex (41–43). Hence, T20 binding may be stabilized through an interaction of its WNWF sequence with residues from the gp41 fusion peptide. 5H-ex variants with N-HR segments extending further toward the gp41 N terminus are currently under development to test this possibility. Alternatively, other factors independent of the T20/gp41 interaction might stabilize peptide binding during HIV-1 membrane fusion. T20 has been shown to interact with lipid membranes much more avidly than peptides similar to C37 (1, 44). This enhanced membrane interaction appears to be linked to aromatic residues in the WNWF sequence that critically determines T20 inhibitory potency (1, 2, 44). The results suggest that T20 binding to gp41 might be additionally stabilized through interactions of the peptide tail with the target cell membrane. In this manner, T20 could inhibit at lower concentrations than it binds to 5H-ex, as observed in these studies.
Although T20 variants with the T639I substitution bound 5H-ex with significantly higher affinity, they did not inhibit wild type HIV-1 with higher potency. The poor correlation between binding affinity and inhibitory potency is similar to results obtained in a study of 5-Helix inhibition (33). Both T20 and 5-Helix target transient intermediate gp41 conformations during the fusion process. As a consequence, their inhibitory potencies are influenced by nonequilibrium parameters such as the lifetime of the sensitive intermediate state and the rate of inhibitor association (20, 21, 33, 45). These kinetic factors are the main determinants of 5-Helix antiviral activity (33), and, as a result, 5-Helix binding affinity can be altered by 4 orders of magnitude without significantly affecting the inhibitory potency. The similarity in IC50 values (2–3 nm) for T20, T20NYI, and even C37 despite the wide range of KD values (650 fM to 30 nm) is consistent with C-peptide inhibitory activity being restricted by kinetic factors.
The higher affinity T20 variants were found to be 4-fold more potent against T20-resistant Env. The results imply that resistance emerges at least partially through a mechanism of binding destabilization. However, the 4-fold improvement in inhibitory activity was small compared with the observed 40-fold enhancement in binding affinity. In fact, even C37, with its subpicomolar affinity, was unable to fully overcome the resistance phenotype (Fig. 6, D and E). These findings suggest that the mechanism of escape from C-peptide inhibition is more complex than mere affinity disruption. One possibility is that the N-HR mutations may reduce the rate of C-peptide association, thereby decreasing the amount of inhibitor able to bind during the lifetime of the sensitive intermediate state. Future interaction experiments using mutant 5H-ex variants might help to elucidate the full mechanism of C-peptide resistance.
The inhibitory activity of 5H-ex illuminates an important property of C-HR exposure. Compared with 5-Helix, 5H-ex targets a larger portion of gp41 with potentially higher affinity. However, the IC50 values for 5-Helix and 5H-ex are equal, implying that inhibition by 5H-ex is limited by the same kinetic factors that restrict 5-Helix potency. These kinetic factors lead to an inverse relationship between the IC50 value and the rate of inhibitor association (kon) (33). Any steric obstruction hindering access to the C-HR region would necessarily reduce the kon value for the bulkier inhibitor, thereby increasing its IC50. Hence, the identical potencies of 5-Helix and 5H-ex imply that the two inhibitors possess similar kon values. These findings support an access pathway to the C-terminal region of the gp41 ectodomain largely free of steric obstacles.
Supplementary Material
Acknowledgments
We thank M. Kay for kindly sharing his MBP-T20 and MBP-C37 expression constructs; K. Kahle and S. Paul for help in obtaining C37 and T20 inhibition data; R. M. Jones for assistance in setting up the KinExA binding assays; and M. Kay and members of the Root laboratory for helpful discussions and critical manuscript evaluations.
This work was supported, in whole or in part, by National Institutes of Health Grants 5-T32-DK0775 (to K. C.) and R01 GM66682 (to M. J. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: HIV-1, human immunodeficiency virus type 1; Env, HIV-1 envelope glycoprotein; N-HR, N-terminal heptad repeat sequence of the gp41 ectodomain; C-HR, C-terminal heptad repeat sequence of the gp41 ectodomain; R-C37, rhodamine-labeled C37; f-T20, fluorescein-labeled T20; TBS, Tris-buffered saline; MBP, maltose-binding protein; MPER, membrane proximal external region; HPLC, high pressure liquid chromatography; CI, combination index(ices).
Because ΔGo =-RTln(KD), these measured affinities translate roughly into binding energies of 16.8 kcal/mole for C37 and 8.4 kcal/mole for T20. Because the last 12 residues of T20 (not included in C37) do not make contact with 5-Helix, the difference in binding energies is primarily linked to the 13 amino-acid segment found at the N-terminus of C37.
