

Abstract
Marine derivatives are of great pharmaceutical interest as inhibitory compound and search of bioactive compounds from Marine organism which is relatively new to medicinal chemistry. Our main aim in the study is to screen possible inhibitors against CCR5 which acts as co-receptor M-tropic HIV-1, through virtual screening of 122 Marine derived compounds from various organisms known to have biological activity. Homology Model of CCR5 was constructed using MODELLER and the Model was energy minimized and validated using PROCHECK to obtain a stable structure, which was further used for virtual screening of Marine derived compounds through molecular Docking studies using GOLD. The Docked complexes were validated and Enumerated based on the GOLD Scoring function to pick out the best Marine inhibitor based on GOLD score. Thus from the entire 122 Marine compounds which were Docked, we got best 4 of them with optimal GOLD Score. (LAMIVUDINE: 45.0218, BATZELLINE-D: 44.3852.ACYCLOVIR: 43.1362 and THIIOACETAMIDE: 42.7412) Further the Complexes were analyzed through LIGPLOT for their interaction for the 4 best docked Marine compounds. Thus from the Complex scoring and binding ability its deciphered that these Marine compounds could be promising inhibitors for M-tropic HIV-1 using CCR5 as Drug target yet pharmacological studies have to confirm it.
Keywords: CCR5, Marine Derivatives, HIV-I, GOLD
Background
In AIDS therapy, there are currently a number of compounds available for multiple targets approved by the FDA and in clinical trials, e.g. protease inhibitors, reverse transcriptase inhibitors (NRTI, NNRTI) and CCR5, CCR4, fusion inhibitors, among others [1–4]. CCR5 is a member of the serpentine-receptor superfamily 5] and is expressed by monocytes, memory T lymphocytes, and preferably by Th1 cells and NK cells [6–8].C CR5 act as a co-receptor for macrophage tropic(R5) strains of HIV-1 [9, 10] via interacting with viral envelope glycoprotein gp120, causing envelope fusion and viral entry [–13]. Certain Individuals who are homozygous for an internal 32-bp deletion in the gene encoding CCR5 produce a truncated form of the receptor that is not expressed on the cell surface, providing a high degree of resistance to M-tropic strains of HIV-1 [14, 15]. The physiological Ligand of CCR5 has been shown to block the co-receptor activity of M-tropic HIV-1 [11,16– 18].
In contrast, the viral chemokine vMIP-II [19], an antagonist of CCR5, has been shown to have limited virtue as an inhibitor of co-receptor activity [20]. This variation may be ascribed to the binding of these Ligand s to different regions of CCR5, indicating the importance of the N-terminal extracellular region (N-ter) and the second extra cellular loop (ECL2) in co-receptor activity [21,22]. Both the N-ter and the body of CCR5 have been shown to affect the coreceptor activity of CCR5 [23].
The N-ter domain significance has been shown in several studies, which entangle specific acidic and aromatic residues in co-receptor activity [24–26]. Substitution of the N-ter regions of chemokine receptors, such as CCR1 or CCR2b, with that of CCR5 has been shown to allow virus fusion and entry [23,27– 30]. However, replacement of the N-ter region of CCR5 with that of chemokine receptors CCR1 or CCR2b has not been found to abolish co-receptor activity considerably [23, 27–30],indicating that both regions play vital roles in co-receptor activity. Ligand binding and HIV-1 entry are also affected by the posttranslational sulfation of Y10 and Y14 of the N-ter, which have been shown to bind gp120 at micromolar affinities when included in synthetic peptides [31,32], contrasted by a complete lack of binding of the non-sulfated forms. Thus, two regions of CCR5 have been postulated to exhibit important functions in cell fusion, the N-ter domain, and the extracellular loops [29]. Transmembrane- spanning domains 4 [33] and 5 [34] have been shown to play a role in co-receptor function, but it is unclear if these domains play a role in direct interaction with gp120, or are important for regulating receptor conformation.
A small molecule inhibitor of CCR5 co-receptor function, TAK-779 [35], has been shown to require residues mapped to TM1, 2, 3, and 7 [36]. Other compounds such as SCH-C, SCH-351125, and SCH-350581, as well as members of another set of chemically unrelated small molecule inhibitors, 2-aryl-4-(piperidin-1-yl) butanamines and 1,3,4-trisubstituted pyrrolidines, appear to utilize a non-identical overlapping binding site involving TMs 2, 3, 6, and 7 [37,38]. Thus, it is likely that these CCR5 antagonists have an allosteric effect on receptor conformation, in contrast to direct blockade of the binding site for physiologic and pathologic Ligands.
