

Abstract
Background & Aims
MiRNAs are small non-coding RNA molecules that regulate gene expression through partial or complete complementarity with target mRNAs. The function of miRNAs in normal liver physiology is largely unknown. Here we address the role of Dicer1 in the differentiated liver.
Methods
We derived mice lacking Dicer1 function in hepatocytes and assessed the loss of mature miRNA by quantitative PCR. Gene expression microarray analysis was performed on liver RNA from mutant and control mice. Liver sections from mutant and control mice were examined and liver function tests were performed.
Results
Mice lacking Dicer1 function in hepatocytes appeared and behaved normally. Despite the loss of mature miRNAs, hepatic function was maintained, as reflected by normal blood glucose, albumin, cholesterol, and bilirubin. However, mutant mice between 2-4 months of age exhibited progressive hepatocyte damage with elevated serum ALT and AST. Liver mass was increased in mutant mice, as were cellular markers of both proliferation and apoptosis. Microarray analysis indicated large-scale changes in gene expression, with increased expression of many miRNA targets, particularly imprinted genes.
Conclusions
Loss of miRNA processing in the liver at late gestation has a remarkably mild phenotype, suggesting that miRNAs do not play an essential role in hepatic function. However, miRNA deficiency results in hepatocyte apoptosis, hepatocyte regeneration, and portal inflammation. Finally, microarray analysis of gene expression in mutant liver supports a previously hypothesized role for Dicer1 in the repression of imprinted genes.
Introduction
MiRNAs are short non-coding RNA molecules that function through the post-transcriptional cleavage and/or translational repression of cognate target mRNAs (1, 2). Dicer1 is an RNaseIII-type enzyme that cleaves both long double-stranded RNA molecules and hairpin microRNA (miRNA) precursors, and it is required for the processing of all miRNAs (3-6). Previous studies have shown that distinct miRNA signatures can be assigned to particular organs and tumor types, suggesting that miRNAs function in cellular differentiation and growth. Indeed, examples of miRNAs acting in both development and disease have been described in diverse organisms from worms to humans (7, 8).
The liver-specific miRNA, miR-122a, is estimated to account for 70% of the total liver miRNA content, and antisense oligonucleotide-mediated inhibition of its function in adult mice has suggested a role for this miRNA in the regulation of cholesterol and fat metabolism (9-12). Additional studies implicate miR-122a in resistance to hepatitis C virus, and loss of its expression is correlated with the progression of hepatocellular carcinoma (13, 14). With the exception of miR-122a, little is known about the function of other miRNAs in liver physiology.
In this study we address the function of miRNA in the post-natal liver through the global loss of Dicer1 function (and the consequent elimination of the pool of mature miRNA transcripts) in hepatocytes during the perinatal period. Homozygous Dicer1 null mouse embryos die at a very early stage (15). Using the combination of loxP-flanked Dicer1 alleles and a variety of tissue-specific Cre transgenic mouse lines, Dicer1 has been functionally depleted in several organ systems, such as retina, brain, lymphocytes, and skin (16-20). The majority of the previously published studies show severe developmental defects in the organ in which loss of Dicer1 has occurred, often accompanied by increased rates of apoptosis.
Using the well-characterized AlfpCre line (in which the Cre recombinase is expressed under the regulatory control of a proximal element from the mouse Alb promoter and a distal fragment of the mouse Afp enhancer), we have depleted Dicer1 in cells derived from Afp-expressing hepatoblasts (21). In addition to characterizing the histological and physiological effects of the global loss of functional miRNA regulation, we performed microarray analysis on total liver RNA from 4-week-old mutant and control mice to identify the pool of mRNA whose levels are perturbed by the loss of Dicer1 function.
Materials and Methods
Mice
All mice were housed, handled, and euthanized in accordance with federal and institutional guidelines under the supervision of the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee. The AlfpCre strain was provided by Klaus Kaestner (21). The Dicer1flox mouse strain was a kind gift from Matthias Merkenschlager (16).
Serological and Histological analysis
Blood glucose levels were assayed after 5 hours of fasting, using a Contour glucometer (Bayer). Serum biochemical studies were performed by AniLytics (Gaithersburg, MD). A new rabbit anti-CK19 peptide antibody was generated using the peptide antigen HYNNLPTPKAI and was used in standard immunofluorescent detection at 1:1000. Anti-Dicer1 (rabbit, 1:100) was from Santa Cruz Biotechnologies. The anti-Ki67 immunoglobulin (rabbit, 1:100) was from AbCam. The TUNEL assay was from Chemicon and was performed according to the manufacturer's recommendation.
Hepatocyte purification
Hepatocytes were isolated from P40 mice by perfusion of collagenase (0.05% collagenase type I, Worthington) via the inferior vena cava. Cells were recovered and subjected to Percoll (Amersham Biosciences AB) gradient purification as described (21).
MiRNA quantification
Reverse transcription was performed using the Taqman miRNA reverse transcription kit (Applied Biosystems) and miRNA-specific RT primers from the appropriate Taqman miRNA Assay Kit (Applied Biosystems). Real-time quantitative PCR was carried out on a Stratagene Mx3000P using Taqman miRNA assay kits, with the Taqman Master Mix. MiRNA levels were normalized to U6 RNA using the RNU6B TaqMan assay. Loss of the Dicer1 transcript upon Cre-mediated deletion was monitored using PCR primers within the floxed region, and normalized to Hprt. Sequences of the primers used for RT-PCR are available upon request.
