

Abstract
R-spondin 4 is a secreted protein mainly associated with embryonic nail development. R-spondins have been recently identified as heparin-binding proteins with high affinity. Proteoglycan binding has been associated with both the TSR and the C terminal basic amino acid rich domains. In this paper, molecular modelling techniques were used to construct the model of R-spondin 4 TSR domain based on the structure of the F-spondin TSR domain 4 (30-40¢ sequence identity). Beside a positively charged surface in the TSR domain, presence of the basic amino acid rich domain which could forms a continuous heparin binding surface may explain the high affinity of R-spondins for heparin. Our results provide a framework for understanding the possible regulatory role of heparin in R-spondins signalling.
Keywords: TSR domain, R-spondin 4, heparin binding, molecular modelling
Background
R-spondin 4 gene plays a key role in the embryonic nail development. Recent reports identified R-spondin 4 gene as responsible for congenital anonychia which is a rare autosomal-recessive disorder characterized by the absence of finger and toenails [1]. The human R-spondin 4 protein gene, member of R-spondin family (R-spondin 1-4), consists of five coding exons corresponding to predicted structural domains. Predicted domains include the N-terminal signal peptide sequences encoded by exon 1, the two adjacent cysteine rich furin like domains encoded by exon 2 and 3 and the single thrombospondin TSP1 domain that is encoded by exon 4. The C terminal basic region encoded by exon 5 is of varying length and scores as a nuclear localization signal [1].
The two cysteine rich domains are thought to be responsible for binding to the wnt signalling pathway. Recent reports have suggested that the R-spondin plays a positive modulatory role in Wnt ligand activity presumably by directly interacting with Wnt ligands [1] and [2]. The thrombospondin TSP 1 domain (designated TSP-1 domain in the pfam database or TSR elsewhere) was found for the first time in the thrombospondin protein where it is repeated three times [3]. Since then, the TSR domain has been identified within numerous protein families like complement factors, TRAP proteins of Plasmodium and F-spondin. No single function is attributable to all TSRs. TSR has been involved in the regulation of cell proliferation, migration and apoptosis in a variety of physiological and pathological settings, such as wound healing, inflammation and angiogenesis [4].
The TSR domains (» 60 amino acids) are characterized by conserved Cys, Trp and Arg residues. The NH2-terminal portion of the TSR domain contains two or three tryptophan residues separated by two to four amino acids each [5]. Most TSRs have six cysteine residues; however, those found in some complement factors and in the malaria proteins contain less cysteine.
The TSR and the basic amino residues rich domains were recently shown to be responsible for binding of R-spondin to heparin or HSPGs (heparan sulfate proteoglycans) [2]. HSPG have numerous biological functions. They are found predominantly at the cell surface and in the extracellular matrix, where they mediate cell interactions and modulate growth factor binding and activity [6].
The three-dimensional structure of R-spondin 4 has not been determined until now and the structural study of its TSR domain may provide additional information about its molecular recognition of heparin. In this article, we present a model of the human R-spondin 4 TSR domain generated using molecular modelling techniques. Sequence analysis within the resulting structural model allowed the characterization of essential features of TSRs (namely the conserved tryptophans, arginines, and cysteines) and the prediction of a glycosaminoglycan binding site within this domain.
Methodology
Template selection
The human R-spondin 4 TSR domain (residues from 138 to 197) (accession number Q2I0M5), was submitted to PDB-Blast [7] and fold recognition servers (mGenTHREADER [8] and PHYRE [9]) separately. Inspection of the template-target alignment generated by these servers shows that the best ranked templates belong to the same group within the TSR family. All cysteine residues in the template-target alignment produced by PHYRE were conserved in number and position. So this latter alignment was chosen for model building.
Model building, evaluation and structural analysis
The predicted model was constructed using the MODELLER program [10] based both on the structure of F-spondin TSR domain 4 (pdb: 1vex) [11] and the alignment generated by PHYRE program. The model obtained was analyzed by Ramachandran's map using PROCHECK [12]. Structural analysis was performed and figures representations were generated with SwissPDBViewer [13].
Multiple sequence alignment
The Multiple sequence alignment was constructed by aligning the TSR domains of human R-spondin 4, the TSR domains 1, 2, 3, 4 and 5 of human F-spondin (Q9HCB6), the TSR domain of TRAP from Plasmodium falciparum (P16893) and the TSR domains 1, 2 and 3 of human Thrombospondin-1 (P07996). The alignment using the ClustalW tool [14] was obtained on the basis of the conservation of the six cysteines as suggested previously [15]. Manual adjustments based on PHYRE results were performed and the result was shown in Figure 1.