Similar to plant derived compounds for the therapeutic uses, Marine derived compounds do also have similar kind of effect from the pharmacological point of perspective and hence it’s been used in cure for many deadly diseases. Marine sources were not utilized to any extent until the mid 20th centaury the last 30 years has seen an explosive growth of natural products from Marine organisms and these products are invariably described with some sort of biological activity. The search of bioactive compounds from Marine organisms relatively a new field. However, the biodiversity of Marine environment far exceeds that of its terrestrial counter partner, So the oceans represents an enormous resource for new biologically Active compounds. There are 15 Marine natural products currently in clinical trails (Modern Drug discovery 2002) up to date. Our research work focus on virtual screening of such Marine compounds targeting HIV-1.
The parameters used for genetic algorithm were population size (100), selection pressure (1.1), number of operations (100,000), number of islands (5), niche size (2), migrate (10), mutate (95) and cross-over (95). The default speed selection was used to avoid a potential reduction in docking accuracy. Fifty genetic algorithm runs with default parameter settings were performed without early termination. To estimate the protein-Ligand complexes, the scoring function, the entire sets of 122 Marine therapeutic compounds were docked against Modeled CCR5 using GOLD [Genetic Optimization of Ligand Docking] version 2.1.2 was used to Screen for the possible inhibitory Marine compounds against CCR5 using GOLD Score. [44]. LIGPLOT [45] was constructed for analyzing the interaction with in the Best Docked complex.
Methodology
Homology Modeling of CCR5
The Homology Model of Human CCR5 with Swiss-Prot ID: P51681 was generated using MODELLER [39] with ClustalW alignment of Bovine Rhodopsin sequence [accession number P02699] and its crystal structure [PDB: 1F88] as Template [40] chosen from PDB BLAST hit. The obtained Model was validated using PROCHECK [41] and final Energy minimized using CHARMm to obtain stable structure for further studies.
Active site Identification
After the final Model was built, the possible Active sites of CCR5 were identified using Discovery Studio 2.0 [42] using the option Find site from Receptor Cavities.
Ligand Generation and Optimization
Marine Therapeutic compounds were gathered from Marine bioactive database [43] and 3D structures were generated using Discovery Studio 2.0[42] further the compounds were submitted for Ligand Minimization using CHARMm Forcefield with Smart Minimizer Algorithm Steps 1000 and Optimized.
Virtual Screening of Marine compounds against CCR5 using Molecular Docking
The Ligand s, including all hydrogen atoms, were built and minimized using Discovery studio 2.0 and GOLD [Genetic optimization for Ligand Docking] version 2.1.2 [44] was used for Docking the entire 122 set of Marine compounds [43]. The Active site was defined as the collection of protein residues enclosed within a 15 Å radius sphere. The annealing parameters for Van der Waals and hydrogen bonding were set to 4.0 Å and 2.5 Å respectively.
Results and Discussion
A confidence level sequence identity gives a reliable alignment between the target sequence and template structure. Our PDB BLAST Hit for CCR5 target sequence gave a best hit of Rhodopsin crystal structure (PDB: 1F88) .The Initial Model thus generated using MODELLER. Structurally conserved regions (SCRs) for the Model and the template were determined by multiple sequence alignment. Coordinates from the reference protein (1F88) to the SCRs, structurally variable regions (SVRs), N-terminal and C-terminal were assigned to the target sequence based on the satisfaction of spatial restraints. All side chains of the Modeled protein were set by rotamers. Thus, the obtained Model was refined by Energy minimization using CHARMm force field and the final stable structure of CCR5 is obtained (Figure 1).
Figure 1.
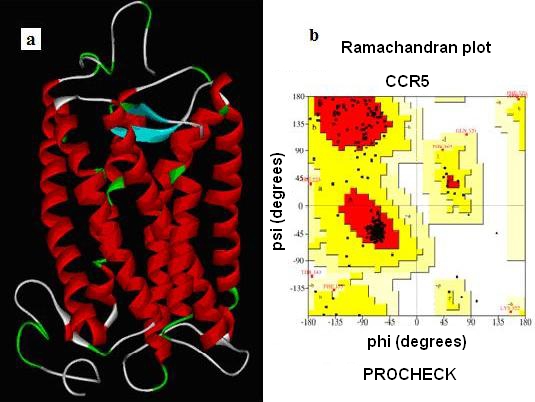
(a) The final model of stable structure of CCR5; (b) Validation of the model using Ramachandran plot computed with the PROCHECK program with 86.4¢ of the residues in favored and allowed regions.
