

Abstract
The export protein CRM1 is required for the nuclear export of a wide variety of cancer-related “cargo” proteins including p53, c-Abl, and FOXO-3A. Leptomycin B (LMB) is a highly specific inhibitor of CRM1 with significant in vitro potency, but limited in vivo efficacy due to toxicity. We now report a series of semi-synthetic LMB derivatives showing substantially improved therapeutic windows. Exposure of cancer cells to these compounds leads to a rapid and prolonged block of nuclear export, and apoptosis. In contrast to what is observed in cancer cells, these agents induce cell cycle arrest, but not apoptosis, in normal lung fibroblasts. These new nuclear export inhibitors (NEIs) maintain the high potency of LMB, are up to 16-fold better tolerated than LMB in vivo, and show significant efficacy in multiple mouse xenograft models. These NEIs demonstrate the potential of CRM1 inhibitors as novel and potent anticancer agents.
Keywords: CRM1, leptomycin B, nuclear export, p53, cancer
INTRODUCTION
Transport of macromolecules across the nuclear membrane is fundamental to the proper functioning of a living cell. For example, the ability to localize to the nucleus is essential for transcription factor activation, and spatial separation of proteins is commonly used as a mechanism for preventing spontaneous signal activation. Many important tumor suppressors and transcription factors protect cells by regulating cell growth and apoptosis, and their cytoplasmic localization can serve as an inactivation mechanism resulting in uncontrolled growth and the onset of disease (reviewed in (1)).
One strategy to prevent cytoplasmic localization of important transcription factors is to inhibit the proteins responsible for their nuclear export. The exportin CRM1 is absolutely required for the nuclear export of a wide variety of cancer-related “cargo” proteins including p53, c-Abl, and FOXO-3A (2). CRM1 recognizes its cargo proteins through their leucine-rich nuclear export sequence. The CRM1-cargo complex is then actively transported across the nuclear pore complex to the cytoplasm where the cargo is released after RanGAP-catalyzed GTP hydrolysis.
The role of CRM1 as an exportin was first elucidated through the use of a highly cytotoxic polyketide natural product known as leptomycin B (LMB) (2, 3). LMB was found to be very potent in vitro against various cancer cell lines (IC50 values in the 0.1 to 10 nM range). LMB’s potent activity stems from highly specific binding and inhibition of CRM1, thereby blocking CRM1-mediated protein export from the nucleus. LMB covalently inhibits CRM1 through a Michael addition of Cys528 onto LMB, and mutation of this cysteine residue prevents the covalent interaction and provides resistance to LMB (4, 5).
Based on its nanomolar potency against cancer cell lines, the in vivo activity of LMB was examined in a number of murine xenograft cancer models. It was found to show only modest efficacy (6). Despite its relatively narrow therapeutic window in mouse tumor models, a single Phase I trial of LMB was performed. Its clinical development was subsequently discontinued due to the significant toxicity observed without apparent efficacy (7).
Notwithstanding its initial failure in the clinic, LMB could serve as the paradigm for a novel class of cancer therapeutics. These compounds would derive their activity by preventing cytoplasmic localization and inactivation of important tumor suppressors that are dependent on CRM1 for nuclear export, such as p53. It has been estimated that roughly 50% of cancers maintain wild type p53 (8). In many of these cases, the tumor suppressor function is compromised by overexpression or inactivation of cellular factors that regulate the levels of p53 in the nucleus or lead to its enhanced export out of the nucleus (1). When p53 is activated in the nucleus, it can promote either cell-cycle arrest or apoptotic cell death depending on the environment and level of cell stress. p53 function is negatively regulated in part by an MDM2-dependent pathway that results in both nuclear export and ubiquitin-dependent degradation of p53. In many cancer cells, such as human papilloma virus (HPV)-positive cancers, aberrant cytoplasmic localization and/or degradation of p53 prevents the activation of pathways that would lead to cell death (9–11). Consequently, a means of re-localizing the anti-oncogenic wild type p53 to the nucleus in these aberrant cell types is a promising approach to regaining control of cell proliferation (12). In fact, previous work has demonstrated that treatment with LMB and actinomycin D leads to the accumulation of transcriptionally active p53 in the nucleus of HPV-positive cervical cancer cell lines resulting in apoptotic death (9). Furthermore, when human keratinocytes were treated with LMB, induction of apoptosis was selectively induced in primary cells expressing the HPV oncogenes (13). Such potent antitumor effects are not limited to HPV-positive cancers. LMB treatment of prostate cancer cells (14) as well as neuroblastoma cell lines (15) induces p53 activation leading to growth arrest and induction of apoptosis.
To establish the potential utility of nuclear export inhibitors as anticancer drugs, we synthesized derivatives of LMB and now report a series of NEIs with substantially improved therapeutic windows. These new NEIs maintain the high potency of LMB and are up to 16-fold better tolerated than LMB in mouse models. We show that exposure of cancer cells to these compounds leads to a rapid and prolonged block of nuclear export, which is further associated with an increase in multiple markers of apoptosis. In contrast to what is observed in cancer cells, these agents induce cell cycle arrest, but not apoptosis, in normal lung fibroblasts. These novel CRM1 inhibitors show significant efficacy in multiple mouse xenograft models, including models of colon and cervical cancer. Identification of molecules such as these that target CRM1 but with a wider therapeutic window than LMB is of great interest as a potential novel anticancer therapy.
MATERIALS AND METHODS
Cell lines and Materials
The U20S RevGFP cell line was obtained from the laboratory of Prof. Pamela Silver (Dana Farber Cancer Institute, Boston, MA). All other cell lines were obtained from the ATCC (Manassa, VA). A general method for formation of 24-LMB-amides is described in the Supplementary Materials.
Cytotoxicity assays
Cytotoxicity assays were performed using the CellTiter-Glo Luminescent Cell Viability Assay (G7573, Promega, Madison, WI) according to the manufacturer’s instructions. Approximately 5,000 cells per well were treated with drug either continuously or for a one hour pulse and cell viability was measured using the assay after 72 hours. For the short exposure experiments, the cells were washed with pre-warmed media after one hour and then maintained in drug-free media until viability was assessed at 72 hours. Luminescence was measured on a Wallac Victor2 multilabel plate reader (Perkin Elmer, Waltham, MA).
