

Abstract
The nucleocapsid protein (N) of the severe acute respiratory syndrome coronavirus (SARS-CoV) packages the viral genomic RNA and is crucial for viability. However, the RNA-binding mechanism is poorly understood. We have shown previously that the N protein contains two structural domains—the N-terminal domain (NTD; residues 45 to 181) and the C-terminal dimerization domain (CTD; residues 248 to 365)—flanked by long stretches of disordered regions accounting for almost half of the entire sequence. Small-angle X-ray scattering data show that the protein is in an extended conformation and that the two structural domains of the SARS-CoV N protein are far apart. Both the NTD and the CTD have been shown to bind RNA. Here we show that all disordered regions are also capable of binding to RNA. Constructs containing multiple RNA-binding regions showed Hill coefficients greater than 1, suggesting that the N protein binds to RNA cooperatively. The effect can be explained by the “coupled-allostery” model, devised to explain the allosteric effect in a multidomain regulatory system. Although the N proteins of different coronaviruses share very low sequence homology, the physicochemical features described above may be conserved across different groups of Coronaviridae. The current results underscore the important roles of multisite nucleic acid binding and intrinsic disorder in N protein function and RNP packaging.
Severe acute respiratory syndrome (SARS) is the first pandemic of the 21st century that spread to multiple nations, with a fatality rate of ca. 8%. The disease is caused by a novel SARS-associated coronavirus (SARS-CoV) closely related to the group II coronaviruses, which include the human coronavirus OC43 and murine hepatitis virus (6, 18). Traditional antiviral treatments have had little success against SARS during the outbreak, and vaccines have yet to be developed (35).
Coronaviruses are positive-sense single-stranded RNA (ssRNA) viruses. The coronavirus genomic RNA is encapsidated into a helical capsid by the nucleocapsid (N) protein, which is one of the most abundant coronavirus proteins (19). The N protein has nonspecific binding activity toward nucleic acids, including ssRNA, single-stranded DNA, and double-stranded DNA (33). It can also act as an RNA chaperone (39). However, the mechanism of binding of the N protein to nucleic acids is poorly understood.
The SARS-CoV N protein is a homodimer composed of 422 amino acids (aa) in each chain. The N protein can be divided into two structural domains interspersed with disordered (unstructured) regions (Fig. 1A) (2). The N-terminal domain (NTD; also called RBD) serves as a putative RNA-binding domain, while the C-terminal domain (CTD; also called DD) is a dimerization domain (13, 36). Both the NTD and the CTD bind to nucleic acids through electropositive regions on their surfaces (3, 13, 32). All coronaviruses share similar domain architectures at both the sequence and structure levels. No structure of N protein or any of its domains in complex with nucleic acids is available.
FIG. 1.

(A) Schematic of the domain architecture of the SARS-CoV N protein. Structured domains are shown as balls, and unstructured regions are shown as lines. (B) Protein constructs used in the current study. Numbers represent the amino acid residue range relative to the full-length N protein (NP). Sumo-1-FL contains a Sumo-1 tag (shown as an oval), followed by the flexible linker of the N protein between residues 181 and 246.
The functions of the disordered regions in the SARS-CoV N protein have not been clearly defined, although some evidence suggests that they are involved in protein-protein interactions between the N protein and other viral and host proteins (11, 20, 22, 38). A previous report has shown that part of the C-terminal disordered region with a polylysine sequence also binds to RNA (21). Unlike the structural domains, the disordered regions of the different coronaviruses share little sequence homology. However, they share a common physicochemical property: they are highly enriched in basic residues. Intrinsic disorder coupled with an abundance of positive charges leads to the possibility of nonspecific binding to nucleic acids (34). These findings prompted us to investigate the role of intrinsically disordered (ID) regions in the RNA-binding mechanism of the SARS-CoV N protein.
Here we tested all three disordered regions of the SARS-CoV N protein and found that they are all involved in RNA binding. The central region, in particular, had a large impact on binding behavior as monitored by electrophoretic mobility shift assays (EMSA). Small-angle X-ray scattering (SAXS) and nuclear magnetic resonance (NMR) results show that this central region is a flexible linker (FL) that connects the two structural domains in an extended conformation. Our results provide new insights into the functional coupling of intrinsic disorder, RNA binding, and oligomerization.
MATERIALS AND METHODS
Protein expression and purification.
