

Abstract
Analysis of the antitumor immune response after gene transfer of a foreign major histocompatibility complex class I protein, HLA-B7, was performed. Ten HLA-B7-negative patients with stage IV melanoma were treated in an effort to stimulate local tumor immunity. Plasmid DNA was detected within treated tumor nodules, and RNA encoding recombinant HLA-B7 or HLA-B7 protein was demonstrated in 9 of 10 patients. T cell migration into treated lesions was observed and tumor-infiltrating lymphocyte reactivity was enhanced in six of seven and two of two patients analyzed, respectively. In contrast, the frequency of cytotoxic T lymphocyte against autologous tumor in circulating peripheral blood lymphocytes was not altered significantly, suggesting that peripheral blood lymphocyte reactivity is not indicative of local tumor responsiveness. Local inhibition of tumor growth was detected after gene transfer in two patients, one of whom showed a partial remission. This patient subsequently received treatment with tumor-infiltrating lymphocytes derived from gene-modified tumor, with a complete regression of residual disease. Thus, gene transfer with DNA–liposome complexes encoding an allogeneic major histocompatibility complex protein stimulated local antitumor immune responses that facilitated the generation of effector cells for immunotherapy of cancer.
Keywords: cancer, gene therapy, direct gene transfer, immunotherapy
Although melanoma is treatable in its early stages, recurrent or metastatic lesions are resistant to standard forms of therapy. Melanoma is among the fastest increasing malignancies in its incidence rates and the most lethal of all primary cutaneous neoplasms. An intriguing aspect of melanoma is its inherent immunogenicity. Depending on the treatment, 10–20% of these tumors respond to some form of immunotherapy (summarized in ref. 1), but approaches involving administration of cytokines and/or adoptive T cell transfer have shown limited efficacy in metastatic disease. Recently, molecular genetics has progressed to the extent that it can be applied more readily to the treatment of human malignancy. Several human gene transfer protocols have been designed to monitor safety, toxicity, gene expression, and the immune response to tumors based on animal models that have used genes encoding cell surface antigens, immunomodulatory cytokines, or T cell costimulatory molecules to enhance the immune response to tumors.
We have developed a gene transfer approach for the treatment of human malignancies that uses the immunogenicity of a transplantation antigen to stimulate immune reactivity. This approach relies on direct gene transfer into tumors in vivo. Expression of a gene encoding a foreign histocompatibility protein (class I) signals the immune system to respond to the transplantation antigen, and more importantly, this stimulation leads to immune recognition of the unmodified tumor cells (2). In animal models, this approach has led to a significant reduction in tumor growth and complete regression in some cases (2). A major advantage of this approach is that DNA can be introduced directly into growing tumors. In contrast to other gene transfer strategies for cancer, direct gene transfer eliminates the need to remove cells from the patient and propagate them in the laboratory. This approach also reduces the delay between the time of diagnosis to initiation of treatment. Modifications of the plasmid and lipid have subsequently been made to improve gene delivery and expression, including addition of the β-2 microglobulin gene to allow synthesis of both chains of the class I major histocompatibility complex (MHC) genes in tumor cells that are unable to express this gene product. In addition, a cationic lipid formulation that improves transfection efficiency was developed (3, 4). In this study, we describe a human clinical study to evaluate the safety and efficacy of gene expression of this DNA–liposome complex and to further characterize the immune response to progressive melanomas.
MATERIALS AND METHODS
Clinical Protocol and Study Design.
Ten patients with stage IV melanoma unresponsive to all standard treatments were enrolled based on guidelines of the clinical protocol (5) and admitted to the Clinical Research Center. Informed written consent was obtained according to The Committee to Review Grants for Clinical Research and Investigation Involving Human Beings of the University of Michigan Medical School, the Recombinant DNA Advisory Committee of the National Institutes of Health, and the Food and Drug Administration.
The borders of a cutaneous tumor nodule were identified for treatment as measured before, during, and after treatment. Staging was performed by computerized tomography immediately before the procedure. Group I (n = 3) received a total of three injections of ≈0.6 ml of DNA–liposome complex [3 μg of DNA:4.5 nM dimyristyloxypropyl-3-dimethyl-hydroxyethyl ammonium (DMRIE)/dioleoyl phosphatidylethanolamine (DOPE)] biweekly into the tumor (9 μg cumulative dose). Group II (n = 3) received three injections of 30 μg of DNA:4.5 nM DMRIE/DOPE biweekly within the same nodule (90 μg cumulative dose). Group III (n = 3) was injected three times biweekly with 100 μg of DNA:148.5 nM DMRIE/DOPE, and Group IV (n = 3) was treated similarly with 300 μg of DNA:450 nM DMRIE/DOPE. All patients received a total of three treatments with a 2-week interval between treatments. Patient 1 received three courses of treatment (groups I, II, and III) with an interval of 9 weeks between escalations, and patient 2 received two treatments (groups I and II) with an interval of 8 weeks.
Vector Production, Preparation, and Administration of DNA–Liposome Complex.
A eukaryotic expression vector plasmid encoding HLA-B7 and β-2 microglobulin was prepared by insertion of an HLA-B7 gene cDNA, an internal ribosome entry site, and β-2 microglobulin into a plasmid using the Rous sarcoma virus enhancer/promoter and bovine growth hormone polyadenylylation site as described (5, 6). Batch preparations of clinical grade DNA and the DMRIE/DOPE cationic liposome were kindly provided by Vical (San Diego).
For gene transfer, a 22-gauge needle was used to inject the DNA–liposome complex, which was prepared as follows. Ten minutes before delivery, 0.1 ml of plasmid DNA (0.05–50 mg/ml) in lactated Ringer’s solution was added to 0.1 ml of DMRIE/DOPE liposome solution (0.15–15 μM). The DNA–liposome solution (0.6 ml) was injected into each nodule under sterile conditions at the bedside after administration of local anesthesia (1% lidocaine) using a 22-gauge needle.
