

Abstract
A total synthesis of brevetoxin A is reported. Two tetracyclic coupling partners, prepared from previously reported advanced fragments, were effectively united via a Horner—Wittig olefination. The resulting octacycle was progressed to substrates that were explored for reductive etherification, the success of which led to a penultimate tetraol intermediate. The tetraol was converted to the natural product through an expeditious selective oxidative process, followed by methylenation.
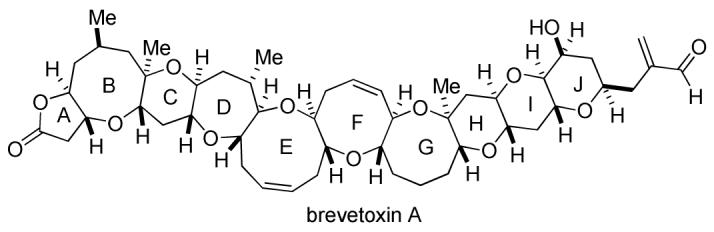
The exquisite structures of marine polycyclic ether natural products have captured the imagination of synthetic chemists for over two decades. The structures of the polyether ladder toxins characteristically contain a linear series of trans fused ether rings of varying sizes from five to nine members with assorted methyl and hydroxyl substituents appended. As novel technologies for the convergent preparation of these targets have emerged, a number of total syntheses of the ladder toxins have been completed.1 The structure of brevetoxin A (1), a representative member of this class, was first elucidated in 1986 by Shimizu2a,b, and co-workers by X-ray analysis and independently determined by Nakanishi through spectroscopic studies.2c Brevetoxin A (1) contains ten rings (including five-, six-, seven-, eight, and nine-membered oxacycles) fused in a linear array adorned by 22 tetrahedral stereocenters. A metabolite of Karenia brevis, brevetoxin A is a toxic component of the infamous red tide phenomenon, which has been responsible for massive fish kills as well as neurotoxic shellfish poisoning and bronchial irritation in humans.3 The potent activity of brevetoxin A is attributed to strong binding to the α subunit of the voltage-sensitive sodium ion channels effecting an increase in the mean channel open time and inhibiting channel inactivation.3a To date, the landmark total synthesis reported by Nicolaou stands as the only completed synthesis of this intriguing target.4
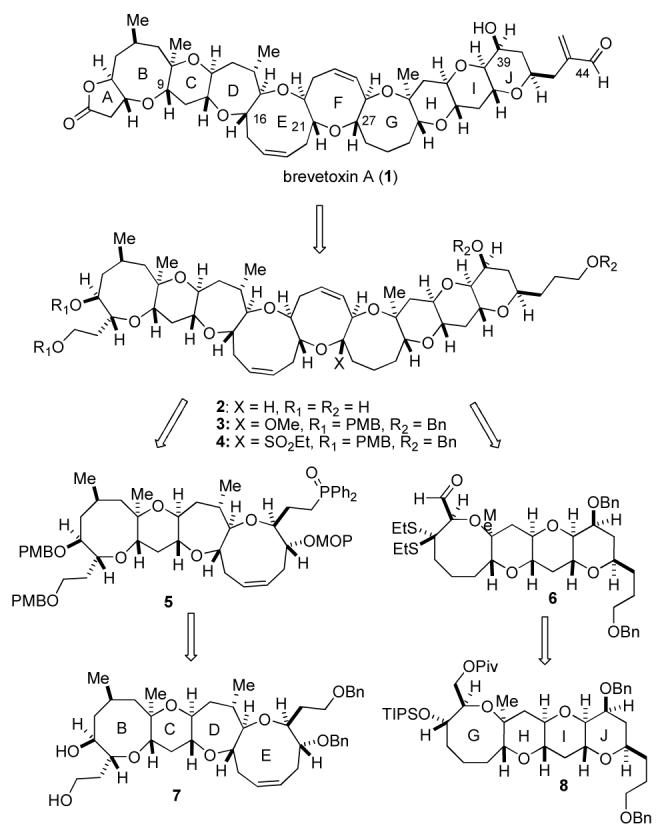