Cell extracts and Western blot analysis
For extracts from cell culture, cells were washed with PBS and resuspended in RIPA buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM EDTA, 1 % (w/v) Nonidet NP40, 0.1% (w/v) SDS, 12 mM sodium deoxycholate, 0.9 mM Na3VO4, Roche Complete Protease Inhibitor Cocktail). Cells were disrupted by several passages through a syringe, centrifuged for 15 min, 40,000g, 4C, and stored at −20C until analysis by SDS-PAGE and western blotting. Antibodies used include p53 (Bp53-12, sc-263) and GAPDH (sc-32233) from Santa Cruz Biotechnology (Santa Cruz, CA), and cleaved caspase-3 (#9664, Cell Signaling, Danvers, MA).
Immunofluorescence
Approximately 5 × 103 cells were seeded onto black, tissue culture treated, optical bottom 96 well plates (Nalge Nunc, Rochester, NY) and grown overnight at 37°C. Media was removed and replaced with drug-containing media. For recovery experiments, cells were treated for one hour and washed with pre-warmed media, followed by incubation in media without drug for the desired recovery period. Plates were fixed with 3.7% formaldehyde in PBS for 20 minutes at room temperature and washed three times in PBS. Cells were permeabilized and blocked in 10% donkey serum, 0.1% Triton X-100 in PBS for 30 minutes at room temperature. Goat-anti-RanBP1 (sc-1160, Santa Cruz Biotechnology, Santa Cruz, CA) antibodies were used at a 1:50 dilution. Mouse-anti-p53 antibodies (sc-126, Santa Cruz Biotechnology, Santa Cruz, CA) were used at a 1:50 dilution. Fluorescent secondary antibodies from Jackson Immunoresearch (West Grove, PA) were used at a 1:200 dilution. Cells were stained with 1 μg/mL Hoechst 33342 (Invitrogen, Carlsbad, CA). Images were acquired and analyzed using a Cellomics ArrayScan Vti with Molecular Translocation software (Pittsburg, PA). A minimum of 500 cells per well was analyzed.
Cell Cycle Analysis
Cells were harvest by trypsinization, washed in PBS, and fixed at a final concentration of 70% ice cold ethanol. Cells were stored at −20°C, washed in PBS, and then stained in propidium iodide/RNase buffer (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Analysis by flow cytometry was performed with a Becton Dickinson FACSCalibur (San Jose, CA).
Apoptosis assays
Annexin V staining was performed using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Analysis by flow cytometry was performed with a Becton Dickinson FACSCalibur (San Jose, CA). Caspase levels were analyzed using the Caspase Glo 3/7 Assay (Promega, Madison, WI) according to the manufacturer’s instructions. Approximately 4,000 cells were treated with serial dilutions of compound for 72 hours, and luminescence was measured on a Wallac Victor2 multilabel plate reader (Perkin Elmer, Waltham, MA).
Animals
Five to 8-week-old female nu/nu nude and C57Bl/6 mice were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN) or Taconic (Oxnard, CA). Food and water were supplied ad libitum. All work in mice was done in accordance with guidelines established by our Institutional Animal Care and Use Committee (IACUC). The animal facility at Kosan is accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care).
Determination of maximum tolerated dose (MTD) of NEIs
C57Bl/6 mice (7–10 mice per dose group) were administered vehicle or escalating dose levels of NEIs intravenously (i.v.) via the lateral tail vein either once or on a weekly schedule. Mice were monitored at least three times a week for body weight change and clinical symptoms for 2–6 weeks. MTD was defined as the highest dose level (of each drug for each schedule) that induced no greater than 20% body weight loss and/or less than or equal to10% mortality.
Xenograft tumor models
Human cancer cell lines were grown in their recommended medium and harvested. The cells were resuspended in PBS. For tumor inoculation, cancer cells (3–10×106 cells/mouse; 0.1mL/mouse) were subcutaneously implanted into the hind flanks of female nu/nu athymic mice. Animals were monitored at least twice a week for the duration of the study. Drug treatment was usually initiated when tumors had reached approximately 100–150mm3. For efficacy studies with large tumors, tumors were allowed to reach >450mm3 before the drug treatment was initiated. Tumor bearing mice were randomized and dosed intravenously (i.v.) via the lateral tail vein with either vehicle or NEIs on the indicated schedule. The NEIs were formulated in 4% Cremophor, 6% propylene glycol, 10% ethanol, 80% saline. Tumor dimensions were measured using calipers and tumor volume was estimated using the following formula: volume = (length × width × width)/2. Statistical analyses were carried out with the use of the GraphPad Prism 4 software (San Diego, CA). All reported p values were from two-way ANOVA tests and were considered to be statistically significant at p<0.05.
RESULTS
NEIs are potent cytotoxic agents
LMB is a polyketide natural product, which rapidly induces cytotoxic effects in cancer cell lines via the covalent inhibition of CRM1. For most cancer cell lines, in vitro IC50 values for a 72 hour exposure are in the sub-nanomolar range (Table 1 and data not shown). We reasoned that the covalent interaction of LMB with CRM1 would result in effects of the drug that persist beyond the initial drug exposure. To confirm this hypothesis and in order to more closely mimic in vivo drug exposures, we performed cytotoxicity assays with a short drug exposure period. Drug treatment was followed by continued incubation in drug-free media and subsequent measurement of cytotoxicity after 72 hours. With this assay, LMB exposure as short as one hour still results in substantial cytotoxicity. IC50 values under such assay conditions are below 5 nM (Table 1), supporting the conclusion that the effects of the drug are rapid and long lasting.
Table 1. Nuclear export inhibitor analogs retain the potency of LMB with better in vivo tolerance.