Figure 1.
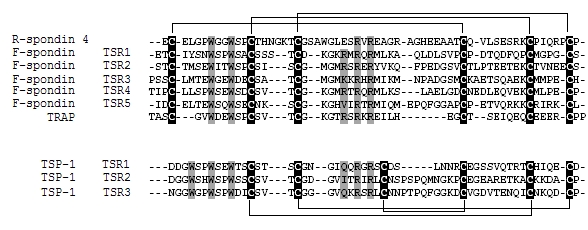
Multiple sequence alignment of human R-spondin 4 TSR domain, F-spondin TSR domains (1, 2, 3, 4 and 5), Thrombospondin Related Anonymous Protein (TRAP) from Plasmodium falciparum TSR domain and human Thrombospondin-1 (TSP-1) TSR domains (1, 2 and 3). The potential locations of disulfide bonds are shown above the alignment (F-spondin group) and below it (TSP-1 group). Tryptophan, arginine residues forming CWR layered structure are highlighted in grey. Cysteine residues implicated in disulfide bond formation are highlighted in black.
Electrostatic potential calculations
The electrostatic potential was calculated with the Poisson-Boltzmann module of SwissPDBViewer utilizing the following parameters: solvent dielectric constant of 80 and protein dielectric constant of 4. The electrostatic potential was then mapped to the molecular surface with a solvent probe radius of 1.4 Å. In figure 3, Blue, white and red colors indicate positive neutral and negative electrostatic potential.
Figure 3.
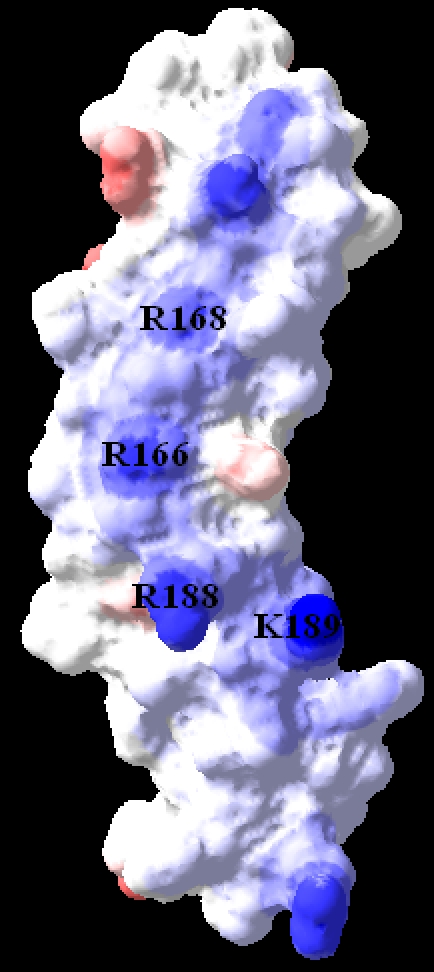
Electrostatic potential surface representation of the R-spondin 4 TSR domain (front face) with positively charged residues labeled.
Results and discussion
Structural analysis
To create a model of R-spondin 4 TSR domain, we submitted the R-spondin 4 sequence (from residue 138 to residue 197) to PDB-Blast, mGenTHREADER and PHYRE servers. The BLAST search against the structures in Protein Data Bank (PDB) identified the F-spondin TSR domain 4 (pdb: 1vex) as the best hit with sequence identity of 30¢. MGenTHREADER identified TSR domain of malaria TRAP protein (pdb: 2bbx), F-spondin TSR domain 4 (pdb: 1vex), and F-spondin TSR domain 1 (pdb: 1szl) as top hits with 40.8, 32.1 and 24¢ of sequence identity respectively while PHYRE server scored the F-spondin TSR domain 4 (pdb: 1vex) as the best template with a lower E-value (1.7e-07) and 32¢ sequence identity.
The multiple sequence alignment (Figure 1) shows that the disulfide bond pattern within the TSR R-spondin 4 is the same as that found in F-spondin rather than that found in TSP-1. These latter's comprise the two major group of the TSR family. The disulfide bond pattern in group 1 (TSP-1) and group 2 (F-spondin) differ mainly by the N-terminal disulfide bond. In group 1, the bond is formed between cysteines within loop between strand B and C.