The planned approach for the total synthesis of brevetoxin A5 focused on a versatile endgame that would exploit the selective manipulation of tetraol 2 (Scheme 1), which would derive from mixed methyl ketal 3 via stereoselective reductive etherification. Ketal 3 would be obtained through the stereoselective Horner—Wittig coupling6 of phosphine oxide 5 and aldehyde 6. This route was attractive not only because it allowed for optimal convergence by simplifying the natural product into two halves of similar complexity, but also because it found precedent in the strategy previously reported by Nicolaou.4 Further, it was reasoned that the dithioketal moiety of aldehyde 6 could serve as a stabilized precursor to mixed ketal 3, or lead to sulfone 4 in the event that formation or reductive etherification of mixed ketal 3 proved problematic. The Horner—Wittig coupling partners 5 and 6 would be obtained from advanced fragments 7 and 8, respectively. The BCDE fragment 7 and GHIJ subunit 8 had been previously prepared in significant quantities through similar highly convergent [X+2+X] strategies based on a Horner—Wadsworth—Emmons coupling of the B and E ring units (and the G and J subunits) and subsequent construction of the central CD and HI rings.5a,b
Scheme 1.
Retrosynthetic Analysis of Brevetoxin A
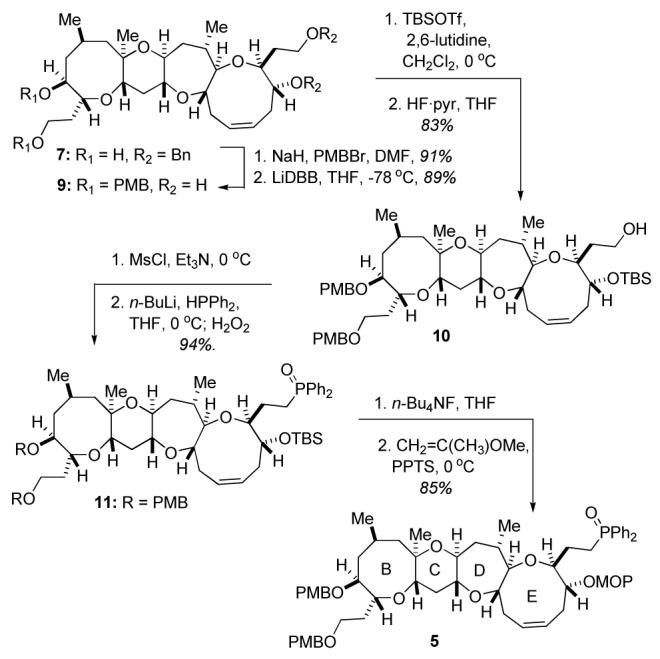
The conversion of diol 7 to phosphine oxide 5 (Scheme 2) commenced with protection of diol 7 as the bis-p-methoxybenzyl ether and subsequent reductive cleavage of the benzyl ethers with LiDBB to form diol 9.7 Diol 9 was protected as the bis-TBS ether, whereupon selective cleavage of the primary TBS ether with HF·pyr afforded alcohol 10. Alcohol 10 was smoothly transformed to phosphine oxide 11 via a sequence which involved mesylation of the alcohol, nucleophilic displacement of the mesylate to provide the alkyl diphenylphosphine, and finally, oxidative workup of the phosphine with H2O2.4,6 Cleavage of the TBS ether with n-Bu4NF and formation of the methoxypropyl (MOP) acetal delivered the required phosphine oxide 5 in high yield.8
Scheme 2.