Microarray Analysis
Liver RNA from P28 control (AlfpCre-; Dicer1flox/flox) and mutant (AlfpCre+; Dicer1flox/flox) mice was prepared using the mirVana™ miRNA Isolation Kit (Ambion) according to the manufacturer's instructions for total RNA purification. Sample quantification and quality assurance were performed using a NanoDrop ND-1000 spectrophotometer and an Agilent 2100 Bioanalyzer (Agilent Technologies). 100 ng of total RNA per sample was amplified and biotinylated using the MessageAmp Premier kit (Ambion). Samples (n=5 for each) were hybridized to Affymetrix GeneChip Mouse Genome 430 2.0 Arrays in the Children's Hospital of Philadelphia Nucleic Acids Core Facility and analyzed with the assistance of the Penn Bioinformatics Core. The raw probe intensities were normalized using the GCRMA method (22) and flags (Absent, Present or Marginal) were calculated (ArrayAssist Lite, v3.4, Stratagene). Probe sets marked as Present in fewer than 3 biological samples were excluded from further analysis. The significance of the log2-transformed, GCRMA-normalized signal intensities was determined using the Statistical Analysis of Microarray (SAM) add-in for Microsoft Excel, v3.02 (23). A cut-off false discovery rate of 1% was applied. Gene Set Enrichment Analysis was performed using the GSEA tool (24, 25). The microarray data has been deposited at the NCBI GEO repository under the accession number GSE11899.
Results
Mice lacking liver miRNA appear normal
Mice in which Dicer1 was deleted in hepatoblast-derived cells were generated by crossing the AlfpCre transgenic line to the Dicer1flox conditional strain (16, 21), to obtain AlfpCre; Dicer1flox/flox animals (“mutants”). Mutants were born alive at the expected Mendelian frequency and were completely indistinguishable from control (AlfpCre-) littermates with respect to birth weight, appearance, and behavior. Furthermore, the mice continued to develop and mature normally, and both male and female mutant mice were fertile.
Dicer1 is functionally depleted by P0 in AlfpCre; Dicer1flox/flox mice
The normal appearance of the mutant mice raised concerns that the deletion of the Dicer1 locus was incomplete. To address this question, we assayed levels of Dicer1 mRNA, mature miRNA transcripts, and known miRNA target mRNA levels by qRT-PCR. Levels of the Dicer1 transcript were first significantly decreased by E18.5 (Fig.1A), and were maximally reduced by P0. Correlating with the depletion of Dicer1 transcript, levels of the hepatocyte-specific miR-122a were strongly decreased. Levels of the widely-expressed miR-20a (26) were moderately decreased (although this did not reach statistical significance), while levels of the granulocyte lineage miRNA miR-223, were not significantly changed (Fig.1B).
Figure 1. Functional depletion of Dicer1 in liver.

A: Wild-type Dicer1 transcript levels in total liver RNA from mutant and control animals were assayed by qRT-PCR and normalized to Hprt transcript levels. Control levels are scaled to 1. By P0, wild-type Dicer1 transcript levels in mutant liver are 9% of those in control liver (p<0.05), and remain at 12% at P28 (p<0.0002). B: MiRNA transcript levels in total liver RNA from mutant and control animals were assayed by qRT-PCR, and normalized to U6 RNA transcript levels. Levels of miR-122a are reduced by 93% in liver lacking functional Dicer1 (p<0.01). (C) Expression of Dicer1 and selected miRNA was assayed in total RNA isolated from P40 purified hepatocytes and normalized either to Hprt (Dicer1) or U6 RNA (miRNA). (D) Transcript levels of selected experimentally validated targets of miR-122a regulation in P40 purified hepatocytes, normalized to Hprt. C, D (control, n=3; mutant, n=5). Mutant, AlfpCre+; Dicer1flox/flox. Control, AlfpCre-; Dicer1flox/flox.
Since liver RNA contains a significant contribution from non-parenchymal cells, which are predicted to be unaffected by the AlfpCre transgene, it is likely that the residual levels of miRNA-20a and miRNA-223 detected in Fig. 1B were not of hepatocyte origin. To address this, we purified hepatocytes from P40 mutant and control mice and assessed the levels of Dicer1 and selected miRNAs (Fig. 1C). As expected, the decrease of both Dicer1 and miRNA transcripts was more apparent in the absence of non-parenchymal RNA, with fold-decreases in transcript levels as follows: Dicer1 18.6 fold (p<10−4); miR-122a, 48.3 fold (p<0.04), miR-194, 133.1 fold (p<0.04), miR-20a 19.4 fold (p<0.08), miR-30a, 9.3 fold (p<0.06). Furthermore, levels of experimentally validated miRNA-122a targets were increased in Dicer1-depleted hepatocytes as follows: AldoA, 2.7 fold (p<0.01); Gpx7, 25.8 fold (p<0.01); Hfe2, 3.2 fold (p<3×10−5); Tmed3, 5.2 fold (p<10−4) (Fig. 1D).
