

Abstract
Computational biology has the opportunity to play an important role in the identification of functional single nucleotide polymorphisms (SNPs) discovered in large-scale genotyping studies, ultimately yielding new drug targets and biomarkers. The medical genetics and molecular biology communities are increasingly turning to computational biology methods to prioritize interesting SNPs found in linkage and association studies. Many such methods are now available through web interfaces, but the interested user is confronted with an array of predictive results that are often in disagreement with each other. Many tools today produce results that are difficult to understand without bioinformatics expertise, are biased towards non-synonymous SNPs, and do not necessarily reflect up-to-date versions of their source bioinformatics resources, such as public SNP repositories. Here, I assess the utility of the current generation of webservers; and suggest improvements for the next generation of webservers to better deliver value to medical geneticists and molecular biologists.
Keywords: SNP, bioinformatics, prediction methods, webservers, review
INTRODUCTION
The rapid growth of genomic tools such as single nucleotide polymorphism (SNP) allele genotyping arrays and next-generation DNA sequencing has produced unprecedented amounts of information about the genotypes of individuals in many species. Yet even when association or linkage studies detect statistically significant correlations between a genomic region and a phenotype, the identity of the causative polymorphism often remains unknown. Tracking down functional SNPs is one of the key challenges of modern genetics, and a new branch of computational biology has emerged to support this effort.
The first computational methods designed to predict the biological impact of SNPs appeared almost a decade ago [1–5]. In subsequent years, a variety of methods have been introduced, reviewed in [6–9], and many now provide websites that take SNPs of interest as input and return annotations, including classifications of biological importance [10–18]. Medical genetics and molecular biology researchers are increasingly turning to these methods and websites as an inexpensive way to prioritize SNPs of interest, prior to functional tests [19–29], and even to select tag SNPs for linkage and association studies [30–33]. These methods incorporate material from computer science, applied mathematics and population genetics, including machine learning, probabilistic modeling, statistics, software engineering and phylogeny. To make technical material accessible, specialized terms have been italicized and are defined in a glossary (Table 1).
Table 1:
Glossary of technical terms
| Affymetrix genotyping array | A microarray designed to identify known single nucleotide polymorphisms, given genomic DNA from an individual. Each SNP allele is represented by multiple short oligonucleotide probes, to which the individual's DNA binds through Watson–Crick base pairing, yielding fluorescence intensities. The intensity of these probes can be used to analyze which DNA base is present at a SNP position and whether the individual of interest carries two identical bases (homozygous) or two different bases (heterozygous) at the position. |
| Amino acid column distributions | A probability distribution describing the amino acid residues seen in an individual aligned column of a protein multiple sequence alignment. This distribution characterizes the probability that each of the 20 amino acids found in proteins will appear in the column. |
| Central or common data model | A data integration paradigm used in software engineering. The central or common data model specifies common rules about how software must access data structures. While individual databases have their own data models, called schemas, the central or common data model defines all the data relationships that exist in a particular software environment. Relationships between source data and the central data model are known as metadata. |
| Clade | A definition used in phylogenetic analysis. It is a taxonomic group that contains a single common ancestor and all the descendants of that ancestor. |
| Coefficient of determination | A statistical term, also known as R2. It is the proportion of variability in a data set that is accounted for by a statistical model. |
| Conserved | A property of entities undergoing evolution. Evolution is change over time, through descent with modification. ‘Conserved’ describes the entities that do not change over evolutionary time. For example, if an amino acid residue type is always seen in a particular position of the sequences from a protein family, it is said to be conserved. A ‘conserved SNP’ is a SNP that is at a conserved sequence position. |
| dbSNP reference identifiers | These are identifiers given to SNPs by the dbSNP database at NCBI. They begin with the letters ‘rs’ and are followed by digits. |
| Decision tree | A machine learning method in which attributes to be tested are organized in a tree structure and a decision is made at each branch point. Each series of decisions results in an overall prediction or classification. |
| Delaunay tesselation | A method used in computational geometry to reconstruct a continuous surface or volume from a discrete set of points. |
| Distributed data integration | Software engineering technology in which multiple databases are not integrated exclusively through a central server (a hub surrounded by spokes), but rather through multiple servers. |
| Domain interface | In a protein structure that has more than one protein domain, a region where amino acid residues from two or more domains are close enough to interact (∼6 Å) apart. |
| Executor kernel | Software engineering term used to describe code that controls the execution process of other code and keeps track of system state. |
| Gene Ontology (GO) | A controlled vocabulary, defined by an international consortium of scientists, known as the Gene Ontology Consortium, which describes gene and gene product attributes. It defines a hierarchy of biological processes and molecular functions. |
| Genome correlation structure | General term to describe non-random associations among DNA sequences either on the same chromosome or on different chromosomes. |
| Genomic range | Distance measured according to number of DNA base pairs, or starting and ending coordinates on a chromosome. |
| Haplotype | Regions of genomic DNA on the same chromosome, which are transmitted together from generation to generation. Also used to describe a set of SNPs on a strand of DNA that are statistically associated. |
| HapMap | An international project that has identified human haplotypes in four ethnic populations. |
| HapMap population | One of the four ethnic populations studied by the HapMap project (HapMap): Yoruba Africans, Han Chinese, Japanese, and the ‘CEPH’ families, who are Caucasians from Utah in the United States. |
| Hidden Markov model | A probabilistic model that was first used in speech recognition and has been useful in representation of related groups of biological sequences. The models contain a series of states, each with its own probability distribution, which estimates the probability that a particular amino acid residue (or nucleotide base) will appear at a position. There are also probabilities assigned to transitions between states. These models can be used to represent protein families. |
| Homolog | A biological sequence with the property that it has a common ancestor with a protein sequence of interest. |
| Homologous protein structure | An experimentally determined (through X ray crystallography or nuclear magnetic resonance spectroscopy) protein structure that is a homolog of a protein structure of interest. |
| HTTP servlet | Software engineering term to describe a Java application that runs on a web server and provides server-side processing, such as database access and HTTP requests from a web browser. |
| Illumina bead array | A technology for identifying SNPs using oligonucleotide probes to which an individual's DNA binds, yielding fluorescence. The probes are attached to silica beads. |
| Intelligent agents | Software agents that can do data mining on the internet, either by following rules or by learning and adapting as they see new data. |
| Ligand binding site | Region on a protein structure where amino acid residues are close enough to a ligand to interact with its atoms (∼5 Å). |
| Linkage disequilibrium | Non-random association between regions of DNA either on the same chromosome or not. |
| Machine learning | A research area within artificial intelligence that focuses on algorithms that are able to learn. A common learning task is classifying examples from two or more categories. |
| Multiple sequence alignment | Alignment of three or more sequences that are assumed to be related through descent from a common ancestor. |
| Multiple sequence alignment column | A column in a multiple sequence alignment that represents part of the conserved core structure of a group of related proteins. |
| Neighbor-joining by sequence identity | A method for constructing a phylogenetic tree from a multiple sequence alignment. Sequences are clustered in a bottom-up, iterative algorithm that uses percent sequence identity (fraction of identical positions) as a similarity measure. |