Biochemical and Hemodynamic Monitoring.
To monitor the potential toxicities of the DNA–liposome treatment, biochemical, hematological, and hemodynamic parameters were evaluated. Vital signs and cardiac rhythm were monitored, and subjective complaints of patients were sought and recorded.
Analysis of HLA-B7 Gene Expression.
To confirm recombinant HLA-B7 gene expression within treated tumor nodules, core needle biopsy samples of the injected tumor were analyzed after the gene transfer procedure. Genomic DNA was isolated from biopsy material (7), and PCR for HLA-B7 gene was performed with two primers [sense, 5′-CAG CTG TCT TGT GAG GGA CTG AGA TGC AGG-3′ (HLA-B7); antisense, 5′-TTC CAA GCG GCT TCG GCC AGT AAC GTT AGG-3′ (CITE A)] to generate a 310-bp fragment (see Fig. 1 A and C). For the RNA analysis, these primers were used after reverse transcription with oligo(dT). For analysis of plasmid in blood, the same set of primers was used. RNA was analyzed by PCR after DNase digestion and incubation with reverse transcriptase as described (8). In some cases, Southern blot hybridization of PCR products from the RNA analysis was performed with a probe to internal sequence, derived by digestion of pHLA-B7, described above, with PvuII and BglII by standard methods (9). The control, 293 cells transfected with plasmids in vitro, was used to establish the conditions for DNase digestion (see Fig. 1B). Under these conditions, no PCR signal was detected in the absence of reverse transcription.
Figure 1.
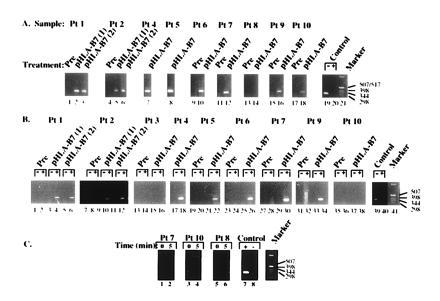
Gene transfer and expression of foreign MHC gene in human melanoma. Size markers (in base pairs) are indicated to the right of each panel. (A) Detection of plasmid DNA in melanoma nodules after direct gene transfer with the DNA–liposome complex. Nucleic acids were isolated from injected nodules and analyzed by PCR (7). Samples were taken at the indicated times, and DNA was extracted according to standard methods (see below). The sensitivity of the PCR analysis is ≈1 copy of recombinant gene per 105 genomes (14). (B) Confirmation of gene expression in tumor nodules transduced by direct gene transfer with HLA-B7. Recombinant HLA-B7 mRNA was detected by using a reverse transcriptase–PCR technique of nucleic acids from biopsy samples. Total RNA was incubated in the presence (+) or absence (−) of reverse transcriptase and analyzed (9). (C) Analysis of blood samples from three patients receiving the highest dose of DNA–liposome complex (300 μg of DNA) before treatment or 5 min after gene transfer as indicated.
Cytotoxic T Lymphocyte (CTL) Frequency of Tumor-Infiltrating Lymphocytes (TIL) and Peripheral Blood Mononuclear Cells Specific for Autologous Tumor.
Limiting dilution analysis (LDA) was adapted (10) to quantify autologous tumor-specific CTL in the peripheral blood of patients, as well as in TIL populations derived from tumor before and after HLA-B7 gene transfer. Dilutions of responder cells were added to round-bottomed microtiter plates along with 2 × 105 irradiated (4000 cGy) autologous tumor cells per well in DMEM complete medium containing 10% human serum and supplemented with 20 units of rIL-2 per ml (final volume = 200 μl). After a 7-day incubation, cytolytic activity was assessed by adding 2 × 103 51Cr-labeled tumor target cells to each microwell. After a 4-hr incubation, 150 μl of supernatant was removed from each well, and the supernatants were assayed for released 51Cr in a computerized gamma counter. Microcultures are considered cytolytic if observed chromium release is greater (mean + 3 SD) than the chromium release observed in control wells that lack responder cells. Specificity of the CTL detected by this assay has been verified by parallel LDA stimulated by autologous tumor but overlaid with allogeneic tumor targets.
Minimal estimates of CTL frequency are obtained according to the Poisson distribution equation as the slope of a line relating the number of responder cells per microwell (plotted on a linear x axis) and the percentage of microwells that failed to develop cytolytic activity (plotted on a logarithmic y axis) (11). The slope of this regression line is determined by computer using χ2 minimization analysis, as described by Taswell (12).
Tumor Preparation and Establishment of TIL Cultures.
Tumor specimens were obtained from the operating room under sterile conditions and processed as described (13). TIL cell cultures were established in X-Vivo-15 (BioWhittaker) media supplemented with 10% human AB serum (Sigma) and 6000 units of interleukin (IL) 2 (Aldesleukin Proleukin, Chiron) per ml of media at 2.5 × 105 nucleated cells per ml in Life cell 3000 tissue culture bags (Baxter Health Care, Fenwall division). By day 7, lymphocyte proliferation was evident, and the culture was diluted 1:2 every 2 or 3 days with X-Vivo-15 supplemented with IL-2 without serum for 21 days.
RESULTS
Ten patients who satisfied the entry criteria of the protocol (5) were included for study in the General Clinical Research Center at the University of Michigan Medical Center. The prior treatments and history of these patients are presented in Table 1. In each case, these patients exhibited progressive disease (stage IV) unresponsive to all conventional forms of therapy. The gene transfer procedure was well-tolerated in each patient, with no acute complications.
Table 1.