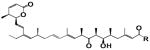
Half maximal growth inhibiting concentration (IC50) was measured for various human cancer cell lines 72h after either continuous drugging or a one hour drug exposure using the Promega Cell Titer Glo assay system. For maximum tolerated dosage (MTD) experiments, female C57Bl/6 mice were used. 7–10 animals per dose group were intravenously administered vehicle or compound as a single bolus injection via the lateral tail vein. The MTD was defined as the maximum dose that induced less than 20% body weight loss for LMB, compound 2, and compound 3. We lost one mouse on day 3 for compound 1, so the MTD for this molecule was defined by mortality. There was no other mortality observed at the MTD dose in this study
| Analog | SiHa (IC50 nM) | HCT-116 (IC50 nM) | SKNSH (IC50 nM) | Mouse MTD | ||||
|---|---|---|---|---|---|---|---|---|
|
1h | 72h | 1h | 72h | 1h | 72h | (mg/kg) | |
| R= ~OH | Leptomycin B | 2.1 | 0.4 | 4.3 | 0.3 | 4.1 | 0.4 | 2.5 |
|
|
1 | 1.9 | 0.4 | 4.4 | 1.5 | 3.2 | 0.9 | 12 |
|
|
2 | 0.4 | 0.5 | 2.6 | 0.6 | 1.7 | 0.45 | 20 |

|
3 | 1.6 | 0.5 | 3.3 | 0.8 | 2.2 | 0.5 | 40 |
In order to determine whether the potential exists to generate analogs that could retain potency but with better in vivo tolerance, we designed a series of semi-synthetic LMB analogs, which we generically term NEIs. This series of LMB derivatives maintain essentially equivalent in vitro cytotoxicity even after a one hour drug exposure (Table 1). We next determined if our semi-synthetic LMB analogs have a wider therapeutic window than LMB by establishing maximum tolerated dosages (MTDs) in mice. The semi-synthetic NEIs were found to be substantially better tolerated when the compounds were intravenously (i.v.) administered as a single bolus injection. The best-tolerated analog from our first series, compound 3, has a >15-fold higher MTD than LMB (2.5 mg/kg for LMB and 40 mg/kg for compound 3, Table 1). MTDs determined after administration of a single dose were found to be equivalent to those determined for a weekly dosing schedule (data not shown). We focused on LMB and compound 3, to further characterize the mechanism of action and therapeutic potential of this class of compounds.
Short exposures to NEIs cause an extended block of nuclear export
In order to define the kinetics of nuclear export inhibition for this class of compounds, we examined the subcellular localization of a protein, RanBP1, which is dependent on CRM1 for nuclear export. In untreated SiHa (cervical cancer) cells, RanBP1 is localized to the cytoplasm (Fig. 1a), but treatment with LMB or compound 3 causes rapid accumulation of RanBP1 in the nucleus (Fig. 1b). Using RanBP1, we demonstrated that nuclear export is completely blocked by 30 minutes after exposure to 10 nM of compound 3 (Fig. 1c). Furthermore, consistent with the covalent nature of the drug interaction with CRM1, the nuclear export block persists well beyond the initial drug exposure. Upon removal of the drug, nuclear export remains inhibited and does not completely recover until approximately 24 hours after the initial exposure (Fig. 2). Similar results for both extent of nuclear export block and time frame of recovery was seen with exposure to 5 or 10 nM LMB (Fig. 2 and data not shown). This long recovery time suggests that recovery from nuclear export block requires de novo CRM1 synthesis, and this is supported by the lack of nuclear export recovery in cells incubated in the presence of the protein synthesis inhibitor, cycloheximide, after removal of the NEIs (Supplementary Fig. 1). The kinetics of nuclear export inhibition and recovery are consistent among multiple cell types tested including HCT-116 (colon cancer), U20S (osteosarcoma), A549, NCI-H460 (lung cancers), LNCaP (prostate cancer), and others (data not shown).
Figure 1. Nuclear export is rapidly blocked by treatment with LMB or compound 3.
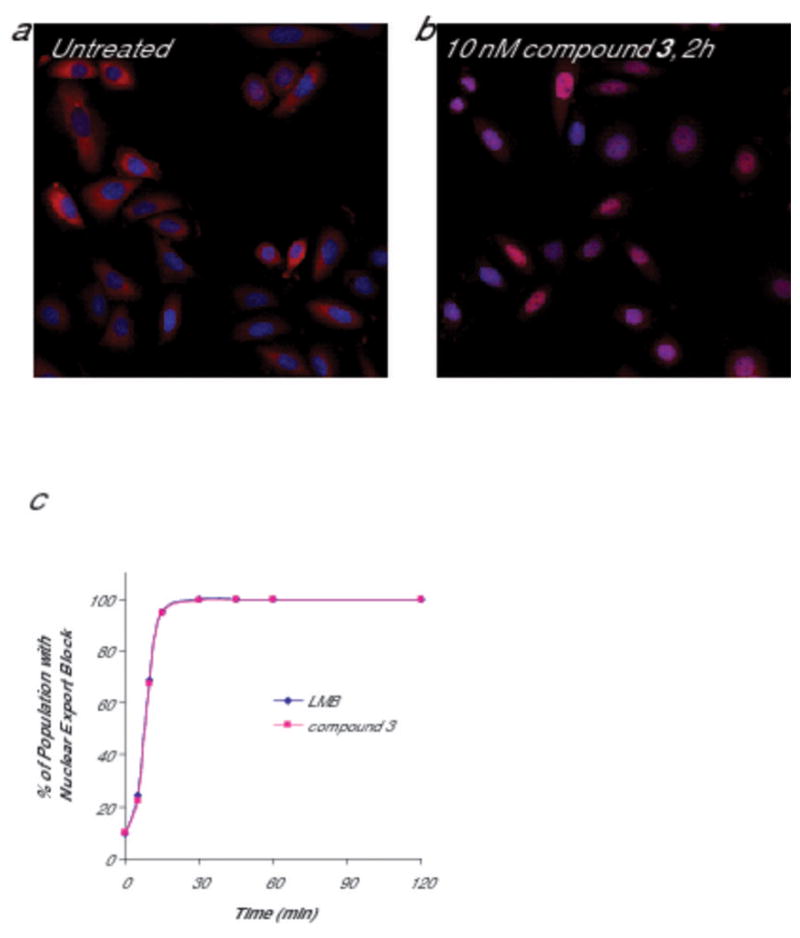
SiHa (human cervical cancer) cells were treated with 10 nM compound 3 or LMB for the indicated timepoints up to two hours. Cells were formaldehyde fixed and stained for RanBP1 (a biomarker for CRM1 inhibition; red) and Hoescht (blue, to define the nucleus). (a) RanBP1 is localized to the cytoplasm in untreated cells. (b) After drug treatment, nuclear localization of RanBP1 is detected in essentially 100% of the cells. (c) For each timepoint, images were acquired and analyzed using a Cellomics ArrayScan Vti with Molecular Translocation software. By 30 minutes, RanBP1 is localized to the nucleus in the entire population. A minimum of 500 cells per well was analyzed.