| Neural network | A machine learning method based on neural organization in the human brain. |
| OMIM | Online Mendelian Inheritance in Man. A database of inherited human mutations in single genes that are known to cause disease. |
| Phylogenetic tree | A tree that represents the evolutionary relationships among a group of nucleotide or protein sequences, assumed to have a common ancestor. |
| Protein domain | A region of a protein that is an independent folding unit. |
| Protein homologs | Proteins that have been inferred to descend from a common ancestor through phylogenetic methods or because their sequences are similar. |
| Protein homology model | A computational model that predicts the 3D Euclidean coordinates of all heavy atoms in a protein of interest, based primarily on an experimentally determined structure of a homolog. |
| Pseudocounts | Technique used in probabilistic modeling in which low probability events are given small probabilities of occurring, even when they are not observed in sample data. Also known as background counts. |
| Random forest | Machine learning method in which hundreds of decision trees (Decision Tree) are combined into an ensemble and a prediction or classification is arrived at by a vote of the entire ensemble. |
| Reconfigurable web wrapper agents | Software engineering tool to automate web browsing sessions using agents, which discover the rules and extract the structure of a web page. |
| Regulatory motifs | Patterns of DNA or mRNA sequence that are the signatures of binding sites for protein or RNA molecules, involved in transcriptional and translational regulation. |
| Sequence profile | Representation of a group of related biological sequences, which estimates the probability that a particular amino acid residue (or nucleotide base) will appear at each position. |
| Sequence weighting | A method used to improve the generalization ability of statistical models of biological sequences. To avoid a group of similar sequences in a data set from dominating the model, sequence ‘subfamilies’ that are overrepresented in the data set are downweighted and sequences that are dissimilar to the rest of the data set are upweighted. |
| Single-marker and two-marker correlations | Metrics of correlation (linkage disequilibrium) between pairs of SNPs (single-marker) and triples of SNPs (two-marker) that are used in selecting the most informative (‘tag’) SNPs for whole-genome association studies. |
| SNP probe libraries | A collection of short oligonucleotides that are used in genotyping microarray and bead technologies. They are designed to bind to pre-designated SNPs, culled from sources such as dbSNP and the HapMap project. |
| Splicing enhancer motif | A probabilistic model of a short sequence of mRNA bases which are the binding target of proteins (‘splicing factors’) involved in splicing. The canonical motif is the most frequently seen sequence of mRNA bases at the binding site of interest. |
| Support vector machine | A machine learning method that is based on ‘decision planes’ to yield maximal linear separation of different classes of data, often through projection into higher dimensions. Reviewed in [77]. |
| Tag SNPs | The most informative SNPs for genome-wise association studies. Tag SNPs have the highest statistical power to detect association. |
| TaqMan | A single-tube PCR (polymerase chain reaction) assay that can be used for SNP genotyping. A SNP is represented by oligonucleotide probes with fluorophores on each end. When the probes hybridize to their targets in sample DNA, a fluorescent dye-specific signal is generated. |
| Web navigation description language | An XML-based language useful in automating data mining over the WWW. |
| Web wrapper agent | Software that automates a user web-browsing session. It visits a website, fills out query forms, and extracts returned data [78]. |
| XML formatted data | Data structured with XML (extensible markup language). XML is a flexible text format with its own grammar, useful for sharing data, primarily over the internet. Like HTML, it has markup symbols, but unlike HTML, these symbols are unlimited and self-defining. |
The SNP function prediction community currently lacks a gold standard. Available methods have been trained and benchmarked on many different data sets (Table 2), and many methods are applicable to only a subset of all SNPs, such as non-synonymous (amino-acid changing) SNPs, or non-synonymous SNPs that can be mapped onto protein structures. Fair assessment of which methods are best is beyond the scope of this review. Instead, I present a survey of available services, discuss trends in the field, and highlight strengths and weaknesses that may be of interest to a potential user of SNP function prediction webservers.
Table 2:
Computational biology SNP prediction webservers fall into three categories
| Name | SIFT | PolyPhen | SNAP | PMUT | PANTHER | nsSNP Analyzer | PhD-SNP | Auto-Mute |
|---|---|---|---|---|---|---|---|---|
| Table 2A: Methods servers | ||||||||
| Computational Methods | conservation among protein homologs | decision tree | Neural network | Neural network | Hidden Markov model of protein family | Random forest | Decision tree coupled to two support vector machines | Random forest, Delaunay tesselation [79] of protein structure. |
| WebsiteURL | http://blocks.fhcrc.org/sift/SIFT.html | http://genetics.bwh.harvard.edu/pph | http://cubic.bioc.columbia. edu/services/SNAP/ | http://mmb2.pcb.ub.es:8080/PMut/ | http://www.pantherdb.org/tools/csnpScoreForm.jsp | http://snpanalyzer.utmem.edu | http://gpcr2.biocomp.unibo.it/~emidio/PhD-SNP/ | http://proteins.gmu.edu/automute/AUTO-MUTE_nsSNPs.html |
| Datatypes | Protein sequences and multiple sequence alignments | Protein sequences, multiple sequence alignments, protein structures | Protein sequences,multiple sequence alignments, predicted protein secondary structures | Protein sequences,multiple sequence alignments, predicted protein secondary structures, protein structure | Protein sequences, hidden Markov models | Protein sequences, multiple sequence alignments, homologous protein structures, protein secondary structure | Protein sequences, sequence profiles | Protein structures |
| Bench-mark or training data | Saturation mutagenesis data sets of two bacterial and one retroviral protein [80–82] | Disease variants and mutagens from SwissProt Variant Pages [83] and presumed netural between-species replace-ments in multiple sequence alignments | The 80 000+ mutations from Protein Mutant Database [84], two bacterial and one retroviral protein [80–82], enzymes with experimentally annotated function in SwissProt [35] | Disease variants from SwissProt Variant Pages [83] and presumed netural between-species replace-ments in multiple sequence alignments | Human Gene Mutation Database [85] for disease mutations and dbSNP [53] for presumably neutral variants | Selected mutants from SwissProt Variant Pages [83] that are mapped onto homologous protein structures | SwissProt Variant Pages [83] | The 1790 disease and neutral variants from SwissProt Variant Pages [83] that can be mapped onto PDB [36] structures |
| Batch input of SNPs? | Yes | No | Yes | Yes | Yes | Yes | No | Up to five SNPs at one time |
| Name | SNP@Domain | PolyDoms | MutDB | Snap | StSNP |
|---|---|---|---|---|---|
| Table 2B: Meta servers | |||||
| Computational Methods | Integration of data from multiple sources | Integration of data from multiple sources | Integration of data from multiple sources | Integration of data from multiple sources | Integration of data from multiple sources |
| Website URL | http://snpnavigator.net | http://polydoms.cchmc.org | http://mutdb.org | http://snap.humgen.au.dk | http://glinka.bio.neu.edu/StSNP |
| Datatypes | Protein multiple sequence alignments, protein structures, predicted functional effects, disease annotations | Protein multiple sequence alignments, protein structures, GO categories [86], disease annotations, pathways, interacting protein networks, mammalian phenotypes, predicted functional effects. Includes synonymous SNPs | Genomic DNA, mRNA transcripts, protein sequence, protein multiple sequence alignments, protein structures, pathways, disease annotations. Includes intronic, untranslated region, and and synonymous SNPs | Genomic DNA, mRNA transcripts, protein sequence, phylogenetic trees, interacting protein networks, diseases, post translational modifications, splice sites | Genomic. DNA, protein sequence, pathways, protein structures, protein homology models |
| Benchmark or training data | N/A | A total of 1338 SNPs from 611 candidate genes with known disease mutations (ftp://ftp.ncbi.nih.gov/snp/Entrez/snp_omimvar.txt) | N/A | N/A | N/A |
| Batch input of SNPs? | Yes | No | Yes | Yes, if in the same gene | Yes |
| Name | PupaSuite | SNP function portal | SNPselect | F-SNP |
|---|---|---|---|---|
| Computational Methods | Integration of data from multiple sources | Integration of data from multiple sources | Integration of data from multiple sources | Integration of data from multiple sources |