After the refinement process, validation of the Model was carried out using Ramachandran plot calculations computed with the PROCHECK program. Altogether 86.4¢ of the residues were in favored and allowed regions (Figure 1). From previous experimental evidence and from Figure 1 we come know that CCR5 receptor has 7 transmembrane helices with extracellular N-terminal end and intracellular C -terminal loop where in the extracellular loop plays an important role in the binding Gp120 to the co-receptor for M-tropic HIV-1 entry and which in intern also inhibitory site for the Antagonist. For the built CCR5 final Model the possible Active sites were identified using Discovery Studio 2.0. Fourteen possible Active sites were obtained using the option Find site from Receptor Cavities. These pockets were compared with the Active site of the template (Bovine Rhodepsin PDB: 1f88) and by careful analysis of the entire predicted 14 Active sites, we found that 12th predicted Active site (shown in Figure 2) with residues Asp2, pro8, Asn13, Tyr14, Tyr15, Thr16, Gln21 of CCR5 were similar to that of the catalytic site in 1f88 and Therefore its chosen as the most biologically favorable site for Docking study and other Identified Active sites were neglected. (Figure 2) A set of 122 Marine Bioactive compounds were gathered from Marine bioactive database and 3D structures were generated using Discovery Studio 2.0 further Energy Minimized using CHARMm Forcefield and using Ligand s preparation option the best conformation were prepared for all 122 Marine compounds.
Figure 2.
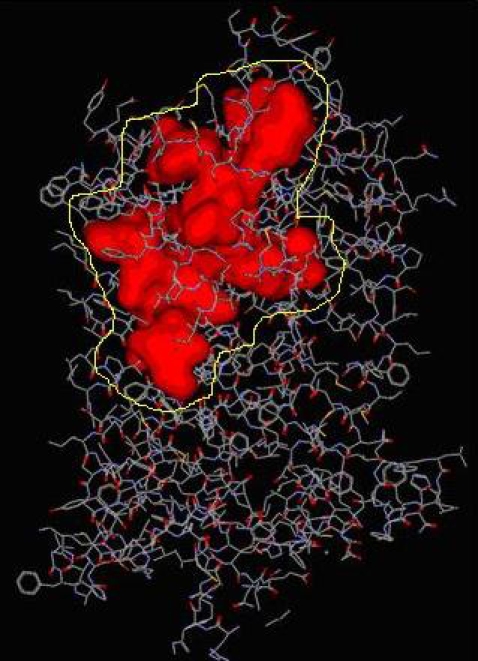
Identified active site of CCR5 where the inhibitors are likely to bind
Docking of Marine Compounds to CCR5
Docking of all 122 Marine compounds with Human CCR5 Model was performed using GOLD, which was set to 50 cycles of run without constraints between the Ligand s and the specific amino acids of the pocket. The algorithm exhaustively searches the entire rotational and translational space of the Ligand with respect to the receptors. The flexibility of the Ligand is given by dihedral angle variations. The various solutions evaluated by a score, which is equivalent to the absolute value of the total energy of the Ligand in the protein environment. The best Four Docking solution's GOLD score for each Compound were considered. It's noted that all the GOLD scores are very similar in all the top three solutions for each Marine compound. The GOLD score and RMSD differences between the consecutive top three runs of each best Docked Marine inhibitor is zero and this indicates the stabilization of particular inhibitor at the Active site. Out of 122 Marine compounds submitted for GOLD Docking run against Modeled Human CCR5 and we obtained 4 Marine compounds with optimal solution with Gold score Shown in Table [Rest of data not shown] (Table 1 in Supplementary material).