Hepatic function is normal in AlfpCre; Dicer1flox/flox mice
We assayed hepatic function by measuring fasting blood glucose, albumin, cholesterol, and bilirubin levels in mutant and control mice at P0, P28, P72 and P101. Control and mutant animals demonstrated virtually identical values (Table 1). Five-hour fasting glucose levels were slightly lower in P28 mutants, but they remained within the normal range and were not different from controls at any other time point.
Loss of Dicer1 leads to progressive liver damage and inflammation
The livers of P28 were grossly normal and indistinguishable from livers of control littermates in shape and size. Similarly, the histology of P28 mutant livers was unremarkable and no adverse effects of the loss of Dicer1 function were apparent (Fig. 2A, B). However, in older animals serum aminotransferase levels rose relative to control animals, with ALT levels more than 3.6 fold higher by P72 (p<10−4) and remaining significantly elevated at P101. While P28 livers appeared normal, by P72, Dicer1 mutant livers had a slightly nodular surface, and in older animals (P101) the mutant livers were significantly enlarged relative to those of control littermates (1.5 times larger as a percentage of total body weight, p<0.02, Table 1).
Figure 2. Dicer1-deficient liver develops progressive hepatitis.
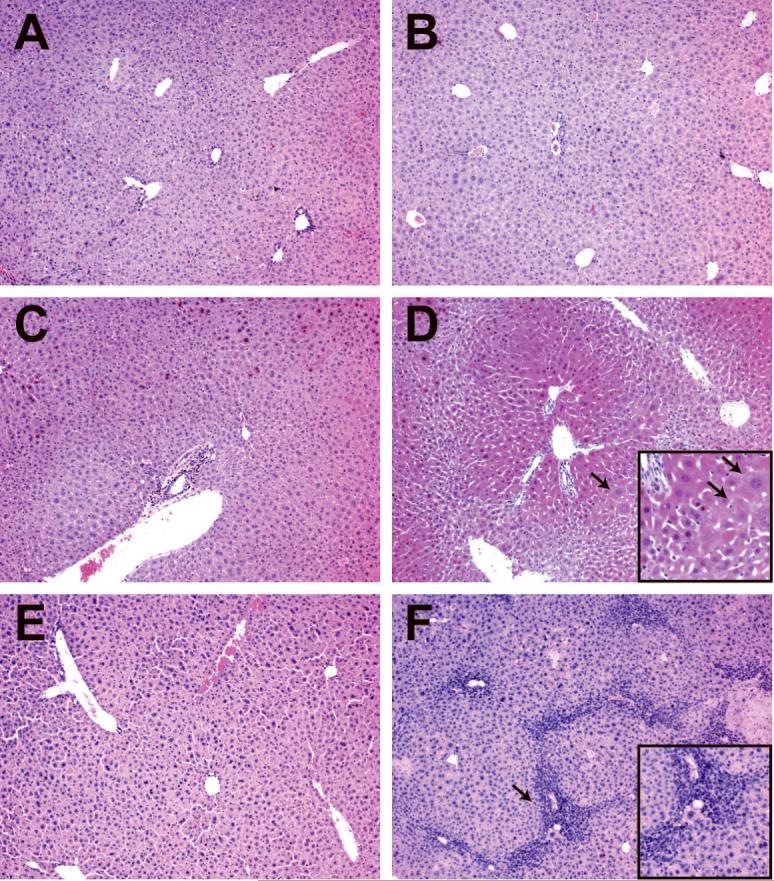
Hematoxylin and eosin stained liver sections. A, C, E: Controls (AlfpCre-; Dicer1flox/flox). B, D, F: Mutant, AlfpCre+; Dicer1flox/flox. A, B: P28. No significant differences were observed between control and mutant tissue. C, D: P72. Hepatocytes close to portal tracts appear to be enlarged (arrows) relative to those close to central veins, resulting in a pseudonodular appearance. E, F: P101. Mutant tissue exhibits extensive infiltration of inflammatory cells, particularly in peri-portal regions (arrows).
Consistent with their surface appearance, P72 mutant livers exhibited a clear tinctorial difference in H&E sections, with a pseudo-nodular histology due to the enlargement of hepatocytes surrounding portal tracts and the compaction of hepatocytes surrounding central veins (Fig 2D). In P72 mutant livers, a significant but variable degree of inflammation was evident relative to control tissue. By P101, there was moderate to severe portal inflammation, and patches of necrosis were seen throughout the liver (Fig. 2F).
Although P72 mutant livers had a nodular appearance, trichrome staining indicated no significant fibrosis in the lobule (Fig 3A-B). In older animals, reticulin staining revealed hepatocyte loss and collapse of the normal hepatic architecture, particularly in areas between closely apposed portal tracts (Fig. 3C-D). Immunofluorescent antibody staining with cytokeratin-19 antibodies revealed both significant ductular proliferation adjacent to portal tracts as well as the presence of isolated cytokeratin-positive cells in the lobule, which were not observed in control tissue (Fig. 3E-F). In addition, the cytokeratin signal in the mutant tissue was much more intense than in control tissue.
Figure 3. Dicer1 mutant livers exhibit hepatocyte loss and increased CK19 expression.