H. K. Steger and M. J. Root, submitted for publication.
References
- 1.Peisajovich, S. G., Gallo, S. A., Blumenthal, R., and Shai, Y. (2003) J. Biol. Chem. 278 21012-21017 [DOI] [PubMed] [Google Scholar]
- 2.Liu, S., Jing, W., Cheung, B., Lu, H., Sun, J., Yan, X., Niu, J., Farmar, J., Wu, S., and Jiang, S. (2007) J. Biol. Chem. 282 9612-9620 [DOI] [PubMed] [Google Scholar]
- 3.Fenouillet, E., and Jones, I. M. (1995) J. Gen. Virol. 76 1509-1514 [DOI] [PubMed] [Google Scholar]
- 4.Chan, D. C., Chutkowski, C. T., and Kim, P. S. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 15613-15617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Root, M. J., and Hamer, D. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 5016-5021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu, M., Blacklow, S. C., and Kim, P. S. (1995) Nat. Struct. Biol. 2 1075-1082 [DOI] [PubMed] [Google Scholar]
- 7.Blacklow, S. C., Lu, M., and Kim, P. S. (1995) Biochemistry 34 14955-14962 [DOI] [PubMed] [Google Scholar]
- 8.Weissenhorn, W., Dessen, A., Harrison, S. C., Skehel, J. J., and Wiley, D. C. (1997) Nature 387 426-430 [DOI] [PubMed] [Google Scholar]
- 9.Chan, D. C., Fass, D., Berger, J. M., and Kim, P. S. (1997) Cell 89 263-273 [DOI] [PubMed] [Google Scholar]
- 10.Tan, K., Liu, J., Wang, J., Shen, S., and Lu, M. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 12303-12308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caffrey, M., Cai, M., Kaufman, J., Stahl, S. J., Wingfield, P. T., Covell, D. G., Gronenborn, A. M., and Clore, G. M. (1998) EMBO J. 17 4572-4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu, S., Wu, S., and Jiang, S. (2007) Curr. Pharm. Des. 13 143-162 [DOI] [PubMed] [Google Scholar]
- 13.Wild, C. T., Shugars, D. C., Greenwell, T. K., McDanal, C. B., and Matthews, T. J. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 9770-9774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang, S., Lin, K., Strick, N., and Neurath, A. R. (1993) Nature 365 113. [DOI] [PubMed] [Google Scholar]
- 15.Furuta, R. A., Wild, C. T., Weng, Y., and Weiss, C. D. (1998) Nat. Struct. Biol. 5 276-279 [DOI] [PubMed] [Google Scholar]
- 16.Munoz-Barroso, I., Durell, S., Sakaguchi, K., Appella, E., and Blumenthal, R. (1998) J. Cell Biol. 140 315-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilgore, N. R., Salzwedel, K., Reddick, M., Allaway, G. P., and Wild, C. T. (2003) J. Virol. 77 7669-7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilby, J. M., Hopkins, S., Venetta, T. M., DiMassimo, B., Cloud, G. A., Lee, J. Y., Alldredge, L., Hunter, E., Lambert, D., Bolognesi, D., Matthews, T., Johnson, M. R., Nowak, M. A., Shaw, G. M., and Saag, M. S. (1998) Nat. Med. 4 1302-1307 [DOI] [PubMed] [Google Scholar]
- 19.Melikyan, G. B., Markosyan, R. M., Hemmati, H., Delmedico, M. K., Lambert, D. M., and Cohen, F. S. (2000) J. Cell Biol. 151 413-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abrahamyan, L. G., Mkrtchyan, S. R., Binley, J., Lu, M., Melikyan, G. B., and Cohen, F. S. (2005) J. Virol. 79 106-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reeves, J. D., Gallo, S. A., Ahmad, N., Miamidian, J. L., Harvey, P. E., Sharron, M., Pohlmann, S., Sfakianos, J. N., Derdeyn, C. A., Blumenthal, R., Hunter, E., and Doms, R. W. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 16249-16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazzarin, A., Clotet, B., Cooper, D., Reynes, J., Arasteh, K., Nelson, M., Katlama, C., Stellbrink, H. J., Delfraissy, J. F., Lange, J., Huson, L., DeMasi, R., Wat, C., Delehanty, J., Drobnes, C., and Salgo, M. (2003) N. Engl. J. Med. 348 2186-2195 [DOI] [PubMed] [Google Scholar]