| Website URL | http://pupasuite.bioinfo.cipf.es | http://brainarray.mbni.med.umich.edu/Brainarray/ Database/SearchSNP/snpfunc.aspx | http://snpselector.duhs.duke.edu/hqsnp36.html | http://compbio.cs.queensu.ca/F-SNP/ |
| Data types | Genomic DNA, mRNA transcripts, protein sequence, haplotypes. Regulatory SNPs, synonymous SNPs, intronic SNPs, untranslated region SNPs, intergenic SNPs, nonsense and frameshift mutations, protein structure, cellular processes, functional sites, evolutionary selection strength dN/dS, and epigenetic effects (triplex DNA regions). Human, mouse and rat included | Genomic DNA, mRNA transcripts, protein sequences, protein structures and homology models, pathways, diseases, and haplotypes, (gene expression is under construction) | Applied Biosystems and Illumina SNP data, genomic DNA and haplotypes | Protein sequences, protein structures protein homology models, mRNA transcripts, predicted functional impact on protein structure, splicing regulation, post translational modifications and evolutionary conservation |
| Benchmark or training data | N/A | N/A | A total of 700 SNPs from 140 genes associated with cardio-vascular disease in [87] | N/A |
| Batch input of SNPs? | Yes | Yes | Yes | Yes, if in same gene or genomic region |
| Name | LS-SNP | SNPeffect | SNPs3D | FastSNP | MutaGeneSys |
|---|---|---|---|---|---|
| Table 2C: Hybrids | |||||
| Computational Methods | Support vector machine | Integration of data from multiple sources | Support vector machine | Decision tree | Identifies indirect correlations between SNPs and mutations from OMIM [13] |
| Website URL | http://karchinlab.org/LS-SNP | http://snpeffect.vib.be/ | http://www.snps3d.org/ | http://fastsnp.ibms.sinica.edu.tw | http://magnet.c2b2.columbia.edu/mutagenesys |
| Datatypes | Protein sequences, multiple sequence alignments, protein homology models, predicted domain interfaces, ligand binding sites, hidden Markov models, genes, pathways, genomic DNA | Predicted changes in protein stability and folding, aggregation and amyloidosis, catalytic sites and binding sites, phosphorylation and glycosylation sites, cellular localization and protein turnover. | Protein sequences, multiple sequence alignments, profiles, protein structures, genes, gene networks, disease candidate genes, GO categories [86], mouse knockout data | Genes, genomic DNA, mRNA transcripts, protein sequences, protein domains | Genomic sequence, haplotypes, linkage disequilibrium data |
| Benchmark or training data | A total of 1457 disease-associated variants from SwissProt [35] which could be mapped to the OMIM database [59] and and 2504 putatively neutral nsSNPs from dbSNP [53] | N/A | A total of 10 263 deleterious mutants in 731 proteins from Human Gene Mutation Database [85] and 16 682 control substitutions in 348 proteins from aligned positions of close orthologs | A total of 1569 SNPs from the SNP500 Cancer database [88] | N/A |
| Batch input of SNPs? | Yes | Yes | Yes | Yes | Yes |
(A) Methods servers primarily disseminate an original computational method for SNP function prediction. (B) Meta-servers pull information from many severs, including general purpose protein and genomic annotation bioinformatics servers and servers from category. (C) Hybrids both disseminate original method(s) and pull information from other servers. Technical terms have been italicized and can be looked up in the Glossary (Table 1).
SNP webservers: strategies and communities
Today's SNP prediction servers generally use one of three strategies (Table 2):
methods servers that disseminate results of original computational method(s);
metaservers that pull information from many servers, including general purpose protein and genomic annotation bioinformatics servers; and
hybrids that both disseminate original method(s) and pull information from other servers.
All of these servers are built on top of an infrastructure of general bioinformatics resources that curate SNPs, genomic and protein sequences, protein structures, interactions, pathways and regulatory elements (such as sites important for transcription factor binding and accurate splicing). The relationships among SNP webservers and other bioinformatics resources can be represented as a directed graph (Figure 1). Partitioning the graph with an algorithm based on local modularity [34] yields three main communities, which can be loosely defined as: the protein community (ellipse), connected to the large, core bioinformatics databases UniProt [35], Protein Data Bank (PDB) [36], Structural Classification of Proteins (SCOP) [37], Biomolecular Interaction Network Database (BIND) [38], Molecular Interactions Database (MINT) [39], Gene Ontology (GO) [40], Kyoto Encyclopedia of Genes and Genomes (KEGG) [41] and BioCarta (http://www.biocarta.com); the regulatory community (trapezoid) connected to webservers that predict post-translational modifications, splicing enhancers and repressors and transcription factor binding sites (TFBSs); and a regulatory plus linkage disequilibrium community (rectangle), which is connected to the HapMap webserver (http://www.hapmap.org). Core resources such as National Center for Biotechnology Information (NCBI) databases (http://www.ncbi.nlm.nih.gov), UCSC Genome Browser [42] (http://genome.ucsc.edu) and Ensembl [43] (http://www.ensembl.org) are used by all three communities. A singleton server (white rectangle), the SNP Function Portal [14], is connected to both protein and linkage disequilibrium communities, and perhaps represents an emergent fourth community. Protein community webservers primarily predict the biological importance of non-synonymous SNPs, using properties such as evolutionary conservation of amino acid sequence, protein structure and protein binding interactions. These properties are often combined in ‘black box’ machine learning algorithms—neural networks, support vector machines and random forests—yielding predictions that are difficult to understand from a biological point of view. The regulatory community primarily harvests predictions from external servers that specialize in identification of regulatory motifs. Although these methods were not designed specifically for SNPs, they can be used, at least in theory, to predict the effect of the SNP on normal patterns of regulation. The third community contains websites connected to resources that provide information about genomic linkage disequilibrium structure.
Figure 1:

Directed graph of relationships among SNP prediction webservers and their bioinformatics sources. A heuristic partition of the graph identifies three communities. They are loosely defined as (1) focus on protein properties (ellipse); (2) focus on regulation (trapezoid); and (3) connect to HapMap and consider linkage disequilibrium among SNPs (rectangle). SNPFunctionPortal is a singleton, perhaps an emerging community (white rectangle) that is equally connected to the protein and LD communities. LD = linkage disequilibrium.
The ‘protein community’ is the largest and the oldest. But the general landscape is shifting towards inclusion of regulatory SNPs and consideration of inter-SNP associations through linkage disequilibrium (Figure 2a). The landscape may also be shifting away from methods servers towards meta-servers and hybrids (Figure 2b).
Figure 2:

Trends in scope of SNP webservers. (a) Prior to 2006, protein-based servers that only handle non-synonymous SNPs were predominant. Newer servers include regulatory SNPs and annotate associations among SNPs through linkage disequilibrium estimates. (b) The earliest servers implemented original computational methods, but the current trend is towards meta-servers and hybrids. Dates associated with each server are based on date of first journal publication, unless an alternate date is documented on its website.
The webserver graph (Figure 1) shows that there is not much feedback to the servers from their sources, although this may change with time. There is currently one exception—a feedback loop connecting two SNP servers in the regulatory/linkage disequilibrium community—SNPeffect [13] and PupaSuite [12]. These servers are synchronized and describe their relationship as a joint effort to cover both protein and regulatory related SNPs. Such relationships may become more common in the next generation.
FIELD TESTING OF CURRENT WEB SERVERS
To assess their usability and scientific utility, I evaluated 22 severs by submitting to each a set of SNPs that were reported to be associated with disease in recently published medical literature. All submissions were done using Firefox 2.0.0.13 on Windows XP Professional Edition. The field tests were done during the week of 28 April 2008. One server returned no results and inquiry emails went unanswered. It was eliminated from the assessment (Pmut [44]). Detailed descriptions of all field tests are provided (Supplementary Tables S2, S3 and S4) with the main results summarized in this section. Since all of these websites were designed by bioinformaticians, it is not surprising that all of them require some bioinformatics expertise on the part of the user. For each server tested, I provide an assessment of the expected user skill set. General definitions of basic bioinformatics skills and expert bioinformatics skills are also provided (Table 3).