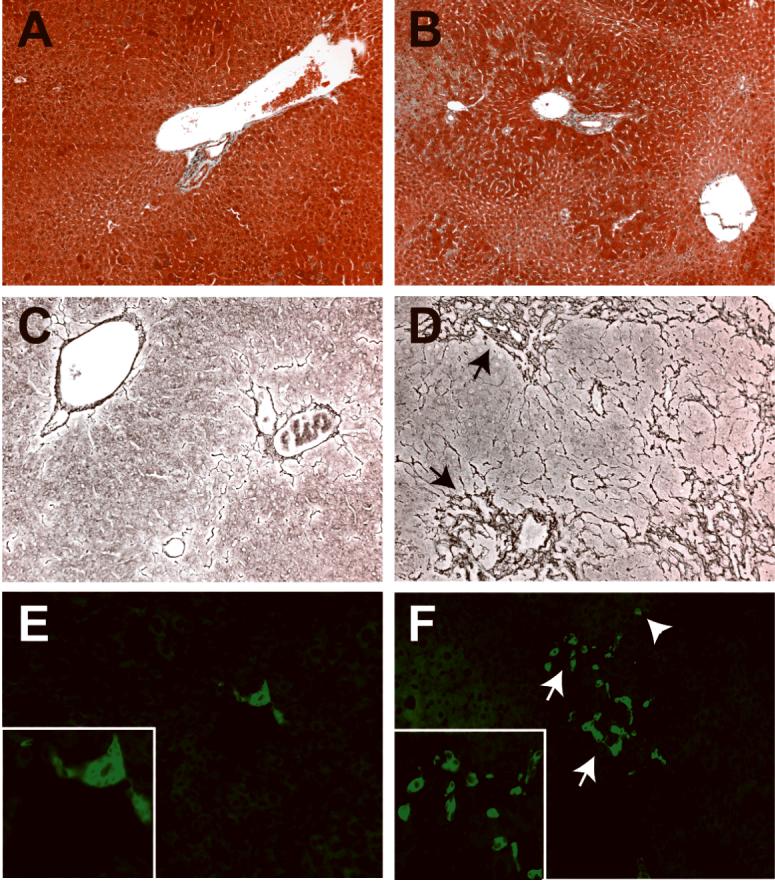
A, C, E: Controls. B, D, F: Mutants. A, B: Trichrome staining of P72 liver sections reveals that despite the pseudonodular appearance of the mutant tissue, no fibrosis is evident at this age. C, D: Reticulin staining of P101 mutant liver sections to highlight the sinusoidal architecture reveals extensive compaction in peri-portal regions (arrows), indicating loss of hepatocytes. E, F: CK19 immunofluorescent antibody staining of P101 liver sections reveals a ductular reaction (arrows), with increased numbers of CK19-positive cells, both in portal areas and occasionally within the lobule (arrowhead). The intensity of CK19 in the mutant sections was increased relative to controls.
Microarray analysis reveals extensive changes in gene expression in the absence of Dicer1
We performed microarray analysis to identify gene expression changes associated with the Dicer1 mutant phenotype. Total RNA from control and mutant P28 livers was subjected to a single round of amplification, biotin labeled, and used to probe a GeneChip expression microarray. Statistical analysis of the resulting microarray data with a false discovery rate (FDR) of 1% indicated that the expression of over 1,600 genes was significantly altered. Consistent with the generally mild influence of miRNA on mRNA levels, most of these expression changes were less than 2-fold; 130 genes were up-regulated by at least 2-fold, while 155 genes were down-regulated to a similar extent. Gene set enrichment analysis (GSEA) using gene sets from the KEGG, BioCarta, and GenMAPP databases revealed no pathways with significant enrichment at an FDR of 25% (24, 25). In contrast, GSEA using the miRNA target gene motif gene set (27) revealed significant up-regulation of 111 out of a total of 200 sets of predicted miRNA targets (Fig. 4A). No miRNA target gene set was found to be down-regulated, consistent with the role of miRNA as repressors of gene expression. These findings further confirm the loss of Dicer1 activity and support the bioinformatic approach used by Xie and colleagues to define the miRNA target gene sets (27).
Figure 4. Microarray analysis of gene expression in Dicer1 mutant liver.
