

Abstract
![]()
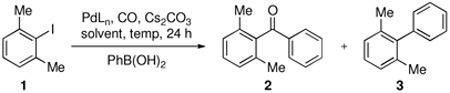
A mild and general protocol for the carbonylative cross-coupling of sterically hindered ortho-disubstituted aryl iodides is reported. Carbonylative Suzuki-Miyaura couplings of a variety of aryl boronic acids provide an array of substituted biaryl ketones in modest to excellent yield. A carbonylative Negishi coupling that utilizes alkynyl nucleophiles is also described.
Aryl ketones and flavanoids are common scaffolds in many natural products and biologically active small molecules.1 Many of these compounds possess substitution at both positions ortho to the ketone moiety. Chemists have most often turned to reactions in the synthome2 that rely upon strongly acidic or basic conditions in order to install the carbonyl functional group. For example, the Friedel-Crafts acylation,3a Fries rearrangement,3b–d and additions of nucleophiles into a variety of acyl electrophiles, including N-methoxy-N-methyl amides,3e have allowed access to ortho-disubstituted aryl ketones. However, obtaining sterically more encumbered ketones is notoriously difficult via the Friedel-Crafts acylation,4 and the Fries rearrangement is limited to phenol derivatives.
An alternative method for the construction of aryl ketones is the three component coupling of aryl halides, carbon nucleophiles, and carbon monoxide that was pioneered by Heck.5 This process is one of the most efficient and direct routes to aryl ketones as it forms two carbon-carbon bonds in a single operation, thereby alleviating the need to introduce the ketone function in a stepwise fashion. The carbonylative coupling has since been further developed to include a range of carbon nucleophiles,6 including tin,7 copper,8 boron,9 zinc,10 aluminum,11 magnesium,12 and silicon.13
During the course of an ongoing synthetic project, we required a reliable method for preparing aryl ketones bearing two ortho substituents. While there are a plethora of methods for synthesizing simple aryl ketones via carbonylative cross-coupling,5d to the best of our knowledge there is only one example of a carbonylative cross-coupling involving an ortho-disubstituted aryl halide with a carbon nucleophile.9b We discovered, however, that the direct application of this protocol to the problem with which we were confronted did not lead to the desired ortho-disubstituted aryl ketone. It was thus necessary to develop a new procedure that would enable efficient carbonylative cross-coupling of different ortho-disubstituted aryl halides with a variety of boronic acids and other nucleophilic partners. We now report the results of some of our findings.
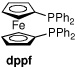
Toward developing a more general process for preparing hindered diaryl ketones, we examined the carbonylative cross-coupling of 2,6-dimethyliodobenzene (1) with phenylboronic acid under a variety of conditions (Table 1). In initial experiments we found that Cs2CO3 and dioxane was the optimal base/solvent combination. Several common phosphine containing catalyst systems were next examined.14 Use of Pd(PPh3)4 and PdCl2(dppf) as catalysts at elevated temperatures and pressures led to consumption of all the starting material; however, the direct coupling product 3 was the major product (entries 1–2) in each case.
Table 1.
Carbonylative cross-coupling of 2,6-dimethyliodobenzene and phenylboronic acida
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | solvent | CO (psi) | temp (°C) | 1:2:3b (% yield)c |
| 1 | Pd(PPh3)4 | dioxane | 60 | 140 | 0:1:8 |
| 2 | PdCl2(dppf) | dioxane | 60 | 140 | 0:1:8 |
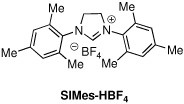
| 3d | Pd(OAc)2/SIMes-HBF4 | dioxane | 60 | 140 | 1:6:2 (50%) |
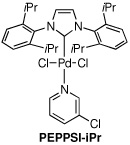
| 4 | PEPPSI-iPr | dioxane | 60 | 140 | 0:6:1 (82%) |
| 5 | Pd(PPh3)4 | PhCl | balloon | 80 | 4:1:10 |
| 6 | PdCl2(dppf) | PhCl | balloon | 80 | 1:6:21 |
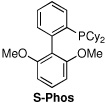
| 7d | Pd(OAc)2/S-Phos | PhCl | balloon | 80 | 2:1:7 |
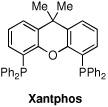
| 8e | Pd(OAc)2/Xantphos | PhCl | balloon | 80 | 1:3:4 |
| 9 | PEPPSI-iPr | PhCl | balloon | 80 | 1:25:0 (95%) |
 |
 |
 |
|||
 |
 |
||||
Selected examples. Reaction conditions: 3 mol % Pd catalyst, 1.0 mmol 2,6-dimethyliodobenzene, 2.0 mmol phenylboronic acid, and 3.0 mmol Cs2CO3 in the indicated solvent (5 mL) at the indicated temperature and CO pressure for 24 h.