Clinical profiles of patients and tumors, and summary of the presence of RNA, recombinant HLA-B7, and increase in CD3+ cell infiltration after gene transfer
| Patient no. | Patient I.D. | Age/ Sex | HLA haplotype | Previous treatment | Site | DNA dose, μg (group) | RNA* | HLA-B7† | CD3+ infil- tration |
|---|---|---|---|---|---|---|---|---|---|
| 1 | G.M. | 50/M | A2, 19; B35−, w6; C4,−; DR1, 12, 52; DQ1, 3, 7 | BCG; chemotherapy; IL-2; TIL; limb perfusion with melphalan, tumor necrosis factor, and interferonγ | Left thigh | 3(I), 30(II), 100(III) | + | ++ | +++ |
| 2 | A.P. | 46/F | A1, 3; B8, 27, w6, w4; C2, 7; DR13, 15 | Surgery, radiation, chemotherapy, activated primed T cells, and IL-2 | Right lateral thigh, right gluteal, right shoulder | 3(I), 30(II) | + | ind. | +++ |
| 3 | A.H. | 54/M | A1, 2; B8, w6; C7; DR1, 15; DQ1, 6, 51 | Surgery, chemotherapy, and IL-2 | Left chest | 3 | − | + | ++ |
| 4 | I.M. | 72/F | A2, 29; B8, −, w6; DR3, −, 52 | Surgery | Left knee | 30 | + | + | ND |
| 5 | G.L. | 40/F | A11, 30; B13, 44, w4, w6; C3, 6; DR2, 7, 53; DQ1, 2, 5, 51 | Surgery, and limb perfusion with tumor necrosis factor and interferon α | Right thigh | 100 | + | ind. | − |
| 6 | D.M. | 52/F | A2, −; B13, −, w4; C6, −; DR7, 8, 53; DQ2, 4 | Surgery and interferon | Scalp | 100 | + | +/− | ND |
| 7 | F.B. | 68/F | A23, 24; B13, 44, w4, w6; C4, −; DR7, −, 53; DQ2, 3 | Surgery and radiation | Left back | 100 | + | ++ | + |
| 8 | M.S. | 45/F | A3, 29; B44, 60; DR7, 13, 52, 53; DQ1, 2 | Surgery | Right thigh | 300 | ND | +/− | ++ |
| 9 | G.B. | 29/M | A3, 26; B18, 62, w6; C3, 5; DQ2; DR52, 53 | Surgery and radiation | Right neck | 300 | + | +/− | ++ |
| 10 | J.C. | 34/M | A1, 11; B44, −, w4; C5, −; DR1, 7, 53 | Surgery, cisplatinum therapy, radiation, Dartmouth regimen, and chemotherapy | Left chest wall | 300 | − | ND | ND |
I.D., Identification; M, male; F, female; BCG, Bacille Calmette–Guérin; ND, not determined; ind., indeterminate.
RNA was detected by reverse transcriptase–PCR.
HLA-B7 was detected by immunostaining.
A sample of the treated tumor nodules was obtained by a core needle biopsy 1–3 days after the second or third intratumoral injection of HLA-B7 DNA–liposome complexes. This tissue was analyzed for the presence of plasmid DNA and mRNA encoding HLA-B7 and β-2 microglobulin, and HLA-B7 expression. In 8 of 10 treatments, the plasmid DNA was detected within the injected nodule (Fig. 1A).
Expression of recombinant HLA-B7 and β-2 microglobulin mRNA was analyzed in transduced tumors. In seven of nine tumor biopsies, RNA coding for HLA-B7 was detected by PCR after incubation with reverse transcriptase but not in its absence (Fig. 1B). In one of the two cases in which mRNA was not detected (patient 3), an inhibitor of the PCR was present (data not shown). In representative cases, blood samples obtained immediately before and multiple times after injection were analyzed for the presence of plasmid DNA by PCR. In these patients, plasmid DNA was not detected at any time in the blood after gene transfer by PCR (sensitivity < 2 pg/ml), even as early as 5 min after injection of doses of DNA up to 300 μg per injection (Fig. 1C).
HLA-B7 protein expression was detected in biopsy tissue by immunochemical staining using a monoclonal antibody against this gene product. The recombinant protein was detected by these methods at frequencies ranging from 1% to 10% of tumor cells near the site of injection (data not shown). Failure to detect DNA in two patient biopsy samples was related to inhibition of the PCR, and HLA-B7 was immunohistochemically detected in tumor cells of these patients. In patient 3, cross-reactivity of the monoclonal antibody to an endogenous HLA-B haplotype, B40, did not allow definitive confirmation of protein expression, although the recombinant mRNA was readily detected (Fig. 1B; Table 1).
Analysis of serum biochemical parameters revealed no abnormalities induced by gene transfer in these patients, including serum markers of liver, renal, pancreatic, and cardiac function (data are available upon request). No changes from baseline in any of the serum biochemical parameters were found in the acute 3- to 7-day period, and no significant abnormalities found up to 2 months after the initial injection. In addition, myocardial abnormalities were not detected by analysis of creatine phosphokinase or its isoenzymes in the serum of treated patients, and no electrocardiographic changes or arrhythmias were noted. Similar to a previous study in humans with DNA–liposome complexes (15), no increases in anti-DNA antibodies were detected in patients, and there was no clinical evidence of autoimmune phenomena, as indicated by changes in antinuclear antibodies, C-reactive protein, or other immunologic markers. These data support the previous observations that DNA is not highly immunogenic in vivo in humans (15) and an immune response to DNA–liposome complexes is unlikely to limit in vivo gene transfer with this vector or other forms of DNA.