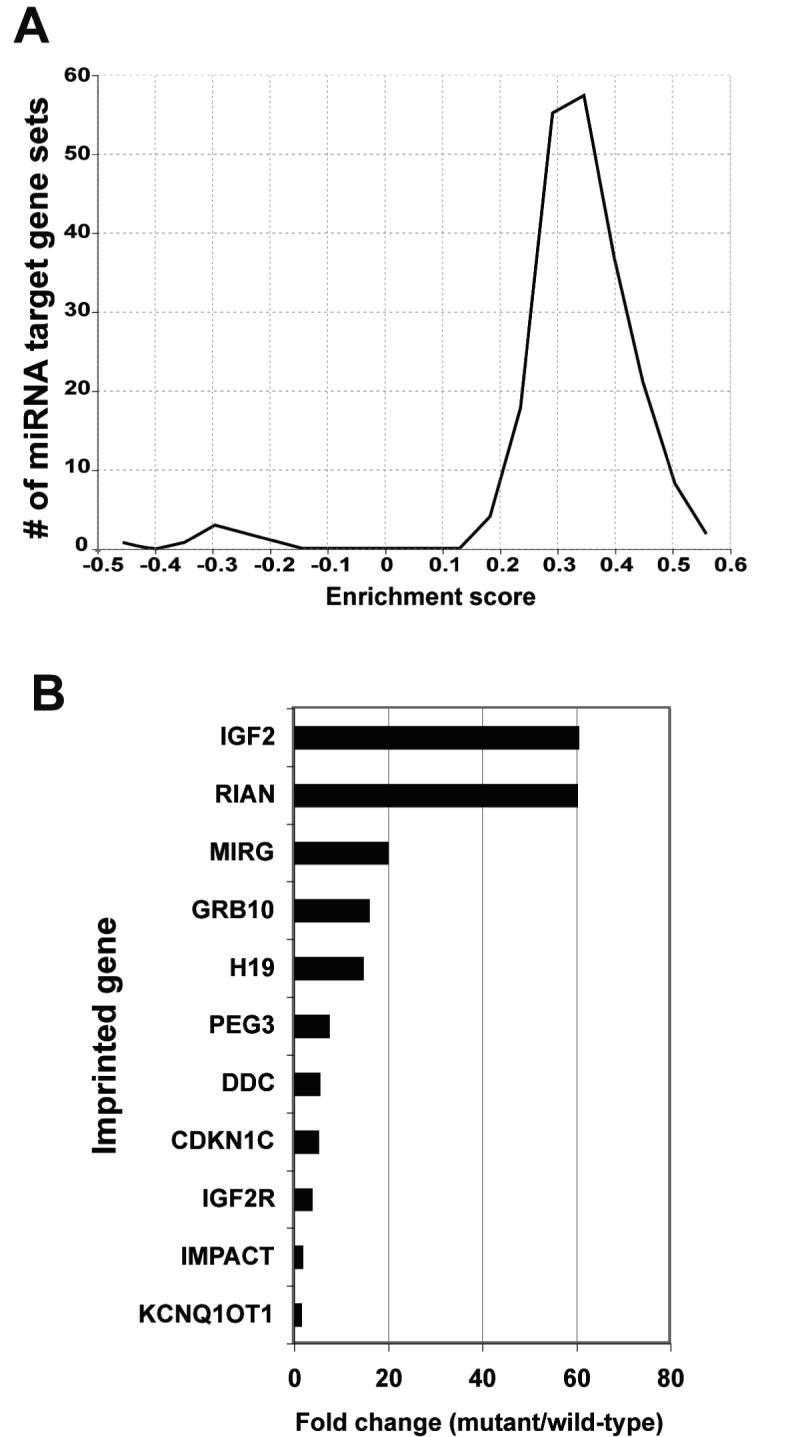
A: Global histogram of gene set enrichment scores for the 192 sets of predicted miRNA target genes. The data is remarkable for the asymmetry between the large number of sets having increased expression and the absence of any sets having decreased expression in Dicer-deficient liver. B: The expression of several imprinted genes is increased in the Dicer1-deficient liver, as determined by microarray analysis (n=5).
Despite the lack of significantly altered pathways as defined by KEGG, BioCarta, and GenMAPP, inspection of the most significantly up-regulated genes revealed a number of gene expression changes which may be of importance with respect to the Dicer1 mutant phenotype. The most highly up-regulated gene was Afp (60-fold), suggesting that the normal loss of Afp expression during hepatic differentiation is dependent on miRNA. The increase in Afp also suggested that the hepatocytes were in a proliferative state.
Among the most highly up-regulated genes in the Dicer1 mutant mice were several that are regulated by methylation-based imprinting, including Glt2, Igf2, H19, Igf2r and Kcnq1ot1 (Fig 4B). To further explore this finding, a gene set comprising 100 known imprinted mouse genes was used in GSEA of the microarray data. This gene set was positively enriched in the mutant mice at an FDR of <6%, suggesting a link between Dicer activity and the silencing of imprinted genes.
Cellular turnover is increased in AlfpCre; Dicer1flox/flox mutant livers
As the P28 microarray data and the increase in Afp led us to expect an increase in cell proliferation, we performed immunohistochemical detection of Ki67, which marks actively dividing cells. The presence of significantly more numerous Ki67-positive cells confirmed increased cellular proliferation in the mutant liver at all post-natal time points (Fig. 5A-C). Furthermore, transcript levels of the growth-associated cyclin, Ccnd1, were elevated in hepatocytes purified from Dicer1-deficient liver (3.6 fold, p<0.001, Fig. 5D).
Figure 5. Dicer1 mutant livers display increased cellular proliferation.