Ratios based on integration of the 1H NMR spectrum of the reaction mixture after work-up.
Isolated yield of 2 after chromatography.
Ligand:Pd (2:1).
Ligand:Pd (1:1).
After several other mono- and bidentate phosphine ligands were found to be ineffective, we decided to probe the utility of N-heterocyclic carbene (NHC) ligands.15 NHC ligands have gained popularity in metal catalyzed cross-coupling reactions for several reasons: (1) the steric bulk that they impart around the metal center facilitates reductive elimination; (2) their strong σ-donating character begets facile oxidative addition; and (3) their greater stability at elevated temperatures relative to phosphine ligands enables their use over a broader range of reaction conditions.16
The first supporting ligand that we studied was SIMes-HBF4, and we were pleased to find that the product distribution now favored the desired ketone 2 (entry 3), although further optimization was clearly necessary. We then discovered that when the commercially available PEPPSI-iPr17 catalyst was used under 60 psi of CO, an 82% yield of ketone 2 was obtained; if the reaction was run under a balloon (1 atm) of CO, the simple biaryl 3 was the sole product. To the best of our knowledge, this is the first example of a carbonylative cross-coupling that utilizes the PEPPSI-iPr catalyst. Because it is more convenient to perform such cross-couplings under a balloon of CO, we conducted a search for modified reaction parameters that were amenable to lower CO pressures. Gratifyingly, after screening several solvents, we found that amounts of the ketone 2 could be observed under a balloon of CO when aromatic solvents were used. When the reaction was performed in toluene, α,α,α-trifluorotoluene, anisole or nitrobenzene, the major product of the reaction was ketone 2; however, significant amounts of starting aryl iodide 1 remained. After some experimentation, we discovered that the optimal catalyst/solvent combination for the carbonylative cross-coupling of 1 with phenylboronic acid employed the PEPPSI-iPr catalyst in chlorobenzene (PhCl) to deliver the ketone 2 in 95% yield (entry 9). Other catalysts, such as Pd(PPh3)4, PdCl2(dppf), Pd(OAc)2/SPhos 18 and Pd(OAc)2/Xantphos19 were also examined using PhCl as solvent, but the major product using each of these catalysts was the biaryl 3.
It is intriguing that PhCl was found to be the best solvent for this carbonylative coupling since the PEPPSI-iPr catalyst has been shown to catalyze Suzuki cross-couplings of aryl chlorides.17 We never detected, however, any benzophenone that would arise from cross-coupling with solvent. One possible explanation for this observation is that a CO ligand is deactivating the palladium catalyst owing to its backbonding ability, thus making oxidative addition more difficult.20
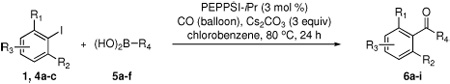
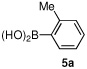
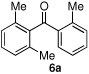
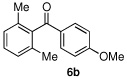
Having optimized the reaction conditions for the cross-coupling of 1 with phenylboronic acid, several different boronic acids and ortho-disubstituted aryl iodides were examined to determine the scope of the process (Table 2). Formation of the trisubstituted benzophenone 6a from 1 and o-tolylboronic acid proceeded in 98% yield (entry 1). The reaction of 1 with 4-methoxyphenyl boronic acid (entry 2) was accompanied by 22% of the corresponding direct coupling product. On the other hand, if this reaction was conducted under 60 psi of CO, the desired biaryl ketone 6b was formed in 92% yield. Use of electron deficient boronic acids was initially more problematic, and only 12% of the desired ketone 6c was isolated from the reaction of 1 with p-cyanophenylboronic acid (entry 3). Electron deficient boronic acids are known to be more prone to side reactions such as homocoupling, and they undergo slower transmetallation than their electron rich counterparts.21 Nevertheless, we found that changing the solvent to dioxane and performing the reaction at elevated temperature and pressure gave the diaryl ketone 6c in 42% yield. Reaction of 1 with 2,6-dimethoxyphenylboronic acid furnished an excellent yield of the tetra-ortho-substituted benzophenone 6d (entry 4). The heteroaromatic ketone 6e was obtained from the reaction of 1 and thiophene-3-boronic acid in 64% yield (entry 5).