Formation of Phosphine Oxide 5a
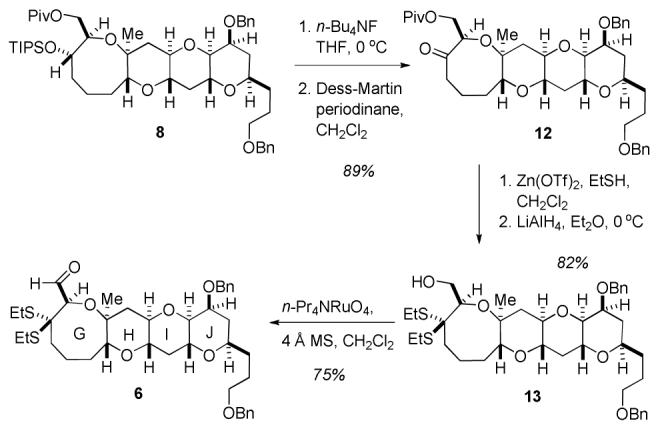
For aldehyde 6, GHIJ tetracycle 8 was treated with n-Bu4NF to cleave the TIPS ether, and the resultant secondary alcohol was oxidized to ketone 12 with Dess-Martin periodinane (Scheme 3).9 Treatment of ketone 12 with Zn(OTf)2 and EtSH produced the dithioketal,10 and reductive cleavage of the pivaloate ester with LiAlH4 delivered alcohol 13. While a host of conditions proved unsuitable for the subsequent oxidation of the primary alcohol to aldehyde 6, due to undesired oxidation of the dithioketal, the use of stoichiometric n-Pr4NRuO4 reproducibly provided the desired aldehyde with minimal byproducts.11
Scheme 3.
Preparation of Aldehyde 6
With both phosphine oxide 5 and aldehyde 6 in hand, their assembly under Horner—Wittig conditions was explored (Scheme 4).6 After some experimentation, addition of 3 equiv of LDA to a solution of phosphine oxide 5 and aldehyde 6 at -78 °C was found to produce 63% of the Horner—Wittig adduct. It is worthy of note that no epimerization of aldehyde 6 was observed, despite the presence of superstoichiometric base. Exposure of the intermediate hydroxy-phosphine oxide to KHMDS provided the desired Z-olefin 14 in 74% yield.
Scheme 4.

Coupling of Tetracyclic Fragments 5 and 6
Having effectively coupled the BCDE and GHIJ fragments, we wished to examine the mixed methyl ketal 3 as a precursor to the targeted nonacycle 16 (Scheme 4). Despite the dearth of literature examples for the conversion of 7-hydroxy ketones or ketals to eight-membered cyclic ketals, we believed that the formation of mixed ketal 3 would be possible due to the structural pre-organization present in olefin 14. In particular, the newly-formed Z-olefin, along with the cyclic conformational constraints about the C21—C22 and C26—C27 bonds, would be expected to assist in the required cyclization event. Additionally, based upon the reported use of (F3CCO2)2IPh in alcoholic solvent to convert dithioketals to dialkyl ketals,12 we reasoned that exposure of olefin 14 to the hypervalent iodine reagent in MeOH would lead to the dimethyl ketal 15, or to mixed ketal 3 directly. In the event, treatment of thioketal 14 with (F3CCO2)2IPh in MeOH rapidly removed the MOP protecting group, and led to a mixture of the expected ketal products in a 4:1 ratio favoring the dimethyl ketal 15.13 Exposure of the unpurified mixture to PPTS and trimethyl orthoformate in toluene provided an 80% overall yield of cyclic mixed methyl ketal 3.
Drawing upon previous successes in our laboratory involving the reductive etherification of precursors to the BCDE and GHIJ fragments,5a,b it was anticipated that treatment of ketal 3 with an appropriate Lewis acid in the presence of a trialkylsilane would deliver the expected nonacycle 16 (Scheme 4). Despite extensive screening of Lewis acids (BF3·OEt2, TiCl4, TMSOTf), solvents, silanes (Et3SiH, Me2PhSiH), and drying agents (4 Å MS, BaO), only traces of the desired reduction product 16 were obtained. Instead, hydrolysis of the ketal, via interception of the oxocarbenium ion by adventitious water, cleavage of the oxocene (producing the C27 methyl ether), and intractable decomposition were repeatedly observed. The use of i-Bu2AlH as the reducing agent also failed to bring about the intended reduction.