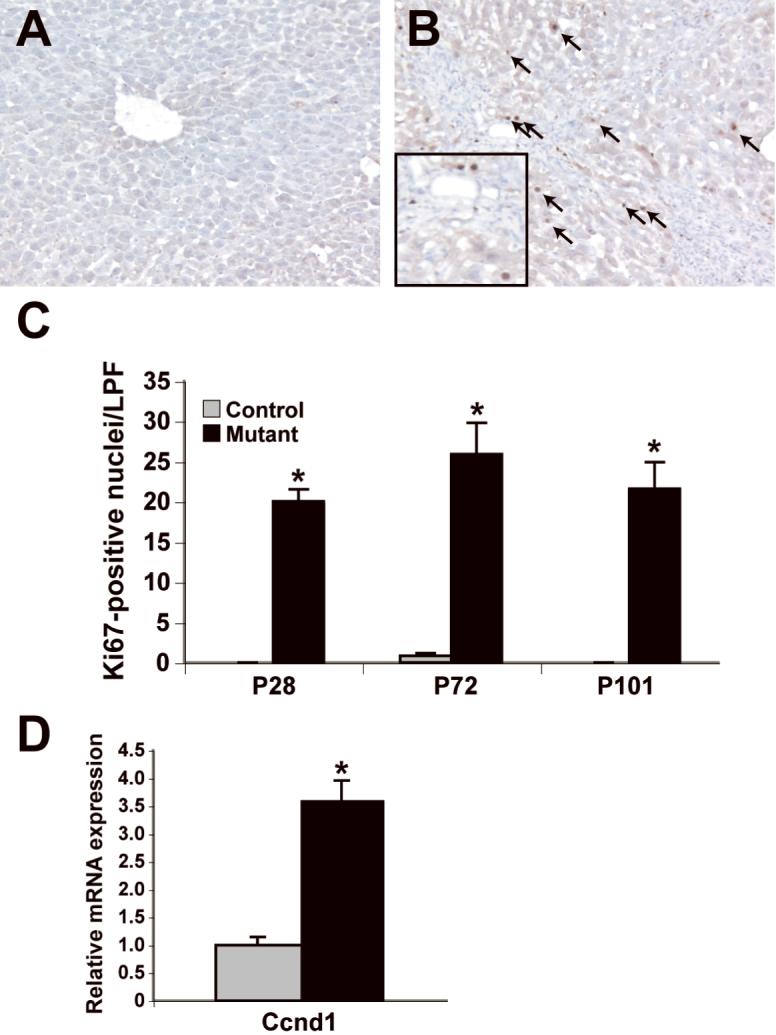
A, B: Ki67 immunohistochemistry on control (A) and mutant (B) liver sections reveals numerous proliferating cells (arrows) in mutant liver (P101 shown). C: Quantification of the results of the Ki67 immunohistochemistry (n=3; *p<0.05). D: Transcript levels of Ccnd1 normalized to Hprt. in P40 purified hepatocytes (control, n=3; mutant, n=5). LPF, low powered field.
To investigate the possibility that the increased cell division was offset by increased apoptosis, we performed TUNEL analysis on mutant and control liver. A significant increase in the number of apoptotic bodies was observed throughout the lobule of Dicer1-deficient liver at all of the post-natal time points examined (Fig. 6A-C). Consistent with this observation, we detected increased mRNA levels of Ccng1 and p53 in hepatocytes purified from Dicer1-deficient liver (14.6 fold, p<3×10−5, and 1.9 fold, p<0.03 respectively, Fig. 6D). Both of these genes are associated with the activation of apoptotic pathways (28).
Figure 6. Dicer1 mutant livers display increased apoptosis.
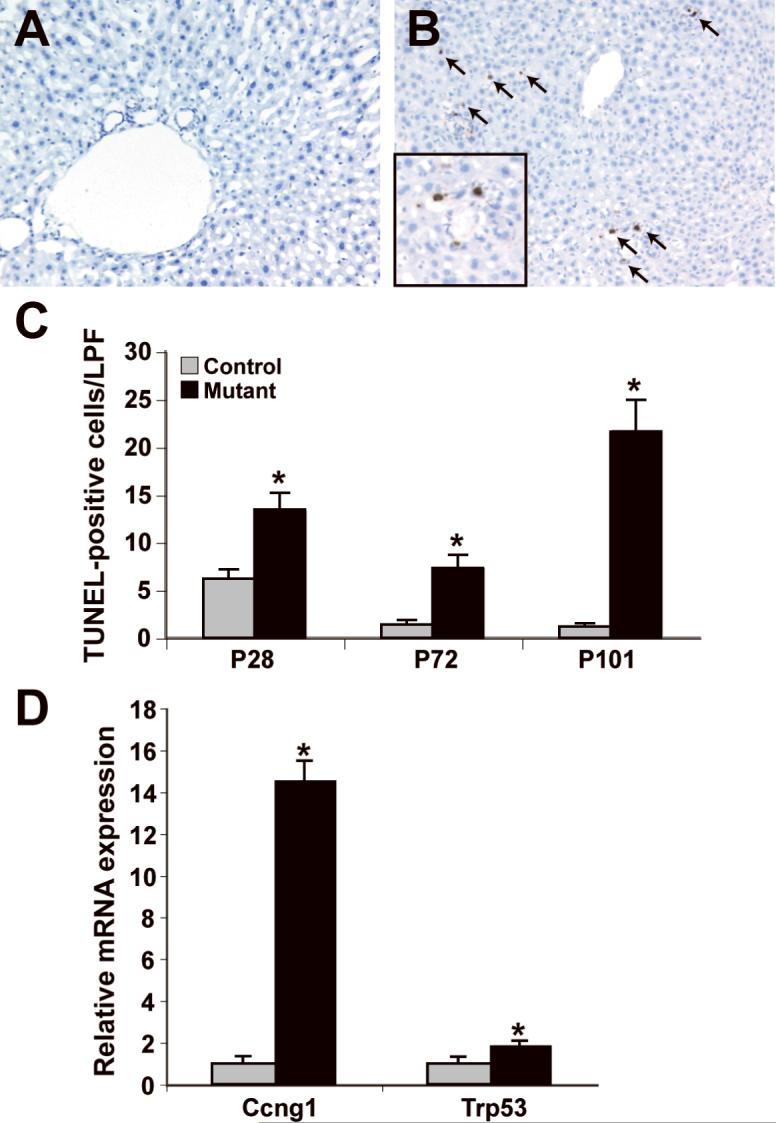
A, B: TUNEL assay of control (A) and mutant (B) liver sections reveals numerous apoptotic bodies (arrows) per low powered field in mutant liver sections (P101 shown). C: Quantification of the results of the TUNEL assay (n=3; *p<0.05). D: Ccng1 and Trp53 expression in P40 purified hepatocytes, normalized to Hprt (control, n=3; mutant, n=5). LPF, low powered field.