To determine whether gene transfer of a foreign MHC gene could alter the T cell response to tumors, immunohistochemical analysis was performed. Tumor biopsy samples were analyzed for the presence of infiltrating T cells before treatment or after gene transfer. Immunostaining with αCD3 in tumor nodules revealed a 77-fold increase in CD3+ cells at the margins of tumors relative to the parenchyma before treatment (Fig. 2A Left). After gene transfer, a 31-fold or ≈2-fold increase in TIL in the tumor parenchyma was detected in patients 1 and 2, respectively (Fig. 3, P = 0.0006, P = 0.037, Student’s t test). This increase was associated with a diversification of T cell receptor use of TIL cultures derived from these nodules (16). Interestingly, in cases in which tumor growth was not affected, this pattern of infiltration was not maintained and decreased 6.4-fold as shown, for example, in patient 1 (Fig. 2A Center vs. Right, P = 0.003, Student’s t test). In contrast, in patient 2, in whom tumor regression occurred, the T cell infiltration was essentially unchanged (Fig. 2B Center and Right, 1.2-fold increase, P = 0.70, Student’s t test), and increasing inflammation and fibrosis were observed, together with an increased degree of tumor necrosis (Fig. 2C). The finding of increased infiltration of CD3+ cells in treated tumors, together with increased antitumor CTL (below), suggested that expression of a foreign MHC gene by direct gene transfer altered the reactivity of the immune system to tumors in patients.
Figure 2.
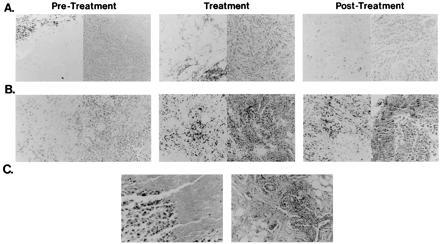
Immunostaining of αCD3 and hemotoxylin and eosin staining of tumor biopsies. Tumor biopsies were obtained before gene transfer (Left), during the gene transfer protocol (Center), or after completion of treatment (Right) in patient 1 (A) and patient 2 (B). Staining with αCD3 (left side of each pair) and hematoxylin and eosin (right side of each pair) is shown. Patient 1 showed a reduced rate of growth without tumor regression, whereas patient 2 experienced a partial remission. The biopsy obtained during treatment occurred 29 days after gene transfer in patient 1 (A Center) and 33 days after treatment in patient 2 (B Center). The posttreatment biopsy was obtained 56 days after gene transfer in patient 1 (A Right) and 114 days after treatment in patient 2 (B Right). Evidence for necrosis, fibrosis, and inflammation was observed after gene transfer in patient 2 (C) 114 days (Left) or 148 days (Right) after treatment. For patient 1, the CD3 count per high power field at the tumor margin was 135 ± 16.7 (Left), and in the tumor was 1.7 ± 1.5 (Left), 53.3 ± 15 (Center), 8.3 ± 5.1 (Right), and for patient 2, CD3+ counts per high powered field in the tumor were 19.5 ± 10 (Left), 37.5 ± 12.7 (Center), and 44.5 ± 15.8 (Right).
Figure 3.
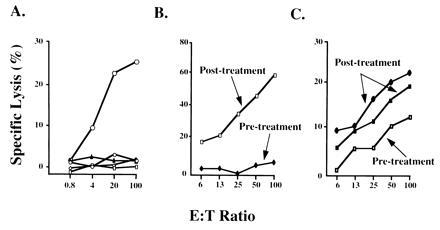
Cytotoxic T cell response of tumor infiltrating lymphocytes to autologous tumor before and after gene transfer. (A) Specificity of T cell lysis of autologous melanoma in patient 2. Lysis of autologous melanoma (□), heterologous melanoma (○), K562 (▵), and YAC-1 (⋄) target cells were analyzed at the indicated effector-to-target ratios. Dose response analysis of cytolytic T cell activity from patients 2 (B) and 9 (C) was performed using autologous melanoma target cells at the indicated effector-to-target (E:T) ratios before or after gene transfer of HLA-B7. (C) Upper line (•) represents lymphocytes derived from an uninjected nodule, whereas the middle line (▪) shows the responsiveness of cells from an injected nodule also derived after treatment, suggestive of a more generalized immune response. In addition, the CTL frequency of TIL specific for autologous tumor was determined by LDA. For patient 9, CTL specific for autologous tumor increased from 1/26,142 before treatment to 1/7,093 after HLA-B7 gene transfer. For patient 2, the CTL frequency of TIL increased from an undetectable level to 1/26,412 after treatment.
With regard to tumor growth, one patient receiving treatment to a subcutaneous nodule (patient 2, group I), showed partial regression of a treated nodule (≈50%). In this patient, at least one metastatic lesion at a distant site, a 3 × 3 cm inguinal lymph node mass, displayed a similar partial regression over the same time period. At the same time, three other distant sites of disease showed no regression in response to DNA–liposome treatment. When one of these latter tumor nodules was subsequently treated, reduction in tumor size was observed, although of lesser degree than the first lesion (10–20%). Together, these data suggest that introduction of the HLA-B7 gene may lead in some cases to antitumor effects at the treatment site and at some distant sites of disease.
The nature of immune cells that infiltrated the tumor in patient 2 after direct gene transfer was investigated further by immunostaining for markers of lymphocyte activation. Before direct gene transfer, there were numerous HLA-DR-positive dendritic cells in and around melanoma tumor cells; only rare, scattered dendritic cells (<10%) expressed either CD80 or CD86 (data not shown). After gene transfer, the injected melanoma sites contained dendritic cells in and around the tumor that were HLA-DR-positive, and numerous peritumoral dendritic cells that were CD80- and CD86-positive were seen (data not shown).
HLA-B7 Gene Transfer Does Not Alter the Frequency of Circulating Tumor-Specific CTL in Peripheral Blood.