In group 2 which is the group of F-spondin (a matrix-associated protein expressed in the floor plate and involved mainly in the neuronal development), one cysteine resides in the loop and the other one in the N-terminal part of the sequence. For these reasons, the F-spondin TSR domain 4 from Rattus norvegicus (1vex) [11] was selected as template to construct the model of R-spondin 4 TSR domain.
The stereochemical quality of the R-spondin 4 TSR model was assessed by PROCHECK, which assigned 77.6¢ of the residues to the Ramachandran's plot most-favoured regions, 22¢ to additionally allowed regions. No residue was found in the generously allowed or in disallowed regions. PROCHECK result is roughly comparable to the corresponding statistics for the structure of F-spondin TSR domain 4 (data not shown). The resulting model (Figure 2) reveals an architecture composed by three antiparallel strands. The first strand (A strand) is irregular, strands B and C form regular β-sheets (Gly162-Arg166) and (Glu186-Cys190 respectively). The fold of the TSR domain is characterized by a cysteine, tryptophan, and arginine residues from the three strands stacked in layers (the CWR layer) and by multiple hydrogen bonds between backbone and side chain atoms.
Figure 2.
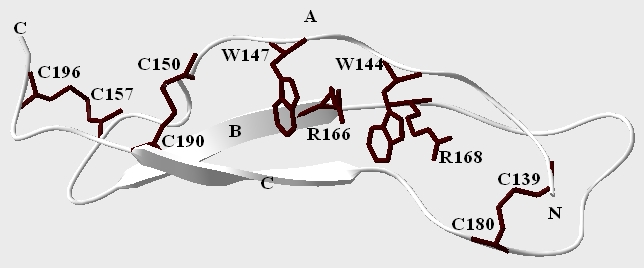
Predicted 3D model of the R-spondin 4 TSR domain. Tryptophan, arginine, and cysteine side chain forming CWR layered structure are shown in dark. The three strands and the two termini are marked with capital letters.
The side chain of two tryptophans (Trp144 and Trp147) make up two tryptophans layers (the W layers) and play a central role in the fold. Two arginines (Arg166 and Arg168) comprise the R layers and their guanidinium groups are alternately stacked between W layers. The alternate stacking of the planar cationic guanidinium groups of the arginines and the aromatic side chains of the tryptophans forms three possible cation-P interactions between (Arg166-Trp144, Arg168-Trp144 and Arg188-Trp147) which may provide a vital stabilization in the structure as estimated by CAPTURE program [16]. Additional stability is brought to the structure by three disulfide bonds one on the N-terminal side (Cys139-Cys180) and two on the C-terminal side (Cys150-Cys190 and Cys157-Cys196) forming the C layer. Altogether the tryptophan, arginine, and cysteine side-chains form a seven-layered stacked structure.
Glycosylation is one of the most abundant and widespread post-translational modifications of proteins. This modification involves the attachment of an alpha-mannosyl residue to the C-2 atom of the tryptophan residue. The Trp residues occur in WXXW patterns, which are the recognition motifs for protein C-mannosylation [17].
The TSR R-spondin 4 has been examined for the presence of C-mannosylation sites and was submitted to NetCGlyc 1.0 available at http://www.cbs.dtu.dk/services/. Results show that tryptophan 144 and 147 residues are predicted to be C-mannosylated.
The O-fucosylation which is a direct addition of O-fucose to serine or threonine is the second type of glycosylation found in TSR domains. The R spondin 4 sequence does not completely match the consensus sequence of WX5C1X2/3(S/T) C2X2G [18] since it contains 5 amino acids between the C1 and S/T instead of 2 or 3 amino acids. Furthermore, Hofsteenge and colleagues [18] showed that TSRs containing a positively charged residue at position immediately prior to the predicted modification site (Ser or Thr) were not O-fucosylated.
Heparin binding prediction
An electrostatic potential map was generated with utilities in DeepView/Swiss PDB Viewer [13]. As shown in Figure 3, the molecular surface shows that the TSR domain of R-spondin 4 contains a large positive charge from the N-terminal side through the central cavity, due to basic amino acids. This is concurrent with the presence of exposed tryptophans from the W layers along with the exposed arginines (Arg166 and Arg168) from the R layers. Other residues that are located at the edge of the C (Arg188, Lys189) strand also help to create a groove-like structure within this positively charged region.