Table 3:
Basic and expert bioinformatics skill levels
| Basic skill level | Basic ‘data types’—sequences and structures of DNA, RNA and protein and their representations in bioinformatics web databases. |
| Retrieving information from bioinformatics web databases. | |
| Pairwise and multiple sequence alignments. | |
| Higher level organization of and relationships between the basic data types—biochemical pathways, biochemical functions, gene and protein families, protein folds, motifs and regulation. | |
| Expert skill level | Algorithms used for biological data search and analysis. |
| Relationship of protein sequence, structure and function. | |
| Protein structure modeling and its limitations. | |
| Sequence alignment and motif-matching algorithms, how to interpret their output scores and also their limitations. | |
| Probability and statistics—extreme value distributions, hidden Markov models, Bayesian networks, profiles, Fundamentals of molecular evolution. | |
| Fundamentals of machine learning methods used in bioinformatics. Neural networks, support vector machines, random forests, decision trees. Strengths and weaknesses of these methods. | |
| Metrics for comparing the performance of classification methods—receiver operating characteristic curves, specificity versus sensitivity. | |
| Skill using a molecular visualization package, such as Chimera [89], PyMol [90] or RasMol [91]. |
Webservers tested in this study implicitly assume that users have familiarity with the concepts listed under the appropriate skill level.
ALS/FTLD study: novel SNPs discovered in sequencing
Novel SNPs are often discovered through DNA sequencing studies that compare individuals with a condition of interest to a control population. In a recent study of familial amyotrophic lateral sclerosis (ALS) with frontotemporal lobal degeneration (FTLD), researchers investigated sequence variation in the gene TARDBP [45]. All coding exons, most of the 5′-untranslated region, and approximately 100 intronic bases upstream and downstream of each exon were sequenced for 259 ALS/FTLD patients and 1127 controls. The TARDBP (NM_007375) variants 869G->C (amino acid change G290A) and 892G->A (amino acid change G298S) were found to be statistically associated with disease, and putatively linked both with loss and/or gain of protein function.
All of the ‘Methods Servers’ are capable of handling novel non-synonymous SNPs, because they offer the ability to submit a protein sequence along with a residue position and amino acid substitution. None of the ‘Hybrid Severs’ or ‘Meta-servers’ allows submission of protein sequences, but one of the ‘Meta-servers’ (FAST-SNP [15]) handles novel SNPs of all kinds, by allowing the user to submit a DNA sequence plus base position and nucleotide substitution. The TARDBP SNPs were submitted to the SIFT [4], PolyPhen [10], SNAP [17], PMUT, PANTHER [18], nsSNPAnalyzer [46], PhD-SNP [47], Auto-mute [48] and FAST-SNP servers. In cases where servers offered a choice of parameter settings, defaults were used. Generally, the servers reported results that were understandable, if accepted on face value. Most predicted that both SNPs are neutral, and the predictions that disagreed with neutrality were low confidence (Table 4). The servers varied widely in terms of communicating prediction reliability. Some have no confidence measures and some have a simple binary (yes/no) confidence measure. The SNAP server provides the most detailed confidence information, including both a reliability index and an estimated accuracy rate for each prediction. In general, the results are qualitative, rather than quantitative, reflecting the current state-of-the-art of webserver-based SNP function prediction.
Table 4:
Field test of novel SNPs discovered in sequencing
| Classification |
Score |
Prediction confidence |
||||
|---|---|---|---|---|---|---|
| G290A | G298S | G290A | G298S | G290A | G298S | |
| SIFT | Affects protein function | Tolerated | 0.03 | 0.41 | Low | Good |
| PolyPhen | Benign | Benign | 1.016 | 0.038 | – | – |
| SNAP | Neutral | Neutral | – | – | 53% | 89% |
| Panther | – | – | – | – | – | – |
| nsSNP analyzer | – | – | – | – | – | – |
| PhD-SNP | Neutral | Disease | – | – | 4 | 0 |
| Auto-mute | – | – | – | – | – | – |
| FAST-SNP | – | – | – | – | – | – |
The TARDBP SNPs 869G->C (amino acid change G290A) and 892G->A (amino acid change G298S) were found to be statistically associated with disease in a case/control study of familial ALS with FTLD and putatively linked to loss and/or gain of protein function. Both SNPs were submitted to eight “Methods Servers” (Table 2A) for SNP function prediction to evaluate required user skills and agreement with associations from the case/control study. Three of the webservers were unable to classify these SNPs for technical reasons (detail in Supplementary Tables 2). FAST-SNP does not classify novel SNPs with respect to overall impact on disease risk, but it predicted that a TFBS might be affected by both of these SNPs. ‘–’ = not provided.
Required user skills
Basic bioinformatics skills (Table 3) such as ability to find and handle data, accession numbers and reference identifiers in web databases such as UniProt, PDB and NCBI.
Bioinformatics expertise (Table 3) is required to understand server errors. Two servers returned error messages that assumed users know about hidden Markov models and protein structural homology.
Bioinformatics expertise is required to think critically about how to interpret server results and their significance.
Interpreting server results
SIFT
In addition to predicting SNP functional impact, SIFT builds a protein multiple sequence alignment of the protein of interest and emails it to the user, allowing alignment analysis with bioinformatics software. I used the SIFT TARDBP alignment to build a phylogenetic tree, using neighbor-joining by sequence identity in JALVIEW [49]. Human TARDBP is located in a distinct clade on this tree. The G290A and G298S SNPs are in a glycine-rich domain that is present only in this clade and appears to be an evolutionary late comer in the TARDBP protein family. Sequence annotations, available through JALVIEW links to European Bioinformatics Institute (EBI) resources, indicate that human proteins in the clade are expressed in brain tissues, rendering plausible the hypothesis that at least one of these SNPs, or a SNP in this protein domain that is in linkage disequilibrium with these SNPs, could contribute to ALS/FTLD, a brain disorder.
FAST-SNP
According to FAST-SNP, the sequence surrounding the SNPs is a significant match to a predicted TFBS, but TFBS predictions are generally not reliable unless the prediction is in a known promoter region. Given that this region is within a coding exon, one should be suspicious of this prediction. FAST-SNP submitted the sequence to three splicing regulatory analysis servers: ESEFinder [50], Rescue-ESE [51] and FAS-ESS [52]. Only one of the three predicted anything. That prediction is that 869G->C (G290A) introduces a significant match (CTAATAG) to the canonical splicing enhancer motif CAGAGGG, which is bound by SF2/ASF proteins. Altogether, this raises the interesting possibility of impact on the regulatory level rather than the protein level.
A user of these SNP methods servers who sees their outputs only on a surface level would conclude that the two ALS/FTD SNPs are neutral. However, a user with bioinformatics expertise (Table 3) might use the server results to suggest testable hypotheses about how these SNPs could affect biological function.
Schizophrenia study: common intronic SNPs
When case–control studies are done with microarray or TaqMan technologies that use SNP probe libraries, researchers may find SNPs in which the frequency differences between cases and controls are statistically significant. These SNPs are not novel, and are already indexed in large databases such as dbSNP [53]. A recent study compared two large schizophrenia populations to ethnically matched controls [54]. Seven SNPs in the introns of PDE4B, which encode a large phosphodiesterase involved in cAMP signaling regulation, were found to be significantly associated with schizophrenia (dbSNP reference identifiers: rs4320761, rs910694, rs1354064, rs1321177, rs2144719, rs1040716 and rs78038).
The ‘schizophrenia SNPs’ were submitted to five servers that handle intronic SNPs: SNPselector [55], PupaSuite, FASTSNP, F-SNP [56] and MutaGeneSys [57] (Table 5). None of the SNPs were predicted to have functional impact by SNPselector, FASTSNP and MutaGeneSys. PupaSuite reported that rs910694 is in a DNA triplex region, a region of DNA with three strands. These regions play a role in repression of transcription, reviewed in [58], thus this SNP could putatively disrupt normal regulation of PDE4B. F-SNP identified three of the SNPs (rs1354064, rs4320761 and rs1040716) as being involved in transcriptional regulation and rs1040716 as being at a position that is conserved among species.