LDA techniques were used to quantify the frequency of autologous tumor-specific CTL in the peripheral blood of five patients (Table 2). Assays were performed before treatment and at 2 weeks after the complete course of HLA-B7 gene transfer. In patients 3 and 7, autologous tumor-specific CTL were rare or not detectable in the peripheral blood by LDA. In patient 6, autologous tumor-specific CTL were detectable at a low frequency in the blood before treatment (1/26,860), and the frequency of these circulating CTL remained unchanged after HLA-B7 therapy (1/26, 909). Patient 9 did show an increase in the frequency of tumor-specific CTL after HLA-B7 therapy, with a pretreatment frequency of 1/59,553 increasing to 1/14,047; however, the cytolytic activity of these cells was weak. In contrast, patient 2 had high frequencies of circulating tumor-specific CTL before (1/6,597) and after (1/8,654) HLA-B7 therapy. In this patient, who received several courses of therapy, the relatively high frequency of tumor-specific CTL was maintained over an 8-month period of observation. Hence, in most patients, HLA-B7 gene transfer did not markedly alter the frequency of autologous tumor-specific CTL in the peripheral blood circulation.
Table 2.
Immunologic effect of HLA-B7 gene transfer on immune function: Autologous tumor-specific CTL frequency in the peripheral blood
| Patient | CTL frequency
|
|
|---|---|---|
| Pretreatment | Posttreatment | |
| 2 (A.P.) | 1/6,597 | 1/8,654 |
| 3 (A.H.) | ND | ND |
| 6 (D.M.) | 1/26,860 | 1/26,909 |
| 7 (F.B.) | (Not done) | ND |
| 9 (G.B.) | 1/59,553 | 1/14,047 |
LDA of CTL specific for autologous tumor was performed on peripheral blood mononuclear cells obtained before treatment and at 2 weeks after the third injection of pHLA-B7. LDA microcultures were stimulated with 104 irradiated autologous tumor cells per well and supplemented with 40 units of IL-2 per ml. After a 7-day incubation, CTL activity was detected by adding 51Cr-labeled autologous tumor cells and assessing 51Cr release. CTL frequencies were determined as described in the text. ND, Not detectable.
Effect of HLA-B7 Gene Transfer on Lysis of Autologous Tumor by TIL.
Two approaches were employed to evaluate the effect of HLA-B7 therapy on the lytic capacity of TIL toward autologous tumor for two patients. First, direct lysis of 51Cr-labeled tumor cells was performed using TIL obtained before and after treatment, with varying effector-to-target ratios. As noted in patient 1, TIL populations generally showed a high degree of specific lysis of an autologous, but not heterologous, melanoma or natural killer target cells in vitro (Fig. 3A). When lymphocyte reactivity was compared before and after gene transfer, TIL obtained from patient 9 before treatment were not cytolytic for autologous tumor cells; however, after treatment, TIL obtained from this patient mediated potent lytic activity (Fig. 3B). A comparable analysis of patient 2 showed similar results; TIL obtained before gene transfer were weakly cytolytic for autologous tumor, and the lytic activity of posttreatment TIL also increased, although not to the same extent as patient 9 (Fig. 3 B vs. C).
In addition, the frequencies of CTL precursors within these TIL populations were determined by LDA. For patient 9, gene transfer increased CTL precursor frequencies 3- to 4-fold after treatment (Fig. 3). Likewise, gene transfer increased the CTL precursor frequency in TIL obtained from patient 2 from an undetectable level to readily measurable levels (Fig. 3). Hence, the frequency of CTL precursor cells paralleled the direct lytic activity of TIL in two of two evaluable patients, and these activities increased to varying degrees among these patients. It is of interest that the marked increase in lytic activity of TIL seen in patient 9 was also accompanied by an increase in the frequency of CTL precursors circulating in the peripheral blood. This cytolytic activity was weak, however, and the relationship between circulating CTL precursors and the lytic activity of TIL was not observed in patient 2, who maintained high numbers of peripheral blood CTL precursors yet a relatively low lytic capacity of TIL.
Adoptive Transfer of TIL in a Responder Patient.
Because patient 2 showed evidence of local and systemic responses to gene transfer of HLA-B7, and TIL cultures showed specific cytolytic activity against autologous tumors, we explored the potential for combined immunotherapy and gene transfer in this patient. Three subcutaneous nodules that received gene transfer with HLA-B7/β-2 microglobulin DNA–liposome complex (30 μg of DNA per nodule) were subsequently removed for TIL isolation and culture. These TIL cultures mediated tumor-specific lysis of autologous tumor in standard cytotoxicity assays. In addition, we documented significantly enhanced release of granulocyte/macrophage colony-stimulating factor, interferon γ, and tumor necrosis factor α in response to autologous tumor stimulation by TIL derived from tumors inoculated with DNA–liposome complexes compared with baseline TIL before gene transfer (Table 3). These TIL cultures derived from the injected nodules were expanded over a 3-week interval, and 1011 cells followed by 10 infusions of IL-2 (180,000 units/kg at 8-hr intervals). This patient showed subsequent partial regression of her residual disease, monitored by CT scan of an enlarged inguinal lymph node 2 weeks after therapy. She was subsequently retreated with the same TIL, which had been cryopreserved and received 1.1 × 1011 cultured cells plus IL-2 2 months after her initial cell infusion. After this second treatment, she manifested a complete regression of her residual disease (Fig. 4). This complete remission has persisted, now more than 21 months after the initial treatment. It is interesting to note that this patient had a shorter duration of response to prior treatments with chemotherapy and immunotherapy (Table 1).
Table 3.