It should be noted that the number of proliferating cells was greater than the number of apoptotic cells at P28 and P72, presumably contributing to the increased liver mass at P101 (Table 1). These results indicated that the loss of Dicer activity results in the loss of mature miRNA, increased hepatocyte turnover, and portal inflammation; however, despite these changes overall hepatocyte function is maintained.
Discussion
MiRNAs represent a relatively recently recognized level of gene control. Estimates of the degree to which miRNAs might be involved in regulating the eukaryotic transcriptome have been largely speculative and mostly computational in origin, but a general consensus has emerged that as much as 20-30% of protein-coding genes may be affected by miRNA-mediated control (27). In part due to the way in which candidate miRNAs have been selected for genetic analysis, these effects have been restricted to particular cell-types or organs (for example miR-1-2 in heart, miR-155 in lymphocytes, and miR-223 in granulocytes) (29-31). Genetic loss of individual miRNAs has confirmed that these molecules play important roles in eukaryotic development.
Initial attempts to address the effect of the global loss of miRNA-mediated regulation were hampered by the fact that null mutations in Dicer1 lead to early embryonic lethality (15). More recent studies in which Dicer1 is conditionally deleted due to the action of cell-, tissue-, or stage-specific Cre-recombinase expression in trans to a floxed Dicer1 allele have yielded similar results; loss of Dicer1 function generally leads to autonomous lethality in the affected cells (17-20, 32). The phenotypic effects of loss of Dicer1 in particular organs are therefore difficult to separate from the secondary consequences of the ablation of the cells in which its function is lost.
Here we show the effects of global loss of miRNA processing in liver. The fact that hepatocytes lacking both Dicer1 and mature miRNA function normally for an extended period of time has permitted us to assess the regulatory consequences in a functioning organ of the loss of miRNA-mediated regulation. The Dicer1-deficient liver undergoes a large-scale alteration in gene expression, with statistically significant (but generally mild) alterations in the expression of 1,602 of 25,997 genes assayed relative to control tissue. It should be noted that the effects of some of the changes in gene expression are undoubtedly indirect. Nonetheless, the microarray data serves as a potentially useful starting point in selecting candidate mRNAs for validation as targets of particular liver-expressed miRNAs.
Interestingly, a striking number of imprinted genes are up-regulated in the absence of Dicer1 function in the liver. Previous studies have suggested a role for Dicer1 in processing long non-coding RNA transcripts that mediate transcriptional gene silencing at imprinted loci (as distinct from post-transcriptional miRNA-mediated silencing) (33). Loss of Dicer1 function in mouse embryonic stem cells has been shown to repress cellular differentiation and to de-repress centromeric silencing (34). Consistent with the latter observation, Dicer1 has also been suggested to function in the processing of the long non-coding transcripts that mediate transcriptional gene silencing in plants and yeast (33, 35). The Dicer1 substrate underlying these mechanisms is proposed to originate from repetitive elements within a single transcript, or from overlapping transcripts derived from opposite DNA strands. Similarly, Dicer1 function has been shown to be required in HCT116 colon cancer cells to maintain promoter hypermethylation. Cells homozygously deleted for Dicer1 exon5 (which encodes the helicase domain) re-express a number of genes that are frequently silenced during tumorigenesis (36). Future studies will be directed towards determining whether the effect on imprinted genes is due to the loss of miRNA regulation or to a more direct mechanistic involvement of Dicer1 in gene silencing.
Although the Dicer1 mutant livers in juvenile mice appear to be minimally affected by the dysregulation of a large number of genes, we detected both increased cell proliferation and increased apoptosis. Associated with these histological findings, we observed higher levels of cyclin D1 mRNA, a marker of hepatocyte proliferation (37) and higher levels of transcripts of both cyclin G1 and p53, which participate in a regulatory loop activated during apoptosis (28).
As the Dicer1 mutant mice age, there is evidence of progressive liver damage, with a moderate ductular reaction, extensive inflammation, and an increasing degree of necrosis. While the molecular etiology of this organ damage is unknown, based on the striking similarities to autoimmune hepatitis in humans, we speculate that the increased apoptosis in mutant liver may lead to an autoimmune response that mediates the escalating destruction of liver tissue.