Figure 2. Nuclear export block persists after removal of NEI.
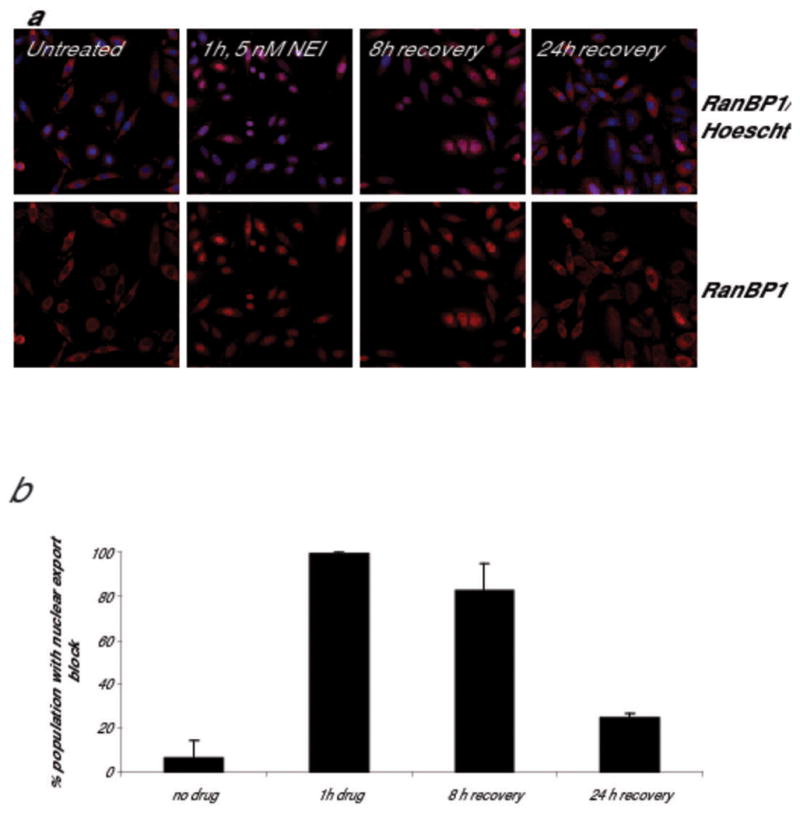
SiHa (human cervical cancer) cells were treated with 5 nM compound 3 for 1 hour. The drug was removed by washing, and cells were allowed to recover in drug-free media for the times indicated. Cells were formaldehyde fixed and stained for RanBP1 (a biomarker for CRM1 inhibition; red) and Hoescht (blue, to define the nucleus). (a) In untreated cells, RanBP1 is localized to the cytoplasm. After one hour of drug treatment with compound 3, nuclear localization of RanBP1 is detected in essentially 100% of the cells. (b) For each timepoint, images were acquired and analyzed using a Cellomics ArrayScan Vti with Molecular Translocation software. A minimum of 500 cells per well was analyzed, and data shown is the average of two independent experiments. Nuclear export remains blocked in >80% of the cell population 8 hours after the drug is removed and a minority (~25%) of the cells remain at least partially blocked 24 hours after the drug is removed.
Nuclear export inhibition leads to apoptosis in tumor cell lines but reversible cell cycle arrest in normal lung fibroblasts
We next explored the downstream effects of treatment with NEIs. After a one hour exposure to compound 3, persistent cell cycle arrest is observed in tumor cell lines (SiHa cells) and in normal human lung fibroblasts (MRC5) (Supplementary Table 1). The treated cells arrest in the G2 and/or G1 phase of the cell cycle, and the percentage of proliferating cells (S phase cells) is dramatically reduced in both tumor cells and normal normal lung fibroblasts, consistent with what has been previously observed for LMB (15, 16). Moreover, although normal lung fibroblasts respond to NEIs with a persistent cell cycle arrest, they remain viable after nuclear export inhibition. These results are consistent with previous work using multiple human normal fibroblast cell lines which demonstrated that cells regain their ability to proliferate after LMB-induced growth arrest upon drug removal and passage by trypsinization (15). Similar results were also observed in experiments using normal primary human keratinocytes, which regain their ability to divide approximately 96 hours after the withdrawal of LMB (13).
In contrast to what was observed in normal lung fibroblasts, inhibition of nuclear export in cancer cells leads to an induction of multiple markers of apoptosis including plasma membrane alterations and caspase activation. SiHa cervical cancer cells were treated for one hour with 10 nM compound 3 followed by drug removal and continued growth. 72 hours after the initial drug exposure, approximately 30% of the population is Annexin V positive, indicative of plasma membrane alterations, which occur in the early stages of apoptosis (Fig. 3a). Similarly, caspase activation is also observed in SiHa cells 72 hours after a one-hour exposure of 25 nM or 500 nM compound 3 or LMB (Fig. 3b and data not shown for 25 nM). Induction of apoptosis is observed in many other human tumor derived cell lines (e.g. HCT-116 (colon), LoVo (colon), LNCaP (prostate), A498 (kidney), and others; data not shown). In contrast, nuclear export inhibition in normal lung fibroblasts does not lead to the induction of apoptosis, consistent with the transient growth arrest observed upon exposure to NEIs (13, 15). Although a control compound, staurosporine, caused dramatic induction of caspases in MRC5, WI-38 and IMR-90 normal lung fibroblast cell lines, no caspase activation was observed in any of these cell lines in response to high doses of NEIs (500 nM compound 3 or LMB, Fig. 3b). Similarly, no Annexin V staining was observed in MRC5 cells exposed to compound 3 (Fig. 3a) or LMB (data not shown)
Figure 3. NEI treatment induces apoptosis in cancer cells but not in normal lung fibroblasts.