Table 5:
Field test of common intronic SNPs
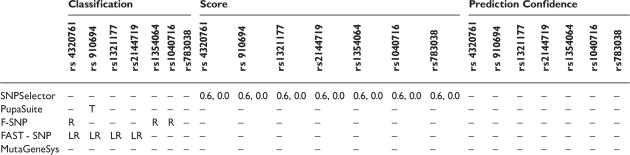 |
The PDE4B intronic SNPs rs4320761, rs910694, rs1354064, rs4320761, rs1321177, rs2144719, rs1040716 and rs78038 were found to be statistically associated with schizophrenia in a case/control study. The SNPSelector score ‘0.6’ means that the SNP is in an intron, but not at a exon–intron junction. The SNPSelector score ‘0.0’ means that the SNP is predicted to not be important for regulation of transcription. R = biologically important because of impact on regulation of transcription. LR = low risk (general assessment with respect to increases in disease susceptibility). T = in DNA triplex region ‘–’= not provided.
Required user skills
Submitting queries to these servers requires no special skills, just dbSNP reference identifiers for a SNP of interest.
Bioinformatics skills are required to understand outputs of SNPselector, PupaSuite and F-SNP, even at a surface level.
Unix skills are required to access SNPselector's results, which are sent by email as a compressed tarball.
F-SNP requires expertise specifically with UCSC Genome Browser tools and terms.
The FASTSNP server outputs are integrated into a decision tree algorithm, which is clearly laid out and understandable to a general user. This feature is not available in FASTSNP's ‘novel SNP’ service.
MutaGeneSys requires some knowledge of statistical genetics, as the user must select a minimum coefficient of determination and has the option of selecting a HapMap population. It reports when a SNP is correlated by linkage disequilibrium with an externally annotated disease-associated SNP, based on OMIM [59]. Both single-marker and two-marker correlations are considered.
MutaGeneSys is a tool aimed at the medical genetics community, where the importance of linkage disequilibrium is well understood. By enabling identification of SNPs that are indirectly associated with disease, it can help users narrow down the number of SNPs likely to have a direct functional effect. The PupaSuite result for rs910694 suggests a testable hypothesis that might explain schizophrenia association.
Esophageal cancer study: mix of common exonic and intronic SNP
Esophageal and esophago–gastric junction adenocarcinomas (EAC and EGJAC) have been linked to acid reflux, obesity and smoking. Risk is also related to exposure to nitrites (found in compounds such as tobacco smoke) that alkylate DNA at the O6 position of guanine [60]. A recent population case–control study in Australia looked at SNPs in DNA-repair genes MGMT, XPD, XRCC1 and ERCC1 to identify possible genetic predispositions to EAC and EGJAC. MGMT (O6-methylguanine-DNA methyltransferase) specifically repairs O6-guanine alkylation damage. Results point to MGMT SNPs rs12268840 (intronic) and rs2308321 (non-synonymous) as being statistically significant in frequency between EAC patients (n = 263) and controls (n = 1337) [60].
The MGMT SNPs were submitted to all 22 servers (those that do not handle intronic SNPs were only queried about rs2308321) (Table 6). None of the servers predicted that rs2308321 has an impact on protein function. Several servers reported that this SNP was found at splicing regulation sites, but only F-SNP predicted that it would impact splicing regulation, because it changes both an exonic splicing enhancer and an exonic splicing repressor. None of the servers predicted functional impact for rs12268840.
Table 6:
Field test of non-synonymous and intronic SNPs
| Classification |
Score |
Prediction confidence |
||||
|---|---|---|---|---|---|---|
| rs2308321 | rs12268840 | rs2308321 | rs12268840 | rs2308321 | rs12268840 | |
| SIFT | Tolerated | – | 0.7 | – | Good | – |
| PolyPhen | Benign | – | 0.378 | – | – | – |
| SNAP | Neutral | – | – | 60% | – | |
| Panther | – | – | 0.24a | – | – | – |
| nsSNP-analyzer | Neutral | – | – | – | – | – |
| PhD-SNP | Neutral | – | – | – | 4b | – |
| Auto-mute | – | – | – | – | – | – |
| SNP@Domain | – | – | – | – | – | – |
| Pmut | – | – | – | – | – | – |
| PolyDoms | – | – | – | – | – | – |
| MutDB | – | – | – | – | – | – |
| Snap | – | – | – | – | – | – |
| StSNP | – | – | – | – | – | – |
| PupaSuite | ESE site | – | 3.13c | – | – | – |
| Snp Function Portal | TFBS | – | – | – | – | – |
| SNPselector | – | – | 1d, 0.0d | 0.6d, 0.0d | – | – |
| F-SNP | ESE site ESR site | – | – | – | – | – |
| LS-SNP | Neutral | – | – | – | High | – |
| SNPEffect | No effect | – | – | – | – | – |
| SNPs3D | Neutral | – | – | – | – | – |
| FAST-SNP | Low-medium risk | Very low-low risk | – | – | – | – |
| MutaGeneSys | No LD to known disease mutations | No LD to known disease mutations | – | – | – | – |
The MGMT SNPs rs2308321 (non-synonymous) and rs12268840 (intronic), found to be significantly associated with esophageal cancers, were submitted to all 22 servers in this study (those that do not handle intronic SNPs were only queried about rs2308321). aPanther score is probability of SNP being deleterious. bMeaning of the PhD-SNP prediction confidence score is not explained on their website. cMeaning of the ESE site prediction score is not explained on their website. dSNPselector score 0.6 describes an intronic SNP that is not at an exon/intron junction. 1.0 describes a non-synonymous (amino-acid changing) SNP. The ‘0.0’ means the SNP is predicted to not be important for regulation of transcription. ‘–’= not provided. ESE = exonic splicing enhancer; ESR = exonic spicing repressor.
Required user skills
The basic skills required to input queries and interpret outputs are the same as described for the TARDBP and ‘schizophrenia SNPs’.
Bioinformatics skills and knowledge of human genome structure allow users to submit advanced input queries. Genomic range is accepted by MutDB [61], Snap [62], PupaSuite, SNP Function Portal [14], F-SNP and LS-SNP [11]. Linkage disequilibrium can be factored into inputs using PupaSuite, SNP Function Portal, SNPselector and MutaGeneSys. In total, 18 distinct input data types are available on the servers tested (Table 5).
Results output of the meta-servers (Table 2B) is generally large, heterogeneous and difficult to integrate without bioinformatics skills. One exception is the FastSNP server, which integrates its harvested data in a decision tree algorithm that is transparent and clearly explained to users.
The only testable hypothesis yielded from these server results was the possibility that splicing regulation of MGMT might be affected by rs2308321. In general, there is poor agreement among servers that harvest predictions of SNP impact on splicing, and the predictions are not associated with clear reliability measures.
Stale data
Most of the tested servers use NCBI's dbSNP [53] database as a primary source of SNP data, but are not up-to-date, increasing the chances that annotations for SNPs of interest will not be available to users. Between 2003 and 2008, dbSNP has been updated, on average 2–3 times per year. Fourteen of the tested servers accept dbSNP rsIDs, and the current dbSNP build is version 129, May 2008. Only one server, FastSNP is using version 128. Seven servers are using version 126; three are using version 125; two are using version 124 and one is still using version 123 (from October 2004) (Supplementary Table 1).
These three field studies suggest a set of desirable features for a SNP webserver:
Options for submission input that require minimal bioinformatics expertise. Even when advanced submission options are available, offering an easy way to input SNPs ensures that a wider community will have access to the server.
Error messages that do not require bioinformatics expertise to understand. Such messages can be confusing and frustrating to users and alienate non-bioinformaticians.
For those with bioinformatics expertise (Table 3), the option to download server outputs such as alignments and protein structure models. The ability to use external bioinformatics software to analyze server output will help bioinformaticians develop testable hypotheses about SNP biological impact.
Quantitative, calibrated measures of prediction reliability. If server output contains many predictions, such as impact on protein structure, impact on exonic splicing, etc., a reliability measure should be provided for each prediction. Without such information, users will have difficulty assessing which prediction is the most likely to be correct.
A method to integrate diverse outputs of heterogeneous data types and to put them in perspective. Without algorithms to integrate and prioritize information available about a SNP, many users will come away with nothing of value.
Ability to handle all kinds of SNPs, possibly through linkouts to other servers. Users will often not know in advance whether SNPs of interest impact protein function or regulation. It is annoying to submit SNPs and find that there is no information about them because you have chosen an inappropriate server.