Borrowing from precedent in the Nicolaou synthesis, attention was turned to the reductive etherification of sulfone 4 rather than the ketal 3 in an attempt to increase the lability of the leaving group at C27.4,14 Removal of the MOP acetal from olefin 14 (Scheme 5) under acidic conditions followed by treatment of the hydroxy-dithioketal with AgClO4 provided the cyclic mixed S,O-ketal 18. Upon obtaining sulfone 4 via oxidation with m-CPBA, reductive etherification with concomitant removal of the PMB protecting groups was readily accomplished, furnishing diol 16 in 85% yield.
Scheme 5.
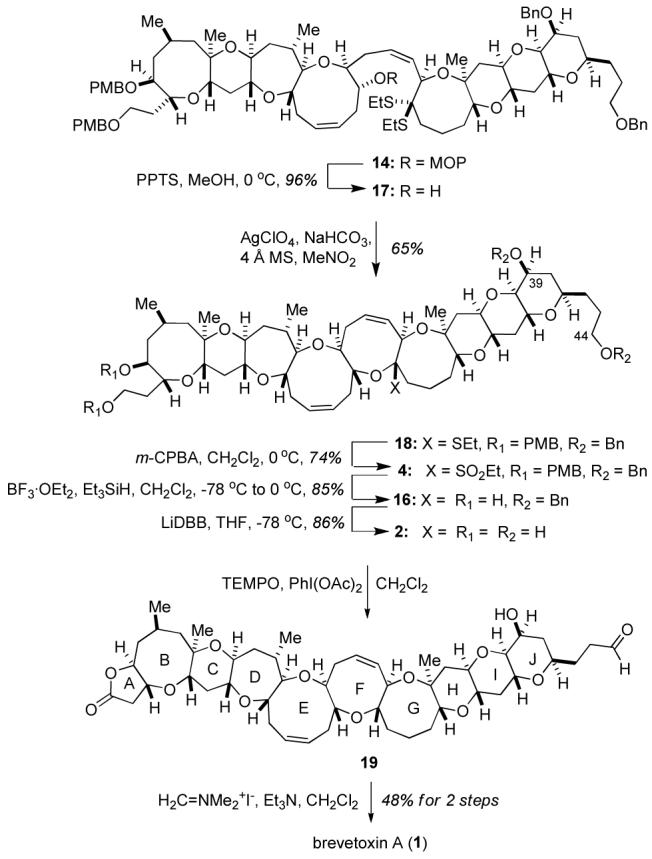
Completion of Brevetoxin A (1)a
Initial attempts to complete the synthesis of brevetoxin A from diol 16 focused on the formation of the A ring lactone followed by removal of the J ring benzyl ethers and functionalization of the J ring side chain. While the A ring lactone could be readily constructed by treatment of diol 16 with n-Pr4NRuO4, subsequent attempts to cleave the J ring benzyl ethers under a host of reductive or Lewis acidic conditions met with failure due to significant amounts of decomposition. Ultimately, a strategy involving the selective oxidation of tetraol 2 was examined. Unlike the A ring lactone derived from diol 16, the J ring benzyl ethers could be readily cleaved from diol16 itself to deliver tetraol 2. Thus, brevetoxin A (1) was accessed in three straightforward operations from diol 16 (Scheme 5). Reductive cleavage of the J ring benzyl ethers, and subsequent exposure of tetraol 2 to PhI(OAc)2 in the presence of catalytic TEMPO15 served to selectively form the A-ring lactone and the C44 aldehyde while leaving the axially-disposed C39 secondary alcohol unaffected. The unpurified decacyclic aldehyde 17 was treated with Eschenmoser′s salt in the presence of Et3N4,16 to complete the synthesis of brevetoxin A (1). Synthetic brevetoxin A (1) was identical in all respects (1H and 13C NMR, IR, HRMS, [α]D) to an authentic sample.4,17
In summary, the total synthesis of brevetoxin A was completed from aldehyde 6 and phosphine oxide 5 through a stereoselective Horner—Wittig olefination to assemble two advanced tetracycles. The uncommon cyclization of a medium ring mixed methyl ketal was accomplished and the mixed methyl ketal was assessed as a substrate for a reductive etherification of an oxocene. Ultimately, a sulfone-based approach proved superior and set the stage for a selective oxidation strategy of tetraol 2, allowing the completion of the second total synthesis of brevetoxin A (1).