Table 2.
Synthesis of ortho-disubstituted aryl ketones via PEPPSI-iPr catalyzed Suzuki coupling at balloon pressurea
 | ||||
|---|---|---|---|---|
| entry | halide | borane | product | yieldb |
| 1 |  |
 |
 |
98% |
| 2 | 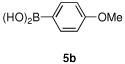 |
 |
72%(92%)c | |
| 3 | 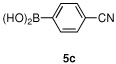 |
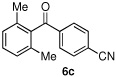 |
12%(42%)c | |
| 4 | 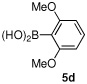 |
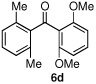 |
95% | |
| 5 | 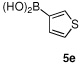 |
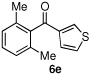 |
64% | |
| 6 | 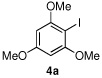 |
5b | 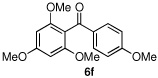 |
52% |
| 7 | 5d | 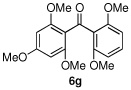 |
33% | |
| 8 | 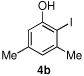 |
 |
 |
51%d |
| 9 | 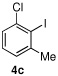 |
5f | 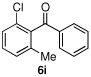 |
89% |
Reaction conditions: 3 mol % PEPPSI-iPr, 1.0 mmol aryl iodide, 2.0 mmol boronic acid, and 3.0 mmol Cs2CO3 in chlorobenzene (5 mL) at 80 °C for 24 h. Reactions were not further optimized unless otherwise noted. The coupling could also be performed on a 1 g scale with no appreciable loss in yield.
Isolated yields are an average of two runs.
Dioxane was used as the solvent; CO pressure increased to 60 psi; temperature increased to 140 °C.
K2CO3 used as base.
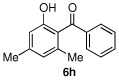
We then turned our attention to the reactions of more electron-rich aryl iodides. In the event, 2-iodo-1,3,5-trimethoxybenzene (4a)22 was converted into several highly oxygenated benzophenones (entries 6 and 7). Despite the modest yields observed in these reactions, these experiments were nonetheless promising as oxidative addition in such cases is presumably disfavored by the highly electron rich nature of the aryl iodide. The more base sensitive 2-iodo-3,5-dimethyl phenol23 was also carbonylatively coupled to phenyl boronic acid under slightly modified conditions employing K2CO3 as the base to give 6h (entry 8). Moreover, the electron deficient aryl iodide 3-chloro-2-iodotoluene underwent facile carbonylative coupling to give the expected ketone 6i in 88% yield (entry 9). The selectivity of the catalyst for the aryl iodide bond in the presence of the aryl chloride bond poses a signifcant advantage, because the aryl chloride can then be further elaborated by subsequent cross-couplings.
We next wished to expand the scope of the cross-coupling to the synthesis of alkynyl ketones, because they are synthetically useful intermediates en route to fused pyranone rings found in natural products.8e,24 Unfortunately, the requisite alkynyl boronic acids were not readily available. Indeed, stable alkynylboranes have been notoriously difficult to prepare due to the lability of the CB bond.25 Nevertheless, we were able to synthesize the alkynylboronic ester 10 from the corresponding lithium acetylide, and we were pleased that upon reaction with 1 under CO, the acetylenic ketone 8a was obtained in modest yield (Table 3, entry 1).
Table 3.