Ability to report other SNPs in linkage disequilibrium with submitted SNPs. If submitted SNPs are indirectly linked to disease, users will benefit by discovering which other SNPs might be responsible, so that their biological impact can be investigated.
Up-to-date data. The dbSNP database is updated several times a year. The number of new human SNP reference IDs ranges widely (e.g. 44 000 in Build 127, over 6 000 000 in the current Build 129). When SNP webservers do not keep up with these updates, users miss out on coverage of thousands to millions of SNPs.
HOW DIFFERENT ARE THE VARIOUS SNP ANNOTATION METHODS?
A review of current literature reveals that medical geneticists are grappling with issues surrounding the meaning of agreement and disagreement among available SNP annotation methods.
In a meta-analysis study that included computational biology nsSNP methods, predictive scores (for 54 nsSNPs in 37 genes) were compared to lung cancer risk odds ratios from 51 published case–control studies, using a non-parametric correlation test (Spearman rank) [19]. The authors designed a summary statistic which combined scores from SIFT, PolyPhen, SNPs3D [16] and PMut and reported that the summary was more highly correlated with the lung cancer risk odds ratios (r = 0.51) than any of the individual scores. The correlation increase was modest with respect to SIFT, the most highly correlated individual score (r = −0.36). The rationale for combining scores produced by different methods was that each method uses a ‘fundamentally different algorithm’, and that when the algorithms agree, predictions are more trustworthy.
In a case–control study of nsSNPs in nucleotide excision repair genes, putatively linked with prostate cancer [63], SIFT and PolyPhen were used to explore the possible biological impact of seven nsSNPs with significant association to prostate cancer and minor allele frequency > 0.05. The methods disagreed on four nsSNPs and for two out of three on which they agreed, a functional nucleotide excision repair capacity (NERC) assay disagreed with both. The authors tried to explain these disparities by suggesting that PolyPhen uses protein structure information, while SIFT uses evolutionary sequence conservation, but this is not generally true, as described below.
Although users may perceive SNP prediction services as a set of fundamentally different methods, there are major similarities ‘underneath the lid’. For example, SIFT, PolyPhen's PSIC (Position Specific Independent Counts) score and ‘SNPs3D Profile SVM’ (support vector machine) all base their predictions on a multiple sequence alignment of the protein of interest and related proteins. Although PolyPhen does use protein structural information when it is available, for the majority of queries, its predictions are based on amino acid residue properties and PSIC sequence alignment scores [64]. Like SIFT, the PSIC score measures the probability that a substituted amino acid will be tolerated, based on the distribution of amino acids in a multiple sequence alignment column. The measures differ mainly in technical details, such as how pseudocounts and sequence weighting are applied. When SIFT and PolyPhen outputs are substantially different, it is probably because different multiple sequence alignments were used to calculate scores, rather than these details. Inferences based on amino acid column distributions are also used in PANTHER, and as input features to machine learners LS-SNP, SNPs3D, SNAP and PMut. While the decision algorithms used by these different methods are not the same, the correlation among their outputs is the result of similarity among their inputs, and is not necessarily ground for increased confidence.
The authors of the lung cancer meta-analysis assumed that the two ‘SNPs3D SVMs’ (‘SVM Profile’ and ‘SVM structure’) could be grouped together because they are more similar to each other than either one is to SIFT. Emphasis on the SVM algorithm caused them to miss the fundamental similarity between ‘SVM Profile’ and SIFT. A better choice for the summary statistic would be ‘SVM structure’, because it is based on protein structure, and provides an orthogonal prediction to methods based on sequence alignment.
As scientists outside of the bioinformatics community attempt to optimize their use of SNP prediction methods, those within the community must make an effort to better communicate the inner workings of these methods and to clarify both their similarities and differences.
SNP WEBSERVERS: CHALLENGES FOR THE NEXT GENERATION
SNP webservers of the first generation were created by bioinformaticians for bioinformaticians. A major challenge for next generation tools is how to deliver utility to medical geneticists and molecular biologists.
Flexible input tools that can handle high throughput data
Users should have the option of entering from one to thousands of SNPs, including novel SNPs. FASTSNP already allows entry of genes of interest and returns a list of all known SNPs, which can then be selected for annotation. But as the number of candidate SNPs of interest increases, manual selections will not be feasible. Users should be able to enter SNP lists in the form that they receive them from sequencing centers (DNA base change, chromosome position and transcript identifier) or to directly submit the output files from Illumina bead arrays or Affymetrix genotyping arrays.
Leverage of genome correlation structure
Users should be able to find out if their SNPs are in linkage disequilibrium with other SNPs having known or predicted functional effects. MutaGeneSys already allows users to select a preferred correlation threshold and find SNPs listed in OMIM that are in linkage disequilibrium with input SNPs. Such capabilities can be expanded to SNPs having predicted functional impact on regulation or protein function.
Other types of genetic variation
The causative mutation sought in association studies may turn out to be a copy number variant, an inversion, deletion, insertion or frameshift. As other kinds of genetic variation are catalogued, it will be useful both to annotate them and to provide information about linkage disequilibrium between SNPs and these variants.
Cis-regulation
Associations between phenotype and intronic, UTR, and/or promoter region SNPs are prominent in case/control and family studies published over the last several years [65-76]. Yet computational methods to predict the effects of these SNPs lag behind those developed for their impact on proteins. We do not know yet how to accurately detect the sequence signals that identify sites important for transcription, splicing, or miRNA binding, or how to score the impact of a SNP on these sites. Advances in basic science and computational analysis of these elements will play an important role in advancing the utility of SNP webservers.
Inference framework
In addition to serving hypotheses about molecular mechanisms, servers should offer the option of integrating multiple hypotheses and molecular features into a decision algorithm. Without such a framework, and given the growing number of known regulatory mechanisms, users have difficulty for making sense of available information, particularly when harvested by meta-servers. FASTSNP already offers a decision tree framework to integrate information into a risk level (1–5).
Dynamic visualization and analysis tools
The outputs of many servers include protein sequence alignments, structural models, model viewers and structural features. But protein and SNP representations using ribbons, balls and sticks, and multiple sequence alignments cannot provide biological insight to anyone but a protein expert, even if the graphics are interactive. We can maximize the utility of these tools by designing them to help users gain intuition about SNP effects, such as the impact of amino acid substitution. Interactive protein structure graphics could be pre-annotated by ‘painting’ according to biologically important attributes, such as electrostatic surface potential, and the tools could allow users to see how these attributes change with amino acid substitution. Interactive multiple sequence alignment graphics could dynamically display relevant statistics, such as probability that a given amino acid substitution is tolerated in an alignment column. New tools could allow users to experiment selecting different amino acids and to view how the tolerance probability changes.
Dynamic data updates
Most current SNP webservers are based in academic labs and are not supported by full-time staff. Furthermore, these servers were designed to store data locally, requiring regular downloads from their primary sources (such as NCBI, UCSC Genome Browser, UniProt, etc.) and subsequent rerunning of annotation pipelines. It is not surprising that most servers are 2 years or more out-of-date. FAST-SNP and SNPit (http://students.washington.edu/hyshen/research.html which is not yet publicly available) have already made progress on this problem. FAST-SNP uses reconfigurable web wrapper agents to fetch HTML pages, extract relevant data, deliver to a Web Navigation Description Language (WNDL) executor kernel and then to its machine learning algorithm, which renders a decision about SNP risk level. SNPit's wrappers are HTTP servlets that accept queries as URLs and return XML formatted data. It uses a BioMediator ‘source knowledge base’, composed of a central data model and rules to translate the source data models into the common data model. These distributed data integration technologies help ensure that data delivered to the user is up-to-date, although there is nothing they can do about stale data at their sources.