Supplementary Material
Acknowledgment
Financial support of this work by the National Institute of General Medical Sciences (GM60567) is acknowledged with thanks. We are grateful to Professor Daniel G. Baden and Dr. Andrea Bourdelais (University of North Carolina at Wilmington) for kindly providing an authentic sample as well as 13C and 1H NMR spectral data of brevetoxin A.
Footnotes
Supporting Information Available. Experimental details and spectral data for new compounds, as well as brevetoxin A. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Fuwa H, Sasaki M. Curr. Opin. Drug Discovery Dev. 2007;10:784. [PubMed] [Google Scholar]; (b) Inoue M. Chem. Rev. 2005;105:4379. doi: 10.1021/cr0406108. [DOI] [PubMed] [Google Scholar]; (c) Nakata I. Chem. Rev. 2005;105:4314. doi: 10.1021/cr040627q. [DOI] [PubMed] [Google Scholar]; (d) Kadota I, Yamamoto Y. Acc. Chem. Res. 2005;38:423. doi: 10.1021/ar040118a. [DOI] [PubMed] [Google Scholar]; (e) Sasaki M, Fuwa H. Synlett. 2004;11:1851. [Google Scholar]; (f) Evans PA, Delouvrié B. Curr. Opin. Drug Discovery Dev. 2002;5:986. [PubMed] [Google Scholar]; (g) Marmsäter FP, West FG. Chem. Eur. J. 2002;8:4346. doi: 10.1002/1521-3765(20021004)8:19<4346::AID-CHEM4346>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (h) Mori Y. Chem. Eur. J. 1997;3:849. [Google Scholar]; (i) Alvarez E, Candenas M-L, Pérez R, Ravelo JL, Martín JD. Chem. Rev. 1995;95:1953. [Google Scholar]
- (2) (a).Shimizu Y, Chou HN, Bando H, Vanduyne G, Clardy JC. J. Am. Chem. Soc. 1986;108:514. doi: 10.1021/ja00263a031. [DOI] [PubMed] [Google Scholar]; (b) Shimizu Y, Chou H-N, Bando H, Van Duyne G, Clardy JC. J. Chem. Soc., Chem. Commun. 1986:1656. doi: 10.1021/ja00263a031. [DOI] [PubMed] [Google Scholar]; (c) Pawlak J, Tempesta MS, Golik J, Zagorski MG, Lee MS, Nakanishi K, Iwashita T, Gross ML, Tomer KB. J. Am. Chem. Soc. 1987;109:1144. [Google Scholar]
- (3) (a).Michelliza S, Abraham WM, Jacocks HM, Schuster T, Baden DG. ChemBioChem. 2007;8:2233. doi: 10.1002/cbic.200700317.Naar JP, Flewelling LJ, Lenzi A, Abbott JP, Granholm A, Jacocks HM, Gannon D, Henry M, Pierce R, Baden DG, Wolny J, Landsberg JH. Toxicon. 2007;50:707. doi: 10.1016/j.toxicon.2007.06.005.Kirkpatrick B, Fleming LE, Backer LC, Bean JA, Tamer R, Kirkpatrick G, Kane T, Wanner A, Dalpra D, Reich A, Baden DG. Harmful Algae. 2006;5:526. doi: 10.1016/j.hal.2005.09.004.Fleming LE, Backer LC, Baden DG.Environ. Health Persp 2005113618 and references therein.