(a) SiHa human cervical cancer cells or MRC5 human normal lung fibroblast cells were treated with either vehicle or 10 nM compound 3 for 1h and incubated for an additional 72 hours. Cells were stained with Annexin V-FITC and propidium iodide and analyzed by flow cytometry to determine the Annexin V positive, propidium iodide negative population. Data is representative of multiple experiments. (b) SiHa (cervical cancer cells, black bars), or human normal lung fibroblast cell lines MRC5 (dark gray bars), WI-38 (light gray bars), or IMR-90 (hatched bars) were treated with either vehicle or a high dose (500 nM) of compound 3 or LMB for 1h and incubated for an additional 72 hours. Staurosporine control treatment was at 1 microM for 72 hours. Cells were analyzed for caspase 3/7 induction using the Promega Caspase 3/7 Glo assay. Data is the average of two independent wells and representative of multiple experiments.
Nuclear export inhibition correlates with sustained p53 overexpression
We demonstrated in multiple p53 wildtype cell lines that inhibition of nuclear export strongly correlates with upregulation of p53. Using an immunofluorescence assay in a p53 wild type osteosarcoma cell line (U20S), we demonstrated that p53 is upregulated and localized to the nucleus in a dose dependent manner, and that p53 upregulation correlated with nuclear export block (Fig. 4a). These results are consistent with previous work that demonstrated that LMB treatment leads to p53 nuclear localization and activation in various cell types (9, 13–15, 17). Furthermore, in the HCT-116 colon cancer cell line a one hour exposure to compound 3 lead to increased cellular p53 expression for up to 48 hours (Fig. 4b). This result establishes that a downstream response to CRM1 inhibition, in this case p53 upregulation, can persist even after the nuclear export block is reversed at ~24 hours (see Fig. 2).
Figure 4. Nuclear export inhibition correlates with sustained p53 overexpression and loss of p53 function negatively affects NEI-induced apoptosis.

(A) A U20S osteosarcoma cell line containing an NES-RevGFP reporter protein (32) was treated with increasing concentrations of compound 3. After 24 hours of continuous drug treatment, cells were formaldehyde fixed and stained for p53 and with Hoescht dye. For each concentration, images were acquired and a minimum of 500 cells per time point were analyzed using a Cellomics ArrayScan Vti with Compartmental Analysis software (Pittsburg, PA.) In untreated cells, the RevGFP fusion protein localizes predominantly to the cytoplasm but treatment with compound 3 causes accumulation of RevGFP in the nucleus (squares). p53 expression levels (open circles) correlate with the level of nuclear export block. Western blot analysis confirms the upregulation of p53 expression in response to nuclear export inhibition. (b) HCT-116 colon cancer cells were treated with 10 nM compound 3 for one hour followed by a recovery period in drug free media. Cell extracts were made at the indicated times and analyzed by SDS-PAGE for p53 expression levels. GAPDH is shown as a control for protein loading levels. (c). SiHa cells were treated with 50 nM compound 3 in the presence or absence of 10 or 30 μM pifithrin-alpha, an inhibitor of p53. After 72 hours, caspase activation was measured using the Promega Caspase 3/7 Glo assay. As a positive control for apoptosis, cells were treated with 1 μM staurosporine.
Previous studies have demonstrated that the cytotoxic effects of LMB are reduced in human neuroblastoma and cervical cancer cell lines when the activity of endogenous p53 is abrogated by overexpression of a dominant negative p53 mutant (9, 15). Consistent with this result, addition of a small molecule inhibitor of p53, pifithrin-alpha (18), blocked compound 3-induced apoptosis in SiHa cells (Fig. 4c). These results demonstrate that p53 can play an important role in mediating NEI-induced apoptosis.
Novel NEIs show robust efficacy in multiple tumor models
Because of the strong induction of apoptosis observed in compound 3 treated cells in culture (see Fig. 3), we harvested tumor samples to determine if this effect was also observed in vivo. HCT-116 tumor bearing mice were treated with a single i.v. dose of either vehicle or compound 3, and tumors were harvested 24 hours later. Consistent with the induction of apoptosis observed in vitro, we observed evidence of increased apoptosis in vivo, as shown by an increase in the levels of cleaved caspase-3 (Supplementary Fig. 2).
We next compared the anti-tumor activity of compound 3 and LMB in mouse xenograft models. Given the strong biological rationale and supporting in vitro data demonstrating NEI induced activation of p53 in HPV infected cells (9, 13), we first studied efficacy in the HPV-positive SiHa cervical cancer model. Tumor regression was induced by weekly compound 3 treatment for three weeks (Fig. 5a), and tumors began to shrink shortly after the mice had been exposed to compound 3 (day 4). At day 25, tumor volumes were still significantly below those observed when dosing began, and, in fact, two mice had no detectable tumors. In contrast, only a growth delay was found in the LMB-treated group (Fig. 5a), consistent with the limited in vivo efficacy previously observed for LMB (6). To further assess the duration of the anti-tumor effect of compound 3, we treated SiHa tumor bearing mice with either vehicle or compound 3 once a week for 2 weeks and then monitored tumor response for a longer period. Compound 3 treated tumors again responded rapidly after the first dose, and 100% of tumors regressed for at least 4 weeks. Furthermore, tumors could not be detected in 3 out of 7 mice in the compound 3 treated group between days 26 to 40. Two of these mice remained tumor free for >450 days and are considered to be therapeutically cured (Supplementary Fig. 3).
Figure 5. Compound 3 inhibits tumor growth in cancer xenograft models.