SUMMARY AND CONCLUSIONS
As a whole, the 22 SNP annotation webservers assessed in this study yielded interesting hypotheses to explain why several SNPs might be statistically associated to either ALS, schizophrenia or esophageal cancer in recent medical genetics studies. However, these hypotheses were not immediately apparent and required bioinformatics expertise to sift out from a wide array of ‘black box’ classifications, technical details and predictive scores spanning evolutionary conservation, protein structure, splicing regulators, transcriptional regulators, etc.
The next generation of SNP annotation webservers can take advantage of the growing amount of data in core bioinformatics resources and use intelligent agents to fetch data from different sources as needed. From a user's point of view, it is more efficient to submit a set of SNPs and receive results in a single step, which makes meta-servers the most attractive choice. However, if meta-servers deliver heterogeneous data covering sequence, structure, regulation, pathways, etc., they must also provide frameworks for integrating data into a decision algorithm(s), and quantitative confidence measures so users can assess which data are relevant and which are not. Without progress along these lines, all of this data will only be useful to bioinformatics experts.
Key Points.
Computational biology methods for SNP annotations can maximize their contributions to medical genetics research by designing services that are easy for researchers who are not bioinformatics experts to use and understand.
Medical geneticists and molecular biologists who are interested in using available SNP annotation web servers can select from: (i) servers that disseminate original methods to predict biologically important SNPs; (ii) metaservers, which yield large amounts of heterogeneous bioinformatics information from external servers; and (iii) hybrids which combine (i) and (ii).
There is more similarity among bioinformatics SNP annotation methods than many users realize.
Developing new algorithms for integrating heterogeneous datatypes is now essential to take advantage of the available information, which can potentially be used to infer the biological impact of SNPs.
SUPPLEMENTARY DATA
Supplementary data are available online at http://bib.oxfordjournals.org/.
FUNDING
Susan G. Komen Foundation award (KG080137).
Supplementary Material
Acknowledgements
The author thanks Dr Melissa Cline for valuable discussions.
Biography
Rachel Karchin is an assistant professor in the Department of Biomedical Engineering and the Institute for Computational Medicine at Johns Hopkins University. Her research focuses on predicting the functional impact of SNPs and tumorigenic somatic mutations on biological systems. She is the originator of LS-SNP, a SNP annotation webserver that suffered from many of the shortcomings described in this review, and her group has just released a substantially upgraded version.
References
- Sunyaev S, Hanke J, Aydin A, et al. Prediction of nonsynonymous single nucleotide polymorphisms in human disease-associated genes. J Mol Med. 1999;77:754–60. doi: 10.1007/s001099900059. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–38. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Chasman D, Adams RM. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J Mol Biol. 2001;307:683–706. doi: 10.1006/jmbi.2001.4510. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Mooney S. Bioinformatics approaches and resources for single nucleotide polymorphism functional analysis. Brief Bioinform. 2005;6:44–56. doi: 10.1093/bib/6.1.44. [DOI] [PubMed] [Google Scholar]
- Steward RE, MacArthur MW, Laskowski RA, et al. Molecular basis of inherited diseases: a structural perspective. Trends Genet. 2003;19:505–13. doi: 10.1016/S0168-9525(03)00195-1. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Thornton JM. Understanding the molecular machinery of genetics through 3D structures. Nat Rev Genet. 2008;9:141–151. doi: 10.1038/nrg2273. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchin R, Diekhans M, Kelly L, et al. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- Conde L, Vaquerizas JM, Dopazo H, et al. PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res. 2006;34:W621–25. doi: 10.1093/nar/gkl071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reumers J, Maurer-Stroh S, Schymkowitz J, et al. SNPeffect v2.0: a new step in investigating the molecular phenotypic effects of human non-synonymous SNPs. Bioinformatics. 2006;22:2183–85. doi: 10.1093/bioinformatics/btl348. [DOI] [PubMed] [Google Scholar]
- Wang P, Dai M, Xuan W, et al. SNP Function Portal: a web database for exploring the function implication of SNP alleles. Bioinformatics. 2006;22:e523–29. doi: 10.1093/bioinformatics/btl241. [DOI] [PubMed] [Google Scholar]
- Yuan HY, Chiou JJ, Tseng WH, et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–41. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–35. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Guo N, Kejariwal A, et al. PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 2007;35:D247–52. doi: 10.1093/nar/gkl869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Hoffman A, Wu X, et al. Correlating observed odds ratios from lung cancer case-control studies to SNP functional scores predicted by bioinformatic tools. Mutation Res. 2008;639:80–8. doi: 10.1016/j.mrfmmm.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plourde M, Manhes C, Leblanc G, et al. Mutation analysis and characterization of HSD17B2 sequence variants in breast cancer cases from French Canadian families with high risk of breast and ovarian cancer. J Mol Endocrinol. 2008;40:161–72. doi: 10.1677/JME-07-0101. [DOI] [PubMed] [Google Scholar]
- Merner ND, Hodgkinson KA, Haywood AFM, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–21. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer U, Neumann C, Thiel J, et al. Screening of functional and positional candidate genes in families with common variable immunodeficiency. BMC Immunol. 2008;9:3. doi: 10.1186/1471-2172-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron J, Holla OL, Laerdahl JK, et al. Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J Intern Med. 2008;263:420–31. doi: 10.1111/j.1365-2796.2007.01915.x. [DOI] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- Bouchet C, Gonzales M, Vuillaumier-Barrot S, et al. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum Mutat. 2007;28:1020–27. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- Conen D, Glynn RJ, Buring JE, et al. Natriuretic peptide precursor a gene polymorphisms and risk of blood pressure progression and incident hypertension. Hypertension. 2007;50:1114–19. doi: 10.1161/HYPERTENSIONAHA.107.097634. [DOI] [PubMed] [Google Scholar]
- Dempster EL, Burcescu I, Wigg K, et al. Evidence of an association between the vasopressin V1b receptor gene (AVPR1B) and childhood-onset mood disorders. Arch Gen Psychiatry. 2007;64:1189–95. doi: 10.1001/archpsyc.64.10.1189. [DOI] [PubMed] [Google Scholar]
- Gorlov IP, Meyer P, Lilogiou T, et al. Seizure 6-like (SEZ6L) gene and risk for lung cancer. Cancer Res. 2007;67:8406–11. doi: 10.1158/0008-5472.CAN-06-4784. [DOI] [PubMed] [Google Scholar]
- Zeitz C, Forster U, Neidhardt J, et al. Night blindness-assocliated mutations in the ligand-blinding, cysteline-rich, and intracellular domains of the metabotroplic glutamate receptor 6 abolish protein trafficking. Hum Mut. 2007;28:771–80. doi: 10.1002/humu.20499. [DOI] [PubMed] [Google Scholar]
- Nitz I, Fisher E, Weikert C, et al. Association analyses of GIP and GIPR polymorphisms with traits of the metabolic syndrome. Mol Nutr Food Res. 2007;51:1046–52. doi: 10.1002/mnfr.200700048. [DOI] [PubMed] [Google Scholar]
- Tocharoentanaphol C, Promso S, Zelenika D, et al. Evaluation of resequencing on number of tag SNPs of 13 atherosclerosis-related genes in Thai population. J Hum Genet. 2008;53:74–86. doi: 10.1007/s10038-007-0220-8. [DOI] [PubMed] [Google Scholar]
- Gong Y, Beitelshees AL, Wessel J, et al. Single nucleotide polymorphism discovery and haplotype analysis of Ca2+-dependent K+ channel beta-1 subunit. Pharmacogenet Genomics. 2007;17:267–75. doi: 10.1097/FPC.0b013e3280105235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lopez J, Mustafa Z, Pombo-Suarez M, et al. Genetic variation including nonsynonymous polymorphisms of a major aggrecanase, ADAMTS-5, in susceptibility to osteoarthritis. Arthritis Rheum. 2008;58:435–41. doi: 10.1002/art.23201. [DOI] [PubMed] [Google Scholar]
- Aaron C. Finding local community structure in networks. Phy Rev E Stat Nonlin Soft Matter Phys. 2005;72:026132. doi: 10.1103/PhysRevE.72.026132. [DOI] [PubMed] [Google Scholar]