- (4) (a).Nicolaou KC, Yang Z, Shi G-Q, Gunzner JL, Agrios KA, Gärtner P. Nature. 1998;392:264. doi: 10.1038/32623. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Bunnage ME, McGarry DG, Shi S, Somers PK, Wallace PA, Chu X-J, Agrios KA, Gunzner JL, Yang Z. Chem. Eur. J. 1999;5:599. [Google Scholar]; (c) Nicolaou KC, Wallace PA, Shi S, Ouellette MA, Bunnage ME, Gunzner JL, Agrios KA, Shi G.-q., Gärtner P, Yang Z. Chem. Eur. J. 1999;5:618. [Google Scholar]; (d) Nicolaou KC, Shi G.-q., Gunzner JL, Gärtner P, Wallace PA, Ouellette MA, Shi S, Bunnage ME, Agrios KA, Veale CA, Hwang C-K, Hutchinson J, Prasad CVC, Ogilvie WW, Yang Z. Chem. Eur. J. 1999;5:628. [Google Scholar]; (e) Nicolaou KC, Gunzner JL, Shi G.-q., Agrios KA, Gärtner P, Yang Z. Chem. Eur. J. 1999;5:646. doi: 10.1038/32623. [DOI] [PubMed] [Google Scholar]
- (5) (a).Crimmins MT, McDougall PJ, Ellis JM. Org. Lett. 2006;8:4079. doi: 10.1021/ol0615782. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT, Zuccarello JL, Cleary PA, Parrish JD. Org. Lett. 2006;8:15. doi: 10.1021/ol0526625. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, McDougall PJ, Emmitte KA. Org. Lett. 2005;7:4033. doi: 10.1021/ol051543m. [DOI] [PubMed] [Google Scholar]; (d) Crimmins MT, Cleary PA. Heterocycles. 2003;61:87. [Google Scholar]
- (6) (a).L. Horner L., Hoffmann H, Wippel HG, Klahre G. Chem.Ber. 1959;92:2499.Buss AD, Warren S.J. Chem. Soc. Perkin Trans 198512307, and references therein.For the use of LDA in Horner-Wittig reactions, see: Earnshaw C, Wallis CJ, Warren S.J. Chem. Soc.,Perkin Trans. I 19793099
- (7) (a).Freeman PK, Hutchinson LL. J. Org. Chem. 1980;45:1924. [Google Scholar]; (b) Ireland RE, Smith MG. J. Am. Chem. Soc. 1988;110:854. [Google Scholar]
- (8).Attempts to prepare the phosphine oxide with the C21 hydroxyl or MOP acetal directly were hampered by purification difficulties and lower yields, respectively.
- (9).Dess DB, Martin JC. J. Org. Chem. 1983;48:4155. [Google Scholar]
- (10).Corey EJ, Shimoji K. Tetrahedron Lett. 1983;24:169. [Google Scholar]
- (11) (a).William P, Griffith WP, Ley SV, Whitcombe GP, White AD. J. Chem. Soc., Chem. Commun. 1987:1625. [Google Scholar]; (b) Griffith WP, Ley SV. Aldrichimica Acta. 1990;23:13. [Google Scholar]
- (12).Stork G, Zhao K. Tetrahedron Lett. 1989;30:287. [Google Scholar]
- (13).The 4:1 ratio of products appears to be the kinetic ratio, since re-subjecting either of the isolated ketal products to the (CF3CO2)2IPh/MeOH conditions did not cause equilibration.
- (14).Nicolaou KC, Prasad CVC, Hwang C-K, Duggan ME, Veale CA. J. Am. Chem. Soc. 1989;111:5321. [Google Scholar]
- (15).Hansen TM, Florence GJ, Lugo-Mas P, Chen JH, Abrams JN, Forsyth CJ. Tetrahedron Lett. 2003;44:57. [Google Scholar]
- (16).Schreiber J, Maag H, Hashimoto N, Eschenmoser A. Angew. Chem. Int. Ed. Engl. 1971;10:330. [Google Scholar]
- (17).See Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.