We did not observe a significant effect on cholesterol metabolism in the mutant animals. Previous studies have implicated miR-122a in regulating cholesterol and lipid metabolism in the liver (10, 12). We suspect that additional miRNAs contribute to the regulation of these metabolic pathways, and that global loss of all miRNAs masks the phenotypes uncovered by the unbalanced inhibition of a single component of the miRNA-mediated regulation. Alternatively, it may be that cholesterol homeostasis is subtly affected in the Dicer1 mutant livers, in which case provocative dietary regimens, such as a high fat diet, might uncover additional phenotypes.
The most highly up-regulated gene in the microarray experiment was Afp, suggesting that the increased proliferation may be in part due to cells reverting to, or remaining in, a hepatoblast-like state. Whether or not the increase in Afp expression is causally related to increased proliferation, together the two factors suggest that Dicer1-deficient livers may be predisposed to hepatocellular carcinoma. Future studies will examine older mice and take advantage of established hepatocellular carcinoma models to determine if malignancy is promoted or retarded.
Our initial interest in eliminating Dicer1 function in the liver was to determine the potential involvement of miRNA regulation in liver development and disease. Although previous characterization of the AlfpCre strain used in this study demonstrated that it is induced during embryonic development and capable of generating Cre-mediated loss of function between E14.5 and E18.5, we found that Dicer1flox; AlfpCre-mediated loss of function did not substantially deplete the wild-type Dicer1 transcript until P0. This may be due to positional effects on the floxed Dicer1 allele or a long half-life of the Dicer1 mRNA. Regardless of the cause, we are unable to conclude from the work presented here that miRNA regulation is dispensable for normal liver embryonic development. We are addressing the developmental consequence of loss of Dicer1 function in ongoing experiments using Cre lines which are anticipated to result in target gene deletion at earlier time points.
Acknowledgments
Support for this work was provided by the Fred and Suzanne Biesecker Pediatric Liver Center, the Children's Digestive Health and Nutrition Foundation, and by NIH grant K08DK070881 to JRF. We are grateful to Matthias Merkenschlager for generously sharing the floxed Dicer1 mouse strain. We thank John Tobias for assistance with, and discussion of, the analysis of the microarray data. Finally, the authors wish to express our gratitude to K.H. Kaestner, and L.E. Greenbaum for critical reading of the manuscript.
Grant support: NIH K08DK070881, Children's Digestive Health and Nutrition Foundation, The Fred and Suzanne Biesecker Pediatric Liver Center)
Abbreviations
- miRNA
microRNA
- E
embryonic day
- P
post-natal day
Footnotes
Financial disclosures: No conflicts of interest exist.
References
- 1.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 4.Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 5.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing. C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knight SW, Bass BL. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001;293:2269–2271. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blakaj A, Lin H. Piecing together the mosaic of early mammalian development through microRNAs. J Biol Chem. 2008;283:9505–9508. doi: 10.1074/jbc.R800002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyd SD. Everything you wanted to know about small RNA but were afraid to ask. Lab Invest. 2008;88:569–578. doi: 10.1038/labinvest.2008.32. [DOI] [PubMed] [Google Scholar]
- 9.Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 10.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 11.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 12.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, Jacob ST, et al. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99:671–678. doi: 10.1002/jcb.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jopling CL, Norman KL, Sarnow P. Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb Symp Quant Biol. 2006;71:369–376. doi: 10.1101/sqb.2006.71.022. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 16.Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, Wilson CB, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damiani D, Alexander JJ, O'Rourke JR, McManus M, Jadhav AP, Cepko CL, Hauswirth WW, et al. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008;28:4878–4887. doi: 10.1523/JNEUROSCI.0828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT, Ullian EM. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28:4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi R, O'Carroll D, Pasolli HA, Zhang Z, Dietrich FS, Tarakhovsky A, Fuchs E. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet. 2006;38:356–362. doi: 10.1038/ng1744. [DOI] [PubMed] [Google Scholar]
- 20.Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. 2007;56:2938–2945. doi: 10.2337/db07-0175. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab. 2005;2:141–148. doi: 10.1016/j.cmet.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Wu Z, Irizarry RA, Gentleman R, Murillo FM, Spencer F. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. 2004. Johns Hopkins University Department of Biostatistics Working Papers. [Google Scholar]
- 23.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RH. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat Methods. 2006;3:27–29. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- 27.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 29.Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008 doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 30.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Schaefer A, O'Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204:1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pauler FM, Koerner MV, Barlow DP. Silencing by imprinted noncoding RNAs: is transcription the answer? Trends Genet. 2007;23:284–292. doi: 10.1016/j.tig.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furuno M, Pang KC, Ninomiya N, Fukuda S, Frith MC, Bult C, Kai C, et al. Clusters of internally primed transcripts reveal novel long noncoding RNAs. PLoS Genet. 2006;2:e37. doi: 10.1371/journal.pgen.0020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ting AH, Suzuki H, Cope L, Schuebel KE, Lee BH, Toyota M, Imai K, et al. A requirement for DICER to maintain full promoter CpG island hypermethylation in human cancer cells. Cancer Res. 2008;68:2570–2575. doi: 10.1158/0008-5472.CAN-07-6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelsen CJ, Rickheim DG, Timchenko NA, Stanley MW, Albrecht JH. Transient expression of cyclin D1 is sufficient to promote hepatocyte replication and liver growth in vivo. Cancer Res. 2001;61:8564–8568. [PubMed] [Google Scholar]