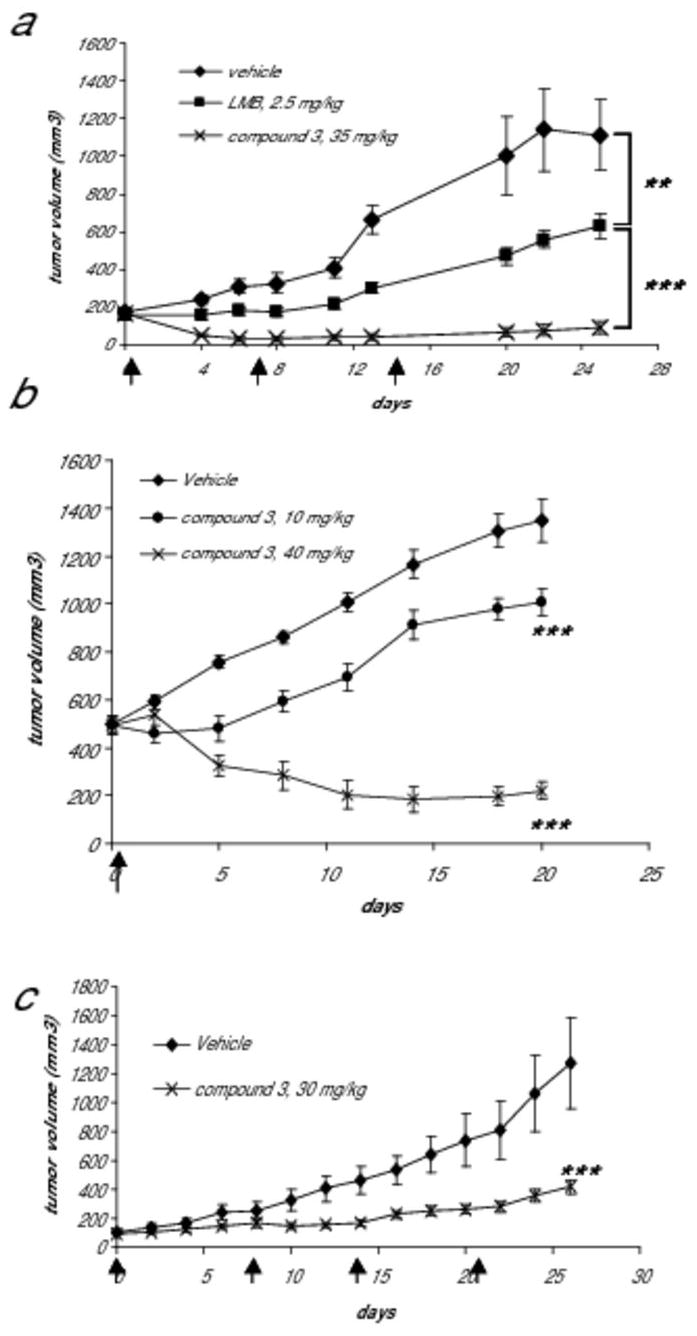
(a) SiHa tumor bearing mice (n=6 each group) were treated i.v. with either vehicle, 2.5 mg/kg LMB, or 35 mg/kg compound 3 using a Q7D schedule for a total of 3 doses (dosing days indicated by arrows). Tumor growth curves of vehicle- or NEI-treated groups are shown. In the compound 3-treated group, 2 mice (~33%) had no detectable tumors after day 6 and remained tumor free at day 25 of the follow up period. **, p<0.01; ***, p<0.001 (based on a 2-way ANOVA test). (b) SiHa xenograft tumors (n=6 each group) were allowed to reach ~500 mm3 before dosing (dosing day indicated by an arrow). A single i.v. bolus dose of compound 3 produced growth inhibition at 10 mg/kg or regression at 40 mg/kg of large SiHa tumors. ***, p<0.001 (based on a 2-way ANOVA test). (c) HCT-116 tumor bearing mice were treated i.v. with either vehicle (n=7) or 30 mg/kg of compound 3 (n=8) using a Q7D schedule for a total of 4 doses (dosing days indicated by arrows). Tumor growth curves are shown. ***, p<0.001 (based on a 2-way ANOVA test).
Many anti-cancer agents are less effective against larger tumors. To assess the anti-tumor activity of compound 3 in mice with a greater tumor burden, we allowed the SiHa tumors to grow to ~500mm3 before treating with a single i.v. injection of compound 3. We found a single administration of compound 3 was able to demonstrate anti-tumor activity in a dose-dependent manner with 10 mg/kg causing tumor growth inhibition and 40 mg/kg causing significant tumor regression (Fig. 5b).
In order to determine if compound 3 is active in other tumor models, we tested for anti-tumor activity using an HCT-116 colon cancer xenograft model. Using this model, we demonstrated that compound 3 significantly inhibited tumor growth when dosed at 30 mg/kg weekly (Fig. 5c). Furthermore, consistent with results in the SiHa model, 30 mg/kg of compound 3 was found to be significantly more efficacious and less toxic than 2.5 mg/kg of LMB (data not shown). 30 mg/kg of compound 3 was found to be similarly efficacious as 35 or 40mg/kg in the SiHa and HCT-116 models but better tolerated (inducing only ~10% body weight loss), and hence 30mg/kg was chosen as an appropriate dose at which to examine the activity of compound 3 in additional xenograft models. Antitumor activity was also observed in NCI-H460 (non-small cell lung cancer), A375 (melanoma), and K562 (chronic myelogenous leukemia) cancer models (Supplementary Table 2). Thus, NEIs like compound 3, which have a significantly improved tolerability relative to LMB, show robust efficacy in multiple xenograft models.
DISCUSSION
In this study, we have synthesized analogs of LMB, the prototypical nuclear export inhibitor, which show potential as novel anticancer therapeutics. LMB itself has significant in vitro potency but is poorly tolerated in vivo (6, 7). The covalent nature of the interaction with CRM1 needs to be considered when comparing compounds. In vitro IC50 values for covalent inhibitors determined after prolonged drug exposure cannot be interpreted as indicators of target binding affinity, as they would be for reversible inhibitors, since covalent inhibitors have essentially infinite binding affinity for their target. The IC50 values for NEIs determined after short drug exposure serve as an indirect measure of the rate of CRM1 inactivation and thereby reflect the relative efficiencies of a series of compounds at inactivating CRM1. The observation that our series of NEIs retain essentially equivalent cytotoxicities to LMB after a one hour exposure (Table 1) demonstrates that they retain the potency of LMB.