Effect of gene transfer on cytokine production by TIL in response to autologous or allogeneic tumor
| Patient | Therapy | GM-CSF,
pg/ml
|
IFN-γ, units/ml
|
TNF-α,
pg/ml
|
|||
|---|---|---|---|---|---|---|---|
| Auto | Allo | Auto | Allo | Auto | Allo | ||
| 1 (G.M.) | Pre | 2,614 | 82 | 2232 | 254 | 2,097 | 24 |
| Post | 3,181 | 711 | 1208 | 1674 | 2,305 | 374 | |
| 2 (A.P.) | Pre | 879 | 0 | 974 | 27 | 0 | 0 |
| Post | 26,632 | 0 | 2258 | 0 | 27,966 | 0 | |
TIL isolated before (pre) or after (post) gene therapy were assessed for cytokine production upon stimulation with irradiated autologous (auto) or allogeneic (allo) tumor cells. After 24 hr, supernatants were harvested and assessed for cytokine concentration by ELISA.
GM-CSF, granulocyte/macrophage colony-stimulating factor; IFN-γ, interferon γ; TNF-α, tumor necrosis factor α.
Figure 4.
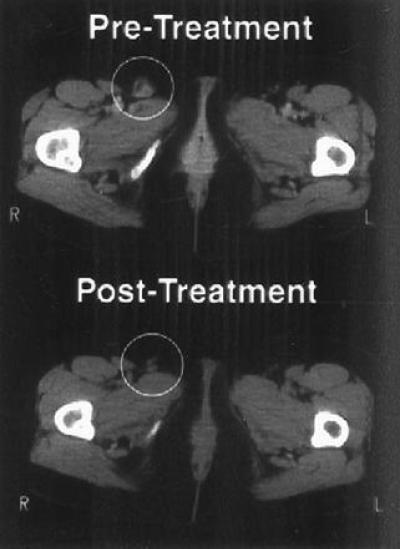
CT scan of right inguinal lymph node before and after gene transfer and TIL adoptive transfer. Serial sections of the pelvic region were obtained, and lymph node size was evaluated before adoptive transfer (Upper) and 9 months after transfer (Lower). A responsive right inguinal lymph node is circled.
DISCUSSION
In this report, expression of an allogeneic MHC gene, HLA-B7, was achieved in patients with melanoma by direct gene transfer with DNA–liposome complexes. Gene expression was localized to the site of injection, and no apparent toxicity or anti-DNA antibodies were associated with this treatment. The immune response to the malignancy induced by this gene was characterized, and tumor growth was assessed. In addition, the potential for combination gene transfer and cellular immunotherapy was evaluated. We found that gene expression can be consistently achieved without toxicity using this method of gene transfer. In addition, changes in the immune response can be induced within tumor nodules subsequent to the gene transfer procedure (Figs. 2 and 3). There were no major changes in circulating peripheral blood lymphocyte reactivity, as might be expected from other model systems (17); however, infiltration of T lymphocytes was observed in six of seven patients. These TIL manifested improved cytotoxicity (Fig. 3), and enhanced tumor-specific release of cytokines (Table 3) and an altered utilization of T cell receptor types (16). Several studies have reported that this tumor-specific release of cytokines by immune lymphoid cells correlates with therapeutic efficacy in adoptive transfer systems (18–20) and stimulated us to evaluate a combined approach to therapy.
Knowledge of the molecular genetics of human cancer has grown exponentially in recent years. Substantial advances have occurred in the complementary disciplines of molecular biology, virology, gene transfer technology, and gene mapping. Together, this information has led to an improved understanding of the genetic basis for human cancer and has stimulated interest in using genetic information to develop new therapies. Although the original expectation was that gene therapy would be limited in scope, primarily for the treatment of inherited genetic diseases, it has become increasingly clear that a variety of acquired diseases, including common diseases such as cancer, cardiovascular disease, and infectious diseases show strong underlying genetic determinants and can be treated by using this approach. Despite the increased understanding of its molecular basis, many malignancies remain unresponsive to standard treatments. The definition of tumor-associated genetic mutations, however, has heightened interest in cancer as a target for gene therapy. Certain neoplasms, such as melanoma or renal cell carcinoma, are relatively more immunogenic, presumably through recognition of mutant gene products that arise in these cells. Conventional approaches to immunotherapy have used cytokines, adjuvants, or adoptive immune cell therapy in animal models (21–24) and in humans (25–27). More recently, the potential of molecular genetic interventions to improve the efficacy of immunotherapy has been explored (15, 28, 29).
In this study, a nonviral gene delivery vector was used. There are several advantages to the use of nonviral vectors for human gene therapy. Although modified viruses have served as useful vectors for ex vivo gene transfer, their ability to interact with endogenous viruses or to recombine has raised concerns regarding safety issues for in vivo gene transfer. Nonviral vectors provide a potentially safer alternative for this approach and include such agents as naked DNA, DNA–liposome, or gold particle–DNA complexes that can mediate gene transfer into tissues and facilitate uptake by cells in vivo (14, 15, 30). The inclusion of appropriate regulatory sequences within plasmid DNA can be used to regulate expression of a variety of gene products. DNA, liposomes, and other components may also be stored stably for long periods of time, and the presence of potential replication-competent viruses in producer cell lines is eliminated. One mode of nonviral gene transfer is the injection of plasmid DNA (31). In this method, the expression of recombinant genes after intramuscular injection is sufficient to induce the expression of proteins that can be immunogenic and provide for protective immunity against the expressed recombinant gene product (32, 33). Plasmid DNA complexed to liposomes has been employed to transfer genes by injection or catheter into tissues where they stimulate localized biologic responses (5, 30, 34). The expression of recombinant gene products can be achieved locally and may help to avoid systemic effects. For example, systemic administration of cytokines in many cases is not well tolerated, but DNA or DNA–liposome complexes can mediate gene transfer into tissues and facilitate gene expression locally at higher concentrations than would be tolerated systemically.