A similar side chain array of conserved arginine and tryptophan side chains has been observed in the structures of TSP-1 as well as in F-spondin and TRAP TSRs [19]. These TSRs domains have a relatively low affinity for heparin suggesting that other residues may contribute significantly to the heparin binding.
In fact, Nam and colleagues [2] showed that besides the TSR domain, the C terminal basic amino acid rich domain is also required for heparin binding. Although the study of this domain was not straightforward, analysis of the primary protein sequence of this basic rich domain revealed the existence of clusters of basic amino acids, that matches the consensus sequence for heparin binding (XBBX where B is basic and X is hydropathic (neutral and hydrophobic) amino acid [20]). It is feasible that the TSR domain and basic amino acid rich domains of R-spondin 4 could form a continuous binding surface for heparin. Similarly, the affinity for heparin of the whole extracellular domain of TRAP (formed by TSR and A-domain) is twice that of TRAP A domain alone.
The 5th and 6th TSRs of F-spondin (containing eight domains, six of them are TSR) involved in the attachment to the extracellular matrix of F-spondin by binding to protoglycans, have more amino acid basic residues on the front face than that of F-spondin TSR domains 1-4 (more amino acid basic residues on the C strand and BC loop).
On the other hand, a search with PredictNLS algorithm (available at http://cubic.bioc.columbia.edu/cgi/var/nair/resonline.pl) identified the fragment QKKGRKDRRPRKDRKLDRRL from 204 to 223 as Nuclear Localization Signal sequence. Involvement of this basic region in the heparin binding may suggest the implication of heparin in the penetration and nuclear uptake of R-spondin 4. Several studies have suggested the role of heparin in regulating nuclear internalization of growth factors such as FGF-1 and FGF-2 [21]. Further studies should be carried out to substantiate these suggestions.
Conclusion
In this paper the seven CWR layered model of R-spondin 4 TSR domain was constructed on the base of similarity to F-spondin TSR domain 4. The positively charged surface in the TSR domain and the contiguous basic amino acid rich region were predicted to form a continuous binding surface for heparin which may explain the high affinity of R-spondin for heparin. Our results suggest that the binding is largely electrostatic in nature.
References
- 1.Blaydon DC, et al. Nat Genet. 2006;38:1245. doi: 10.1038/ng1883. [DOI] [PubMed] [Google Scholar]
- 2.Nam JS, et al. J Biol Chem. 2006;281:13247. doi: 10.1074/jbc.M508324200. [DOI] [PubMed] [Google Scholar]
- 3.Lawler J, et al. J Cell Biol. 1986;103 [Google Scholar]
- 4.Chen H, et al. Matrix Biol. 2000;19:597. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 5.Klar A, et al. Cell. 1992;69:95. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- 6.Bernfield M, et al. Annu Rev Biochem. 1999;68:729. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 7.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones DT. J Mol Biol. 1999;287:797. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 9. http://www.sbg.bio.ic.ac.uk/~phyre/
- 10.Sali A, et al. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 11.Paakkonen K, et al. Proteins. 2006;64:665. doi: 10.1002/prot.21030. [DOI] [PubMed] [Google Scholar]
- 12.Laskowski RA, et al. J Appl Cryst. 1993;26 [Google Scholar]
- 13.Guex N, et al. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 14.Thompson JD, et al. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huwiler KG, et al. Biochemistry. 2002;41:14329. doi: 10.1021/bi026463u. [DOI] [PubMed] [Google Scholar]
- 16.Gallivan JP, et al. Natl Acad Sci. 1999;96:9459. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furmanek A, et al. Acta Biochim Pol. 2000;47:781. [PubMed] [Google Scholar]
- 18.de Peredo Gonzalez, et al. Mol Cell Proteomics. 2002;1 [Google Scholar]
- 19.Tan K, et al. J Cell Biol. 2002;159:373. doi: 10.1083/jcb.200206062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fromm JR, et al. Archives of Biochemistry and Biophysics. 1997;343:92. doi: 10.1006/abbi.1997.0147. [DOI] [PubMed] [Google Scholar]
- 21.Sperinde GV, et al. Biochemistry. 2000;39 doi: 10.1021/bi992243d. [DOI] [PubMed] [Google Scholar]