In order to define the kinetics of nuclear export inhibition for this class of compounds, we wanted to find an endogenous protein dependent on CRM1 for nuclear export and which is ubiquitously expressed. Such a protein could serve as a useful biomarker for nuclear export in all cell types without the need for additional inducers of expression or nuclear import. We chose RanBP1, an accessory protein for RanGAP-mediated dissociation of CRM1-bound cargo proteins upon export to the cytoplasm. The rapid shuttling of RanBP1 into the nucleus makes it a particularly useful tool for the study of NEIs. Other CRM1-dependent cargo proteins that are imported more slowly, not ubiquitously expressed, or that require inducing conditions for their nuclear import are not as useful for the purpose of defining the kinetics of CRM1 inhibition, although they could undoubtedly play a role in mediating downstream cytotoxic effects. Using RanBP1 as a biomarker, our in vitro experiments show that exposure of cancer cells to NEIs leads to a rapid and prolonged block of nuclear export in all cell types tested.
The observation that the induction of p53 is detectable at 24 or 48 hours (Figure 4b) despite the fact that nuclear export has recovered in the majority of the cell population by 24 hours (Figure 2b and data not shown) shows that the downstream effects of nuclear export inhibition can persist longer than the actual nuclear export block. Nuclear entrapment of p53 turns on the p53 transcriptional program, including activation of transcription of p53 itself. As the data show, this leads to a significant increase in p53 protein levels in the cell (Fig. 4, (9, 15, 19)). Once this program is turned on, since it is part of an auto-activation loop, its regulation need no longer depend on nuclear export block and will instead depend on factors such as the stability of p53 and the dynamic point at which p53 levels drop to the range in which the positive feedback loop is no longer engaged. Thus, a downstream consequence of nuclear export inhibition, in this case p53 induction, can persist independently of the original signal which was the nuclear export block.
The inhibition of nuclear export is associated with an increase in multiple markers of apoptosis in cancer cells. In contrast to this, NEIs induce cell cycle arrest, but not apoptosis, in normal lung fibroblasts. Thus, although NEIs cause the inhibition of CRM1 in both tumor and normal cell types, a difference is observed in the downstream consequences of this inhibition. The basis of this difference in response remains under investigation.
We have synthesized novel NEIs that are up to 16-fold better tolerated than LMB in mouse models while retaining significant potency. These results suggest that the limited in vivo efficacy of LMB was likely due to off-target effects since our NEIs retain the potent inhibition of CRM1, but are clearly better tolerated in vivo. The reasons why the novel NEIs are better tolerated are currently under investigation. Areas of exploration include the tissue distribution profile of these molecules as well an investigation of their in vivo on-target and off-target protein binding properties. The better tolerance enables these novel NEIs to be dosed at higher levels in vivo. As figure 5b shows, 10mg/kg of compound 3, which is four-fold higher than the MTD of leptomycin B, demonstrates only modest efficacy whereas a dose of 40mg/kg results in regression. This supports the conclusion that leptomycin’s low MTD limits its efficacy and higher doses are required for robust efficacy for compounds such as these, which show comparable activity in vitro. Doses above 30mg/kg of compound 3 are associated with significant efficacy in multiple mouse xenograft models, thereby validating nuclear export as a potentially useful therapeutic target in cancer.
In this study, we have focused on p53 wild type cancer models including an HCT-116 colon model and a SiHa cervical cancer model. In these models, the p53 tumor suppressor becomes trapped in the nucleus upon inhibition of CRM1. In HPV-positive cancer types, such as SiHa, this prevents the aberrant cytoplasmic localization and degradation of p53 and leads to activation of pathways that cause cell-cycle arrest and apoptotic cell death. We show that such effects are not limited to HPV-positive cancers, as NEIs induce p53 activation (Fig. 4b) and show anti-tumor efficacy in the HCT-116 colon cancer model (Fig. 5c). Furthermore, we have also tested for anti-tumor activity of compound 3 in a variety of other tumor models, including NCI-H460 (non-small cell lung cancer), A375 (melanoma), and K562 (chronic myelogenous leukemia) (Supplementary Table 2). Compound 3 demonstrated anti-tumor activity in all of these models ranging from induction of tumor regression to tumor growth inhibition. Thus, in contrast to the poor in vivo activity of LMB, the NEI analog compound 3 shows robust efficacy in all xenograft models examined to date and is of great interest as a potential cancer therapeutic.
Although we have focused here on p53 wild type cancer types, CRM1 mediates the nuclear export of numerous other proteins that are also important therapeutic targets. Various lines of evidence provide a strong biological rationale for the use of NEIs in multiple cancer types. For example, in chronic myelogenous leukemia (CML), the oncogenic BCR-Abl tyrosine kinase is found in the cytoplasm where it activates a number of mitogenic signaling pathways (20–22). Treatment of CML cells with the tyrosine kinase inhibitor imatinib not only inhibits BCR-Abl but also promotes its shuttling into and out of the nucleus (23). Co-administration of LMB with imatinib was demonstrated to cause nuclear accumulation of BCR-Abl, ultimately resulting in the activation of programmed cell death both in vitro (24) and in ex vivo experiments (24). Given the poor in vivo tolerance of LMB, a significant therapeutic benefit could be gained by combining a potent but better tolerated CRM1 inhibitor with an inhibitor of BCR-Abl. This combination has the potential to overcome the problem of drug resistance by eradicating rather than inhibiting the growth of CML cells.
Similarly, LMB inhibition of CRM1 has been shown to promote the nuclear buildup of the Forkhead family of transcription factors (FOXOs) (25, 26). These transcription factors are regulated by multiple signaling pathways that play critical roles in tumorigenesis including the PI3K/PTEN/Akt pathway (reviewed in (27, 28)). In PTEN-deficient cells, the Akt pathway is activated and FOXO transcription factors are rendered inactive by localization to the cytoplasm. Restoring PTEN function in these cells blocks Akt activity and restores nuclear localization of FOXO and, therefore, its ability to activate downstream factors. Prolonged FOXO residence in the nucleus leads to the induction of pro-apoptotic genes and ultimately to growth arrest and death in PTEN-null tumor cells (29, 30). In addition, it has recently been shown that localization of FOXO to the nucleus in PTEN-null cells inhibits Hif1 transcriptional activity(31), thereby potentially interfering with the ability of PTEN-null cells to survive under hypoxic conditions. It will thus be of great interest to examine the efficacy of NEIs in PTEN-deficient cancers.