Carbonylative cross-coupling of 2,6-dimethyliodobenzene with alkynyl nucleophilesa
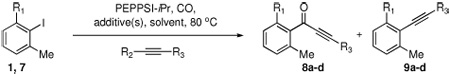 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | additive(s) | solvent | CO (psi) | 1,7:8:9b (% yield) |
| 1 | Me | (iPrO)2B | Cs2CO3 | dioxane | 60 | 4:4:<1 (49%) 8a | |
| 2 | Me | ZnBr | Ph | LiBr | THF/NMP | 60 | 79%c 8b |
| 3 | Me | ZnBr | 4-MeO-C6H4 | LiBr | THF/NMP | 60 | 75%c 8c |
| 4 | OMe | ZnBr | Ph | LiBr, PPh3d | THF/NMP | 170 | 0:3:1 (67%) 8d |
Reaction conditions: 3 mol % catalyst, 1.0 mmol aryl iodide, 2.0 mmol nucleophile, 3.0 mmol additive, 5 mL solvent, 24 h reaction time.
Ratios determined by 1H NMR.
Ratio could not be determined.
3 mol % PPh3 was used.
We then turned our attention toward employing carbonylative Stille (Sn),7 Sonogashira (Cu),8 Hiyama (Si),13 and Suzuki9 couplings that involved air stable potassium organotrifluoroborates (KF3B-R). Despite reasonable experimentation, most of these efforts were rewarded by limited success. In the Sonogashira, Hiyama and Suzuki based protocols, acetylenic ketone 8 was observed in the reaction mixture, but the major product was invariably the direct coupling product 9. Increasing the CO pressure in these experiments did little other than to decrease the amount of starting material that was consumed. On the other hand, the carbonylative Stille coupling delivered a product ratio favoring the desired ketone 8, although unreacted starting material was the major component of the reaction mixture. That the Stille reaction was superior to the others cross-couplings is consistent with the hypothesis that transmetallation is frequently the rate determining step in the Stille reaction.26
There are few examples of carbonylative Negishi (Zn) cross-couplings in the literature, presumably because the higher reactivity of the organozinc reagent tends to deliver the direct coupling product.10 We were thus gratified to find that we obtained the desired acetylenic ketone 8b in 79% yield upon reaction of 1 with an alkynylzinc bromide in the presence of LiBr as an additive (entry 2). Similarly, treatment of 2,6-dimethyliodobenzene with the zinc reagent derived from 4-ethynylanisole furnished a 75% yield of the acetylenic ketone 8c (entry 3). A successful carbonylative cross-coupling was also realized with the more electron-rich 2-iodo-3,5-dimethylanisole27 under slightly modified conditions employing higher CO pressure and PPh3 as an additive to deliver ketone 8d (entry 4).28 In this reaction, it was essential to add PPh3 in order to fully consume the starting material. Although the PEPPSI-iPr catalyst has been shown to be highly efficient in Negishi cross-couplings with a wide range of substrate classes, to the best of our knowledge, this represents the first carbonylative Negishi coupling using the PEPPSI-iPr precatalyst.29 It is noteworthy that the acetylenic ketones produced from this transformation are valuable synthetic intermediates that may be further transformed into many biologically interesting molecules and natural products.
In summary, we have discovered and developed a mild and operationally simple carbonylative Suzuki protocol that enables the efficient cross-coupling of ortho-disubstituted aryl iodides with a range of substituted boronic acids utilizing the commercially available PEPPSI-iPr catalyst. We also found that the carbonylative Negishi cross-coupling of aryl iodides with alkynylzinc reagents delivers acetylenic ketones in good yields. Application of these processes to the synthesis of biologically active natural products will be reported in due course.
Supplementary Material
Experimental procedures, spectral data, and copies of 1H and 13C NMR are available free of charge via the Internet at http://pubs.acs.org
Acknowledgment
We wish to thank the National Institutes of Health General Medical Sciences (GM 31077), the Robert A. Welch Foundation, Pfizer, Inc., Merck Research Laboratories, and Boehringer-Ingelheim Pharmaceuticals for their generous support of this research. N. S. would also like to thank the Arnold and Mabel Beckman Foundation for an undergraduate research fellowship.