To understand the interaction between CCR5 and the four Marine compounds, Complexes were generated using the GoldMine suite for the outputs of Gold solutions. LIGPLOT were run for all the four complexes. It is evident from the analysis of the Docked complex that Marine inhibitors are located in the center of the Active site, and is stabilized by hydrogen bonding interactions and are bound in between 1st and 4th α-helices Extracellular loop arresting the co-receptor activity of CCR5 for the entry of M-tropic HIV-1. (Figure 3) Through the interaction analysis, we know that Gln4, Asn13 and Thy14 are important anchoring residues for CCR5 and are the main contributors towards compound interaction. Though the interaction energy does not include the contribution from the water or the extended compound structure, this preliminary data along with the list of hydrogen bond interactions between the compounds and the Active site residues clearly supports that Gln4, Asn13 andThy14 are more preferred residues in binding.
Figure 3.
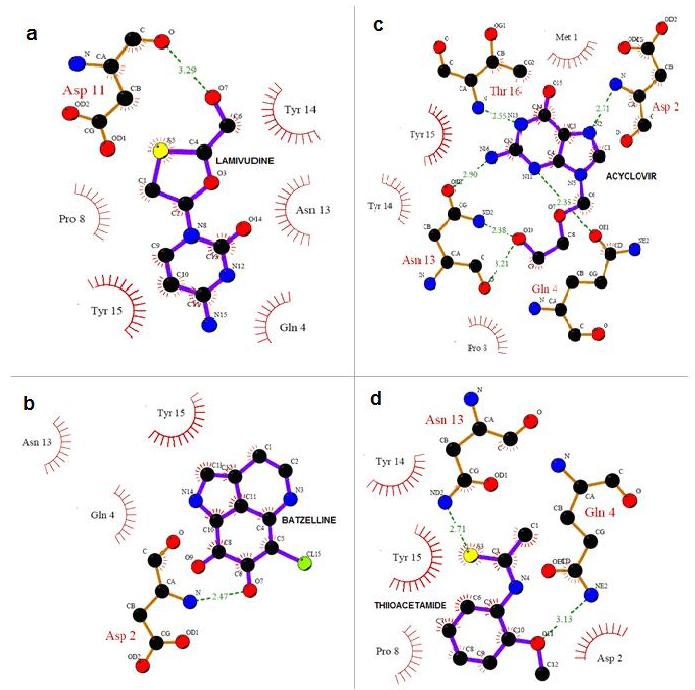
Receptor ‐ ligand interaction is shown. (a) LAMIVUDINE O7 hydrogen bonded with extra cellular loop residue Asp11 OH at a distance of 3.29 Å and Gln4, Pro8, Asn13, Tyr14 and Tyr15 are non-ligand residues involved in hydrophobic contact(s). (b) BATZELLINE O7 hydrogen bonded with Extracellular loop residue Asp2 NH at a distance of 2.88 Å and Gln4, Asn13 and Tyr15 are non-ligand residues involved in hydrophobic contact(s). (c) ACYCLOVIR O10 hydrogen bonded with NH and with OH of Asn13 at a distance of 2.38 Å and 3.21 Å. Like wise NH hydrogen bonded with Gln4 at distance of 2.35 Å where as Met1, Pro8, Thy14 and Thy15 are non-ligand residues involved in hydrophobic contact(s). (d) THIIOACETAMIDE S3 hydrogen bonded with N of Asn13 at distance of 2.71 Å and OH hydrogen bonded with N of Gln4 at distance of 3.13 Å and NH hydrogen bonded with N at a distance of 2.71 Å where Asp2, Pro8, Thy14 and Thy15 are non-ligand residues involved in hydrophobic contact(s).
Conclusion
CCR5 is one of the most recent potent Drug target for HIV1. In this work, we have constructed a 3D Model of CCR5, using the MODELLER software and obtained a refined Model after energy minimization. The final refined Model was further assessed by PROCHECK program, and the results show that the Model was stable and reliable. The stable Model was further used for Virtual Docking of Marine Compounds. Docking results indicate that out of 122 Marine compounds, there were Four inhibitory compounds for CCR5 as Target for HIV-I. As it's well known, hydrogen bonding plays an important role for the structure and function of biological molecules, especially for Inhibition in a complex. Thus our study confirms LAMIVUDINE, BATEZELLINE-D, ACYCLOVIR and THIOACETAMIDE are potential inhibitor for CCR5 as target for HIV-I forming a hydrogen bonding and with non-bonded interaction to act as a drug candidate yet Pharmacological study will yet confirm it to be promising.