- 23.Lalezari, J. P., Henry, K., O'Hearn, M., Montaner, J. S., Piliero, P. J., Trottier, B., Walmsley, S., Cohen, C., Kuritzkes, D. R., Eron, J. J., Jr., Chung, J., DeMasi, R., Donatacci, L., Drobnes, C., Delehanty, J., and Salgo, M. (2003) N. Engl. J. Med. 348 2175-2185 [DOI] [PubMed] [Google Scholar]
- 24.Lawless, M. K., Barney, S., Guthrie, K. I., Bucy, T. B., Petteway, S. R., Jr., and Merutka, G. (1996) Biochemistry 35 13697-13708 [DOI] [PubMed] [Google Scholar]
- 25.Liu, S., Lu, H., Niu, J., Xu, Y., Wu, S., and Jiang, S. (2005) J. Biol. Chem. 280 11259-11273 [DOI] [PubMed] [Google Scholar]
- 26.Root, M. J., Kay, M. S., and Kim, P. S. (2001) Science 291 884-888 [DOI] [PubMed] [Google Scholar]
- 27.Hamburger, A. E., Kim, S., Welch, B. D., and Kay, M. S. (2005) J. Biol. Chem. 280 12567-12572 [DOI] [PubMed] [Google Scholar]
- 28.Eckert, D. M., and Kim, P. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 11187-11192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Louis, J. M., Bewley, C. A., and Clore, G. M. (2001) J. Biol. Chem. 276 29485-29489 [DOI] [PubMed] [Google Scholar]
- 30.Louis, J. M., Nesheiwat, I., Chang, L., Clore, G. M., and Bewley, C. A. (2003) J. Biol. Chem. 278 20278-20285 [DOI] [PubMed] [Google Scholar]
- 31.Doering, D. S., and Matsudaira, P. (1996) Biochemistry 35 12677-12685 [DOI] [PubMed] [Google Scholar]
- 32.Edelhoch, H. (1967) Biochemistry 6 1948-1954 [DOI] [PubMed] [Google Scholar]
- 33.Steger, H. K., and Root, M. J. (2006) J. Biol. Chem. 281 25813-25821 [DOI] [PubMed] [Google Scholar]
- 34.Wang, Z. X. (1995) FEBS Lett. 360 111-114 [DOI] [PubMed] [Google Scholar]
- 35.Tallarida, R. J. (2000) Drug Synergism and Dose-Effect Data Analysis, pp. 1-55, Chapman and Hall/CRC, London
- 36.Chou, T. C., and Talalay, P. (1984) Adv. Enzyme Regul. 22 27-55 [DOI] [PubMed] [Google Scholar]
- 37.Luftig, M. A., Mattu, M., Di Giovine, P., Geleziunas, R., Hrin, R., Barbato, G., Bianchi, E., Miller, M. D., Pessi, A., and Carfi, A. (2006) Nat. Struct. Mol. Biol. 13 740-747 [DOI] [PubMed] [Google Scholar]
- 38.Wexler-Cohen, Y., Johnson, B. T., Puri, A., Blumenthal, R., and Shai, Y. (2006) J. Biol. Chem. 281 9005-9010 [DOI] [PubMed] [Google Scholar]
- 39.Rimsky, L. T., Shugars, D. C., and Matthews, T. J. (1998) J. Virol. 72 986-993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nameki, D., Kodama, E., Ikeuchi, M., Mabuchi, N., Otaka, A., Tamamura, H., Ohno, M., Fujii, N., and Matsuoka, M. (2005) J. Virol. 79 764-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorizate, M., de la Arada, I., Huarte, N., Sanchez-Martinez, S., de la Torre, B. G., Andreu, D., Arrondo, J. L., and Nieva, J. L. (2006) Biochemistry 45 14337-14346 [DOI] [PubMed] [Google Scholar]
- 42.Lorizate, M., Gomara, M. J., de la Torre, B. G., Andreu, D., and Nieva, J. L. (2006) J. Mol. Biol. 360 45-55 [DOI] [PubMed] [Google Scholar]
- 43.Bellamy-McIntyre, A. K., Lay, C. S., Baar, S., Maerz, A. L., Talbo, G. H., Drummer, H. E., and Poumbourios, P. (2007) J. Biol. Chem. 282 23104-23116 [DOI] [PubMed] [Google Scholar]
- 44.Wexler-Cohen, Y., and Shai, Y. (2007) FASEB J. 21 3677-3684 [DOI] [PubMed] [Google Scholar]
- 45.Reeves, J. D., Miamidian, J. L., Biscone, M. J., Lee, F. H., Ahmad, N., Pierson, T. C., and Doms, R. W. (2004) J. Virol. 78 5476-5485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan, D. C., and Kim, P. S. (1998) Cell 93 681-684 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.