References
- 1.For some examples, see:Jiang Y, Tu P. Chem. Pharm. Bull. 2005;53:1164. doi: 10.1248/cpb.53.1164.Nilar, Nguyen L-HD, Venkatraman G, Sim K-Y, Harrison LJ. Phytochemistry. 2005;66:1718. doi: 10.1016/j.phytochem.2005.04.032.Lampe JW, Biggers CK, Defauw JM, Foglesong RJ, Hall SE, Heerding JM, Hollinshead SP, Hu H, Hughes PF, Jagdmann GE, Jr, Johnson MG, Lai Y-S, Lowden CT, Lynch MP, Mendoza JS, Murphy MM, Wilson JW, Ballas LM, Carter K, Darges JW, Davis JE, Hubbard FR, Stamper ML. J. Med. Chem. 2002;45:2624. doi: 10.1021/jm020018f.Rancon S, Chaboud A, Darbour N, Comte G, Bayet C, Simon P-N, Raymond J, Di Pietro A, Cabalion P, Barron D. Phytochemistry. 2001;57:553. doi: 10.1016/s0031-9422(01)00120-0.Ito H, Nishitani E, Konoshima T, Takasaki M, Kozuka M, Yoshida T. Phytochemistry. 2000;54:695. doi: 10.1016/s0031-9422(00)00192-8.Li J-C, Nohara T. Chem. Pharm. Bull. 2000;48:1354. doi: 10.1248/cpb.48.1354.
- 2.Sunderhaus JD, Dockendorff C, Martin SF. Org. Lett. 2007;9:4223. doi: 10.1021/ol7018357. [DOI] [PubMed] [Google Scholar]
- 3.(a) Calloway NO. Chem. Rev. 1935;17:327. [Google Scholar]; (b) Blatt AH. Org. React. 1942;1:342. [Google Scholar]; (c) Bellus D, Hrdlovic P. Chem. Rev. 1967;67:599. [Google Scholar]; (d) Sibi MP, Snieckus V. J. Org. Chem. 1983;48:1935. [Google Scholar]; (e) Sibi MP. Org. Prep. Proced. Int. 1993;25:15. [Google Scholar]
- 4.For a recent example, see:Nakamura H, Arata K. Bull. Chem. Soc. Jpn. 2004;77:1893.
- 5.Heck RF. J. Am. Chem. Soc. 1968;90:5546.Schoenberg A, Bartoletti I, Heck RF. J. Org. Chem. 1974;39:3318.Schoenberg A, Heck RF. J. Org. Chem. 1974;39:3327. (d) For a review on the synthesis of diarylketones by carbonylative coupling, see:Brunet J-J, Chauvin R. Chem. Soc. Rev. 1995;24:89.
- 6.Tamaru Y, Kimura M. Chapter 6. In: Negishi E, de Meijere A, editors. Handbook of Organopalladium Chemistry for Organic Synthesis. 1st ed. Vol. 2. New York: Wiley Interscience: New York; 2002. pp. 2425–2454. [Google Scholar]
- 7.(a) Farina V, Krishnamurthy V, Scott WJ. Org. React. 1997;50:1. [Google Scholar]; (b) Kang S-K, Yamaguchi T, Kim T-H, Ho P-S. J. Org. Chem. 1996;61:9082. [Google Scholar]; (c) Echavarren AM, Stille JK. J. Am. Chem. Soc. 1988;110:1557. [Google Scholar]; (d) Stille JK. Angew. Chem., Intl. Ed. Engl. 1986;25:508. [Google Scholar]; (e) Tanaka M. Tetrahdron Lett. 1979;20:2601. [Google Scholar]
- 8.(a) Sans V, Trzeciak AM, Luis S, Ziolkowski JJ. Catal. Lett. 2006;109:37. [Google Scholar]; (b) Tambade PJ, Patil YP, Nandurkar NS, Bhanage BM. Synlett. 2008:886. [Google Scholar]; (c) Haddad N, Tan J, Farina V. J. Org. Chem. 2006;71:5031. doi: 10.1021/jo060556q. [DOI] [PubMed] [Google Scholar]; (d) Ahmed MSM, Mori A. Org. Lett. 2003;5:3057. doi: 10.1021/ol035007a. [DOI] [PubMed] [Google Scholar]; (e) Torii S, Okomoto H, Xu LH, Sadakane M, Shostakovsky MV, Ponomaryov AB, Kalinin VN. Tetrahedron. 1993;49:6773. [Google Scholar]