Supplementary material
Acknowledgments
This work was supported by CSIR (Council of Scientific and Industrial Research)
References
- 1.Noor MA, et al. AIDS. 2004;18:2137. doi: 10.1097/00002030-200411050-00005. [DOI] [PubMed] [Google Scholar]
- 2.Dando TM, Scott LJ. Drugs. 2005;65:285. doi: 10.2165/00003495-200565020-00010. [DOI] [PubMed] [Google Scholar]
- 3.Veiga AS, et al. J Am Chem Soc. 2004:14758. doi: 10.1021/ja0459882. [DOI] [PubMed] [Google Scholar]
- 4.Princen K, et al. J Virol. 2004. p. 12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samson M, et al. Biochemistry. 1996;35:3362. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- 6.Lee B, et al. Proc Natl Acad Sci. 1999;96:5215. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonecchi R, et al. J Exp Med. 1998;187:129. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallusto F, et al. J Exp Med. 1998;187:875. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connor RI, et al. J Virol. 1993;67:1772. doi: 10.1128/jvi.67.4.1772-1777.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuitemaker H, et al. J Virol. 1992;66:1354. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alkhatib G, et al. Science. 1996;272:1955. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 12.Trkola A, et al. Nature. 1996;384:184. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, et al. Nature. 1996;384:179. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 14.Samson M, et al. Nature. 1996;382:722. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 15.Liu R, et al. Cell. 1996;86:367. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 16.Deng H, et al. Nature. 1996;381:661. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 17.Cocchi F, et al. Science. 1995;270:1811. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 18.Jansson M, et al. Proc Natl Acad Sci. 1996;93:15382. doi: 10.1073/pnas.93.26.15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore PS, et al. Science. 1996;274:1739. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 20.Boshoff C, et al. Science. 1997;278:290. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- 21.Navenot JM, et al. J Mol Biol. 2001;313:1181. doi: 10.1006/jmbi.2001.5086. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, et al. Nature. 1997;387:527. doi: 10.1038/387527a0. [DOI] [PubMed] [Google Scholar]
- 23.Rucker J, et al. Cell. Vol. 87. 1996. p. 437. [DOI] [PubMed] [Google Scholar]
- 24.Blanpain C, et al. J Biol Chem. 1999;274:34719. doi: 10.1074/jbc.274.49.34719. [DOI] [PubMed] [Google Scholar]
- 25.Dragic T, et al. J Virol. 1998;72:279. doi: 10.1128/jvi.72.1.279-285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farzan M, et al. J Virol. 1998;72:1160. doi: 10.1128/jvi.72.2.1160-1164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atchison RE, et al. Science. 1996;274:1924. doi: 10.1126/science.274.5294.1924. [DOI] [PubMed] [Google Scholar]
- 28.Bieniasz PD, et al. EMBO J. 1997;16:2599. doi: 10.1093/emboj/16.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doranz BJ, et al. J Virol. 1997;71:6305. doi: 10.1128/jvi.71.9.6305-6314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picard L, et al. J Virol. 1997;71:5003. doi: 10.1128/jvi.71.7.5003-5011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cormier EG, et al. Proc Natl Acad Sci. 2000;97:5762. doi: 10.1073/pnas.97.11.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farzan M, et al. J Biol Chem. 2000;275:33516. doi: 10.1074/jbc.M007228200. [DOI] [PubMed] [Google Scholar]
- 33.Siciliano SJ, et al. J Biol Chem. 1999;274:1905. doi: 10.1074/jbc.274.4.1905. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, et al. J Biol Chem. 1999;274:28413. doi: 10.1074/jbc.274.40.28413. [DOI] [PubMed] [Google Scholar]
- 35.Baba M, et al. Proc Natl Acad Sci. 1999;96:5698. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dragic T, et al. Proc Natl Acad Sci. 2000;97:5639. doi: 10.1073/pnas.090576697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsamis F, et al. J Virol. 2003;77:5201. doi: 10.1128/JVI.77.9.5201-5208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castonguay LA, et al. Biochemistry. 2003;42:1544. doi: 10.1021/bi026639s. [DOI] [PubMed] [Google Scholar]
- 39.Sali, et al. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 40.Palczewski, et al. Science. 2000;34:439. [Google Scholar]
- 41.Laskoswki, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 42. http://accelrys.com/products/discovery-studio/
- 43. http://www.niot.res.in/m5/mbic/pg/mcdb/antiviralrlist.php
- 44.Jones, et al. J Mol Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 45.Wallace AC, et al. Prot Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.