Expression of HLA-B7 in tumor cells in this study is intended to stimulate recognition of the foreign transplantation antigen by the immune system and the release of cytokines locally which induce a T cell response against the unmodified tumor. We have previously shown that gene transfer of foreign MHC initially stimulates a cytolytic T cell response to the antigen (2, 15). This earlier study made use of a DNA vector and cationic lipid that differed from those described in this report. Most notably, the present vector includes an open reading frame for β-2 microglobulin, needed for surface expression of class I MHC and often missing in human melanoma. Approximately 10–20% of melanomas fail to synthesis β-2 microglobulin in vitro (35–36), and this gene product is required for cell surface expression of class I MHC protein (37). In addition, the DMRIE/DOPE DNA–liposome complex showed less toxicity in preclinical studies (4) and thus allowed much higher doses (>100-fold) to be administered in the present clinical study (2, 15). The previously published study showed successful gene expression, lack of toxicity, and tumor regression in one patient on two independent treatments, with both local and distant tumor regression (15). In this study, despite the 100-fold increase in the maximum dose of the DNA–liposome complex, no toxicity was observed.
Although additional patients will be required to confirm the consistency of the anti-tumor immune response, it is encouraging that some patients in separate clinical studies have shown a response to treatment. In addition to the patients described here and previously (15), 7 of 14 patients treated at an independent site have shown local regression in response to this gene transfer procedure (E. Hersh, personal communication). Further clinical studies will be needed to establish the efficacy of this gene delivery approach for the treatment of melanoma and other cancers; nonetheless, the present study provides insight into mechanisms of generating tumor immunity in humans. These results also suggest that gene transfer approaches can be used in combination with other immune treatments, such as cytokines or adoptive T cell therapy. In a preclinical model, we have demonstrated that in vivo transfer of a foreign MHC gene into a poorly immunogenic murine melanoma resulted in the induction of tumor reactive T cells retrievable in the draining lymph nodes and were capable of mediating tumor regression in adoptive immunotherapy (38). The ability to alter local or regional immune responses by gene transfer and to expand immune effector cells ex vivo may provide an alternative method to eliminate microscopically residual malignancies and complement current immunologic treatments for melanoma. In addition, it is likely that genes encoding antiproliferative genes, or inhibitors of angiogenesis could also complement immunologic approaches. Taken together, these data suggest that direct gene transfer warrants further clinical evaluation to develop its potential to contribute to the understanding and treatment of human cancer.
Acknowledgments
We thank Donna Gschwend for secretarial assistance; Nancy Barrett for preparation of the figures; Judy Stein, Christina Gebstadt, and Lisa Kujawski for help in monitoring patients; and Vical, Inc. for providing plasmid DNA and liposomes for this study. G.J.N. and E.G.N. are members of the Scientific Advisory Board of Vical, Inc. We would also like to thank the patients who participated in this study and Dr. Paul Watkins and the nursing staff at the General Clinical Research Center at The University of Michigan Medical Center. This work was supported in part by grants from the National Institutes of Health (U01-AI33355-01 to G.J.N.; P01 CA59327-01 to G.J.N. and A.E.C.; and RO1 DK42706 to E.G.N.). The clinical study was also supported by a grant from the General Clinical Research Center (M01-RR00042).
Footnotes
Abbreviations: MHC, major histocompatibility complex; DMRIE, dimyristyloxypropyl-3-dimethyl-hydroxyethyl ammonium; DOPE, dioleoyl phosphatidylethanolamine; LDA, limiting dilution analysis; CTL, cytotoxic T lymphocyte; TIL, tumor-infiltrating lymphocytes; IL-2, interleukin 2.
References
- 1.Nabel G J, Nabel E G, Yang Z, Fox B A, Plautz G E, Gao X, Huang L, Shu S, Gordon D, Chang A E. Cold Spring Harbor Symp Quant Biol. 1994;49:699–707. doi: 10.1101/sqb.1994.059.01.081. [DOI] [PubMed] [Google Scholar]
- 2.Plautz G E, Yang Z, Wu B, Gao X, Huang L, Nabel G J. Proc Natl Acad Sci USA. 1993;90:4645–4649. doi: 10.1073/pnas.90.10.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felgner J H, Kumar R, Sridhar C N, Wheeler C J, Tsai Y J, Border R, Ramsey P, Martin M, Felgner P L. J Biol Chem. 1994;269:2550–2561. [PubMed] [Google Scholar]
- 4.San H, Yang Z, Pompili V J, Jaffe M L, Plautz G E, Xu L, Felgner J H, Wheeler C J, Felgner P, Gao X, Huang L, Gordon D, Nabel G J, Nabel E G. Hum Gene Ther. 1993;4:781–788. doi: 10.1089/hum.1993.4.6-781. [DOI] [PubMed] [Google Scholar]
- 5.Nabel G J, Chang A E, Nabel E G, Plautz G E, Ensminger W, Fox B A, Felgner P, Shu S, Cho K. Hum Gene Ther. 1994;5:57–77. doi: 10.1089/hum.1994.5.1-57. [DOI] [PubMed] [Google Scholar]