- 9.For selected examples, see:Ohe T, Ohe K, Uemura S, Sugita N. J. Organomet. Chem. 1988;344:C5.; (b) Ishiyama T, Kizaki H, Hayashi T, Suzuki A, Miyaura N. J. Org. Chem. 1998;63:4726. [Google Scholar]; (c) Andrus MB, Ma Y, Zang Y, Song C. Tetrahedron Lett. 2002;43:9137. [Google Scholar]
- 10.Wang Q, Chen C. Tetrahedron Lett. 2008;49:2916. and references therein. [Google Scholar]
- 11.Bumagin NA, Ponomaryov AB, Beletskya IP. Tetrahedron Lett. 1985;26:4819. [Google Scholar]
- 12.Yamamoto T, Kohara T, Yamamoto A. Chem. Lett. 1976;5:1217. [Google Scholar]
- 13.(a) Hatanaka Y, Fukushima S, Hiyama T. Tetrahedron. 1992;48:2113. [Google Scholar]; (b) Hatanaka Y, Hiyama T. Synlett. 1991:845. [Google Scholar]; (c) Hatanaka Y, Hiyama T. Chem. Lett. 1989;18:2049. [Google Scholar]
- 14.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 15.Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. [Google Scholar]
- 16.For reviews, see:Kantchev EAB, O'Brien CJ, Organ MG. Angew. Chem. Intl. Ed. 2007;46:2768. doi: 10.1002/anie.200601663.; (b) Cavell KJ, McGuinness DS. Coord. Chem. Rev. 2004;248:671. [Google Scholar]; (c) Herrmann WA. Angew. Chem. Intl. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]; (d) Hillier AC, Grasa GA, Viciu MS, Lee HM, Yang C, Nolan SP. J. Organomet. Chem. 2002;653:69. [Google Scholar]
- 17.O'Brien CJ, Kantchev EAB, Valente C, Hadei N, Chass GA, Lough A, Hopkinson AC, Organ MG. Chem. Eur. J. 2006;12:4743. doi: 10.1002/chem.200600251. [DOI] [PubMed] [Google Scholar]
- 18.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 19.Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM. Organometallics. 1995;14:3081. [Google Scholar]
- 20.Maerten E, Sauthier M, Mortreux A, Castenet Y. Tetrahedron. 2007;63:682. [Google Scholar]
- 21.(a) Operamolla A, Omar OH, Babudri F, Farinola GM, Naso F. J. Org. Chem. 2007;72:10272. doi: 10.1021/jo701918z. [DOI] [PubMed] [Google Scholar]; (b) Wong MS, Zhang XL. Tetrahedron Lett. 2001;42:4087. [Google Scholar]
- 22.Orito K, Hatakeyama T, Takeo M, Suginome H. Synthesis. 1995:1273. [Google Scholar]
- 23.Narender N, Reddy KSK, Mohan KVVK, Kulkami SJ. Tetrahedron Lett. 2007;48:6124. [Google Scholar]
- 24.Nakatani K, Okamoto A, Saito I. Tetrahedron. 1996;52:9427. [Google Scholar]
- 25.Brown HC, Bhat NG, Srebnik M. Tetrahedron Lett. 1988;29:2631. and references therein. [Google Scholar]
- 26.Farina V, Krishnan B. J. Am. Chem. Soc. 1991;113:9585. [Google Scholar]
- 27.For synthesis of this compound, see Supporting Information
- 28.Batey RA, Shen M, Lough AJ. Org. Lett. 2002;4:1411. doi: 10.1021/ol017245g. [DOI] [PubMed] [Google Scholar]
- 29.Organ MG, Avola S, Dubovyk I, Hadei N, Kantchev EAB, O'Brien CJ, Valente C. Chem. Eur. J. 2006;12:4749. doi: 10.1002/chem.200600206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, spectral data, and copies of 1H and 13C NMR are available free of charge via the Internet at http://pubs.acs.org