- Wu CH, Apweiler R, Bairoch A, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–91. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande N, Addess KJ, Bluhm WF, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–37. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Ailey B, Brenner SE, et al. SCOP: a structural classification of proteins database. Nucleic Acids Res. 1999;27:254–56. doi: 10.1093/nar/27.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Donaldson I, Wolting C, et al. BIND—the Biomolecular Interaction Network Database. Nucleic Acids Res. 2001;29:242–45. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-aryamontri A, Ceol A, Palazzi LM, et al. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–74. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Yamada T, Hamajima M, et al. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008;36:W423–26. doi: 10.1093/nar/gkn282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, et al. The UCSC genome browser database: update 2007. Nucleic Acids Res. 2007;35:D668–73. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, et al. The ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, et al. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–78. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–16. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Zhou M, Cui Y. nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005;33:W480–82. doi: 10.1093/nar/gki372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–34. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Masso M, Vaisman II. Accurate prediction of enzyme mutant activity based on a multibody statistical potential. Bioinformatics. 2007;23:3155–61. doi: 10.1093/bioinformatics/btm509. [DOI] [PubMed] [Google Scholar]
- Clamp M, Cuff J, Searle SM, et al. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–27. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, et al. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–71. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeo GW, Yeh R, et al. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32:W187–90. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, et al. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–45. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, King DP, Reutiman TJ, et al. PDE4B polymorphisms and decreased PDE4B expression are associated with schizophrenia. Schizophr Res. 2008;101:36–49. doi: 10.1016/j.schres.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Xu H, Gregory SG, Hauser ER, et al. SNPselector: a web tool for selecting SNPs for genetic association studies. Bioinformatics. 2005;21:4181–86. doi: 10.1093/bioinformatics/bti682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–24. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanovich J, Pe’er I. MutaGeneSys: estimating individual disease susceptibility based on genome-wide SNP array data. Bioinformatics. 2008;24:440–2. doi: 10.1093/bioinformatics/btm587. [DOI] [PubMed] [Google Scholar]
- Chin JY, Schleifman EB, Glazer PM. Repair and recombination induced by triple helix DNA. Front Biosci. 2007;12:4288–97. doi: 10.2741/2388. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–17. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doecke J, Zhao ZZ, Pandeya N, et al. Polymorphisms in MGMT and DNA repair genes and the risk of esophageal adenocarcinoma. Int J Cancer. 2008;123:174–80. doi: 10.1002/ijc.23410. [DOI] [PubMed] [Google Scholar]
- Dantzer J, Moad C, Heiland R, et al. MutDB services: interactive structural analysis of mutation data. Nucleic Acids Res. 2005;33:W311–14. doi: 10.1093/nar/gki404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Ma L, Li H, et al. Snap: an integrated SNP annotation platform. Nucleic Acids Res. 2007;35:D707–10. doi: 10.1093/nar/gkl969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockett KL, Snowhite IV, Hu JJ. Nucleotide-excision repair and prostate cancer risk. Cancer Lett. 2005;220:125–35. doi: 10.1016/j.canlet.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Sunyaev SR, Eisenhaber F, Rodchenkov IV, et al. PSIC: profile extraction from sequence alignments with position-specific counts of independent observations. Protein Eng. 1999;12:387–94. doi: 10.1093/protein/12.5.387. [DOI] [PubMed] [Google Scholar]
- Gorman CL, Russell AI, Zhang Z, et al. Polymorphisms in the CD3Z gene influence TCRzeta expression in systemic lupus erythematosus patients and healthy controls. J Immunol. 2008;180:1060–70. doi: 10.4049/jimmunol.180.2.1060. [DOI] [PubMed] [Google Scholar]
- Wei C-L, Cheung W, Heng C-K, et al. Interleukin-13 genetic polymorphisms in Singapore Chinese children correlate with long-term outcome of minimal-change disease. Nephrol Dial Transplant. 2005;20:728–34. doi: 10.1093/ndt/gfh648. [DOI] [PubMed] [Google Scholar]
- Wang G, van der Walt JM, Mayhew G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of ±-Synuclein. Am J Hum Genet. 2008;82:283–89. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GarcÃa-Closas M, Malats Nr, Real FX, et al. Large-scale evaluation of candidate genes identifies associations between VEGF polymorphisms and bladder cancer risk. PLoS Genet. 2007;3:e29. doi: 10.1371/journal.pgen.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Jia Y, Cooper JJ, et al. Common variants in glutamine:fructose-6-phosphate amidotransferase 2 (GFPT2) gene are associated with type 2 diabetes, diabetic nephropathy, and increased GFPT2 mRNA levels. J Clin Endocrinol Metab. 2004;89:748–55. doi: 10.1210/jc.2003-031286. [DOI] [PubMed] [Google Scholar]
- Tokuhiro S, Yamada R, Chang X, et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat Genet. 2003;35:341–48. doi: 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bertolino A, Fazio L, et al. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci USA. 2007;104:20552–7. doi: 10.1073/pnas.0707106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damcott CM, Ott SH, Pollin TI, et al. Genetic variation in adiponectin receptor 1 and adiponectin receptor 2 is associated with type 2 diabetes in the old order amish. Diabetes. 2005;54:2245–50. doi: 10.2337/diabetes.54.7.2245. [DOI] [PubMed] [Google Scholar]
- Muindi JR, Nganga A, Engler KL, et al. CYP24 splicing variants are associated with different patterns of constitutive and calcitriol-inducible CYP24 activity in human prostate cancer cell lines. J Steroid Biochem Mol Biol. 2007;103:334–37. doi: 10.1016/j.jsbmb.2006.12.060. [DOI] [PubMed] [Google Scholar]
- Shan K, Ying W, Jian-Hui Z, et al. The function of the SNP in the MMP1 and MMP3 promoter in susceptibility to endometriosis in China. Mol Hum Reprod. 2005;11:423–27. doi: 10.1093/molehr/gah177. [DOI] [PubMed] [Google Scholar]
- Healy J, Belanger H, Beaulieu P, et al. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007;109:683–92. doi: 10.1182/blood-2006-02-003236. [DOI] [PubMed] [Google Scholar]
- Thompson JF, Wood LS, Pickering EH, et al. High-density genotyping and functional SNP localization in the CETP gene. J. Lipid Res. 2007;48:434–43. doi: 10.1194/jlr.M600372-JLR200. [DOI] [PubMed] [Google Scholar]
- Noble WS. What is a support vector machine? Nat Biotech. 2006;24:1565–67. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- Yuan HY, Chiou JJ, Tseng WH, et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–41. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Berg M, Cheong O, Van Kreveld M, et al. Computational Geometry: Algorithms and Approaches. Berlin, New York: Springer; 2000. [Google Scholar]
- Markiewicz P, Kleina LG, Cruz C, et al. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as ‘spacers’ which do not require a specific sequence. J Mol Biol. 1994;240:421–33. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- Rennell D, Bouvier SE, Hardy LW, et al. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- Loeb DD, Swanstrom R, Everitt L, et al. Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- Yip Y, Scheib H, Diemand A, et al. The Swiss-Prot variant page and the ModSNP database: a resource for sequence and structure information on human protein variants. Hum Mut. 2004;23:464–70. doi: 10.1002/humu.20021. [DOI] [PubMed] [Google Scholar]
- Kawabata T, Ota M, Nishikawa K. The protein mutant database. Nucleic Acids Res. 1999;27:355–57. doi: 10.1093/nar/27.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball E, Howells K, et al. Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet. 2008;45:124–26. doi: 10.1136/jmg.2007.055210. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–20. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer BR, Yeager M, Burdett L, et al. SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006;34:D617–21. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- DeLano WL. San Carlos, CA, USA: DeLano Scientific LLC; 2002. The PyMOL Molecular Graphics System on the World Wide Web. [Google Scholar]
- Sayle RA, Milner-White EJ. RASMOL: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–76. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.