- 6.Lew D, Parker S E, Latimer T, Abai A M, Kuwahara-Rundell A, Doh S G, Yang Z, LaFace D, Gromkowski S H, Nabel G J, Manthorpe M, Norman J. Hum Gene Ther. 1995;6:553–564. doi: 10.1089/hum.1995.6.5-553. [DOI] [PubMed] [Google Scholar]
- 7.Nabel E G, Plautz G, Nabel G J. Proc Natl Acad Sci USA. 1992;89:5157–5161. doi: 10.1073/pnas.89.11.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabel E G, Yang Z, Liptay S, San H, Gordon D, Haudenschild C C, Nabel G J. J Clin Invest. 1993;91:1822–1829. doi: 10.1172/JCI116394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sambrook J, Fritch E F, Maniatis T. Cold Spring Laboratory Harbor Press. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 10.Orosz C G, Horstemeyer B, Zinn N E, Bishop D K. Transplantation. 1989;47:189–194. doi: 10.1097/00007890-198901000-00039. [DOI] [PubMed] [Google Scholar]
- 11.MacDonald H R, Cerottini J C, Ryser J E, Maryanski J L, Taswell C, Widmer M B, Brunner K T. Immunol Rev. 1980;51:93–123. doi: 10.1111/j.1600-065x.1980.tb00318.x. [DOI] [PubMed] [Google Scholar]
- 12.Taswell C. J Immunol. 1981;126:1614–1619. [PubMed] [Google Scholar]
- 13.Chang A E, Yoshizawa H, Sakai K, Cameron M J, Sondak V K, Shu S. Cancer Res. 1993;53:1043–1050. [PubMed] [Google Scholar]
- 14.Stewart M J, Plautz G E, Del Buono L, Yang Z Y, Xu L, Gao X, Huang L, Nabel E G, Nabel G J. Hum Gene Ther. 1992;3:267–275. doi: 10.1089/hum.1992.3.3-267. [DOI] [PubMed] [Google Scholar]
- 15.Nabel G J, Nabel E G, Yang Z, Fox B A, Plautz G E, Gao X, Huang L, Shu S, Gordon D, Chang A E. Proc Natl Acad Sci USA. 1993;90:11307–11311. doi: 10.1073/pnas.90.23.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeBruyne, L. A., Chang, A. E., Cameron, M. J., Yang, Z., Gordon, D., Nabel, E. G., Nabel, G. J. & Bishop, D. K. (1996) Cancer Immunol. Immunother., in press. [DOI] [PubMed]
- 17.Bishop D K, Ferguson R M, Orosz C G. J Immunol. 1990;144:1153–1160. [PubMed] [Google Scholar]
- 18.Goedegebuure P S, Zuber M, Leonard-Vidal L, Burger U L, Cusack J C, Jr, Chang M P, Douville L M, Eberlein T J. Surg Oncol. 1994;3:79–89. doi: 10.1016/0960-7404(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 19.Schwartzentruber D J, Hom S S, Dadmarz R, White D E, Yannelli J R, Steinberg S M, Rosenberg S A, Topalian S L. J Clin Oncol. 1994;12:1475–1483. doi: 10.1200/JCO.1994.12.7.1475. [DOI] [PubMed] [Google Scholar]
- 20.Aruga A, Shu S, Chang A E. Cancer Immunol Immunother. 1995;41:317–324. doi: 10.1007/BF01517220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zbar B, Bernstein I D, Rapp H J. J Natl Cancer Inst. 1971;46:831–839. [PubMed] [Google Scholar]
- 22.Rosenberg S A, Mule J J, Spiess P J. J Exp Med. 1985;161:1169–1188. doi: 10.1084/jem.161.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shu S, Rosenberg S A. Cancer Res. 1985;45:1657–1662. [PubMed] [Google Scholar]
- 24.Yoshizawa H, Chang A E, Shu S. J Immunol. 1991;147:729–737. [PubMed] [Google Scholar]
- 25.Morton D L, Eilber F R, Holmes E C. Ann Surg. 1974;180:635–643. doi: 10.1097/00000658-197410000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg S A, Lotze M T, Yang J C, Aebersold P M, Linehan W M, Seipp C A, White D E. Ann Surg. 1989;210:474–485. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg S A, Packard B S, Aebersold P M, Solomon D, Topalian S L, Toy S T, Simon P, Lotze M T, Yang J C, Seipp C A. N Engl J Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 28.Nabel G J, Chang A, Nabel E G, Plautz G, Fox B A, Huang L, Shu S. Hum Gene Ther. 1992;3:399–410. [Google Scholar]
- 29.Rosenberg S A, Aebersold P, Cornetta K, Kasid A, Morgan R A, Moen R, Karson E M, Lotze M T, Yang J C, Topalian S L, Merino M J, Culver K, Miller A D, Blaese R M, Anderson W F. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 30.Nabel E G, Gordon D, Yang Z, Xu L, San H, Plautz G E, Wu B, Gao X, Huang L, Nabel G J. Hum Gene Ther. 1992;3:649–656. doi: 10.1089/hum.1992.3.6-649. [DOI] [PubMed] [Google Scholar]
- 31.Wolff J A, Malone R W, Williams P, Chong W, Acsadi G, Jani G, Felgner P L. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 32.Liu M, Chen T Y, Ahamed B, Li J, Yau K W. Science. 1994;266:1348–1354. doi: 10.1126/science.266.5189.1348. [DOI] [PubMed] [Google Scholar]
- 33.Sedegah M, Hedstrom R, Hobart P, Hoffman S L. Proc Natl Acad Sci USA. 1994;91:9866–9870. doi: 10.1073/pnas.91.21.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nabel E G, Yang Z, Muller D, Chang A E, Gao X, Huang L, Cho K J, Nabel G J. Hum Gene Ther. 1994;5:1089–1094. doi: 10.1089/hum.1994.5.9-1089. [DOI] [PubMed] [Google Scholar]
- 35.Restifo N P, Kawakami Y, Marincola F, Shamamian P, Taggarse A, Esquivel F, Rosenberg S A. J Immunother. 1993;14:182–190. doi: 10.1097/00002371-199310000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrone S, Marincola F M. Immunol Today. 1995;16:487–494. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 37.Klein J, Juretic A, Baxevanis C N, Nagy Z A. Nature (London) 1981;291:455–460. doi: 10.1038/291455a0. [DOI] [PubMed] [Google Scholar]
- 38.Wahl W L, Strome S E, Nabel G J, Plautz G E, Cameron M J, San H, Fox B A, Shu S, Chang A E. J Immunother. 1995;17:1–11. doi: 10.1097/00002371-199501000-00001. [DOI] [PubMed] [Google Scholar]