In conclusion, the NEIs we have synthesized have enabled us to validate CRM1 as a target for anti-cancer therapeutics. These data show that the limited in vivo efficacy observed for LMB was a result of its poor tolerance. The identification of NEIs that are significantly better tolerated has demonstrated that molecules with this mechanism of action can show robust in vivo efficacy. These results provide strong evidence supporting the development of NEIs as a novel anticancer therapy.
Supplementary Material
Acknowledgments
This work was supported in part by an NIH SBIR grant to SDD (1R43CA109840).
The authors wish to thank Jean Y.J. Wang (University of California, San Diego) for suggesting RanBP1 as a biomarker, Jessica Simmons, Yiqing Zhou, and Yunfei Chen for assistance with in vivo mouse studies, Rishali Gadkari for assistance with in vitro experiments, John Carney for assistance with compound characterization, the Process Sciences Department of Kosan Biosciences for LMB production, Gary Ashley, Ralph Reid, and Chris Reeves for critical reading of the manuscript, and Robert Johnson for his support. This work was supported in part by an NIH SBIR grant to SDD (1R43CA109840).
Footnotes
All authors are current or former employees of Kosan Biosciences, Inc.
References
- 1.Vousden KH, Woude GF. The ins and outs of p53. Nat Cell Biol. 2000;2:E178–80. doi: 10.1038/35036427. [DOI] [PubMed] [Google Scholar]
- 2.Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269:6320–4. [PubMed] [Google Scholar]
- 3.Wolff B, Sanglier JJ, Wang Y. Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol. 1997;4:139–47. doi: 10.1016/s1074-5521(97)90257-x. [DOI] [PubMed] [Google Scholar]
- 4.Kudo N, Matsumori N, Taoka H, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–7. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petosa C, Schoehn G, Askjaer P, et al. Architecture of CRM1/Exportin1 suggests how cooperativity is achieved during formation of a nuclear export complex. Mol Cell. 2004;16:761–75. doi: 10.1016/j.molcel.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 6.Roberts BJ, Hamelehle KL, Sebolt JS, Leopold WR. In vivo and in vitro anticancer activity of the structurally novel and highly potent antibiotic CI-940 and its hydroxy analog (PD 114,721) Cancer Chemother Pharmacol. 1986;16:95–101. doi: 10.1007/BF00256156. [DOI] [PubMed] [Google Scholar]
- 7.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74:648–9. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 9.Hietanen S, Lain S, Krausz E, Blattner C, Lane DP. Activation of p53 in cervical carcinoma cells by small molecules. Proc Natl Acad Sci U S A. 2000;97:8501–6. doi: 10.1073/pnas.97.15.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessis TD, Slebos RJ, Nelson WG, et al. Human papillomavirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proc Natl Acad Sci U S A. 1993;90:3988–92. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 12.Lain S, Lane D. Improving cancer therapy by non-genotoxic activation of p53. Eur J Cancer. 2003;39:1053–60. doi: 10.1016/s0959-8049(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 13.Gray LJ, Bjelogrlic P, Appleyard VC, et al. Selective induction of apoptosis by leptomycin B in keratinocytes expressing HPV oncogenes. Int J Cancer. 2007;120:2317–24. doi: 10.1002/ijc.22591. [DOI] [PubMed] [Google Scholar]
- 14.Lecane PS, Kiviharju TM, Sellers RG, Peehl DM. Leptomycin B stabilizes and activates p53 in primary prostatic epithelial cells and induces apoptosis in the LNCaP cell line. Prostate. 2003;54:258–67. doi: 10.1002/pros.10197. [DOI] [PubMed] [Google Scholar]
- 15.Smart P, Lane EB, Lane DP, Midgley C, Vojtesek B, Lain S. Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. Oncogene. 1999;18:7378–86. doi: 10.1038/sj.onc.1203260. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida M, Nishikawa M, Nishi K, Abe K, Horinouchi S, Beppu T. Effects of leptomycin B on the cell cycle of fibroblasts and fission yeast cells. Exp Cell Res. 1990;187:150–6. doi: 10.1016/0014-4827(90)90129-x. [DOI] [PubMed] [Google Scholar]
- 17.Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–93. doi: 10.1128/mcb.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 19.Lain S, Midgley C, Sparks A, Lane EB, Lane DP. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res. 1999;248:457–72. doi: 10.1006/excr.1999.4433. [DOI] [PubMed] [Google Scholar]
- 20.McWhirter JR, Wang JY. Activation of tyrosinase kinase and microfilament-binding functions of c-abl by bcr sequences in bcr/abl fusion proteins. Mol Cell Biol. 1991;11:1553–65. doi: 10.1128/mcb.11.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McWhirter JR, Wang JY. Effect of Bcr sequences on the cellular function of the Bcr-Abl oncoprotein. Oncogene. 1997;15:1625–34. doi: 10.1038/sj.onc.1201342. [DOI] [PubMed] [Google Scholar]
- 22.Pendergast AM, Quilliam LA, Cripe LD, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–85. [PubMed] [Google Scholar]
- 23.Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7:228–34. doi: 10.1038/84683. [DOI] [PubMed] [Google Scholar]
- 24.Aloisi A, Di Gregorio S, Stagno F, et al. BCR-ABL nuclear entrapment kills human CML cells: ex vivo study on 35 patients with the combination of imatinib mesylate and leptomycin B. Blood. 2006;107:1591–8. doi: 10.1182/blood-2005-05-2123. [DOI] [PubMed] [Google Scholar]
- 25.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol. 2001;21:3534–46. [Google Scholar]
- 27.Arden KC. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp Gerontol. 2006;41:709–17. doi: 10.1016/j.exger.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–82. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramaswamy S, Nakamura N, Vazquez F, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–5. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc Natl Acad Sci U S A. 2008;105:2622–7. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kau TR, Schroeder F, Ramaswamy S, et al. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463–76. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.