Different regions of the SARS-CoV N protein (Fig. 1B) were amplified by standard PCR techniques, subcloned into the pET6H vector, and expressed in Escherichia coli BL21(DE3) cells as previously described (1, 2), with the exception of the Sumo-1-FL construct, which contains a Sumo-1 tag followed by the flexible linker of the N protein between residues 181 and 246. The Sumo-1-FL vector was constructed and expressed with the Champion pET Sumo protein expression system (Invitrogen, CA) by following the manufacturer's protocols. Purification of the N protein fragments followed the procedure previously described (1, 2), except that all buffers contained 0.5 M NaCl. Sumo-1-FL was purified according to the manufacturer's protocol, followed by size exclusion chromatography through a Superdex 75 column (GE Healthcare, CA). 15N-labeled proteins for NMR studies were obtained by replacing Luria broth with M9 medium. The sizes of all protein products were checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Fig. 2) and confirmed by mass spectrometry (Voyager-DE STR; PerSeptive Biosystems, MA).
FIG. 2.
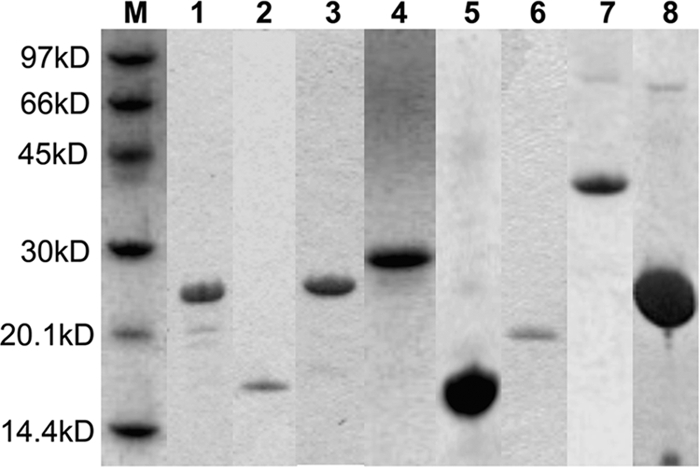
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel strips of the various SARS-CoV NP protein constructs after purification. Almost all constructs appear as a single band in the gel strips, and for the few exceptions, the purity of the main band exceeds 90%. Lanes are labeled in the following order: M, light molecular mass marker; 1, NP1-181; 2, NP45-181; 3, NP45-247; 4, NP181-365; 5, NP248-365; 6, NP248-422; 7, NP45-365; 8, Sumo-1-FL.
EMSA.
Experiments were conducted with 6-aminohexylfluorescein-labeled 20-mer poly(U) ssRNA purchased from Purigo (Taiwan) and freshly prepared protein. Reactions were set up by following previously published protocols (32) but substituting ssRNA for single-stranded DNA. Trial runs were set up to determine the initial protein concentration for each construct. For the Sumo-1-FL construct, the reaction buffer was changed to 50 mM sodium phosphate (pH 7.4)-150 mM NaCl-1 mM EDTA to preserve solubility. In this case, control experiments with pure Sumo-1 (a gift from Mandar T. Naik), NP45-181, and NP248-365 were carried out in parallel. All EMSA studies were executed in triplicate. The total amount of bound ssRNA was calculated by taking the difference in intensity between the control lane band and the corresponding band in each reaction lane. Binding parameters were obtained by fitting the binding isotherms to the equation Y = 1/[1 + (Kd/X)n], using GraphPad Prism (GraphPad Software, CA), where Y is the fraction of ssRNA bound to the protein, X is the protein concentration, Kd is the dissociation constant, and n is the Hill coefficient (32).
NMR spectroscopy.
Samples contained 0.5 to 1 mM protein in NMR buffer (10 mM sodium phosphate [pH 6.0], 50 mM NaCl, 1 mM EDTA, 1 mM 2,2-dimethyl-2-silapentane-5-sulfonate [DSS], 0.01% NaN3, 10% D2O, and Complete Mini protease inhibitor mix [Roche]). Experiments were performed at 30°C unless stated otherwise. Bruker 600-MHz spectrometers equipped with cryoprobes were employed in the experiments. The data acquired were processed with the TopSpin suite (Bruker Biospin, Germany) or iNMR (Nucleomatica, Italy).
Size exclusion chromatography.
Experiments were conducted using an Akta fast-performance liquid chromatography system (GE Healthcare, CA) equipped with a Tricorn 10/300 Superdex 75 column at an elution rate of 0.2 ml/min. Apparent molecular weights of the proteins were estimated from the elution profile calibrated with the LMW gel filtration calibration kit (GE Healthcare, CA). Elution volume and molecular weight have the relationship log(MW) = 6.5404 − 0.1802 EV, where MW is the molecular weight in thousands and EV is the elution volume in milliliters.
SAXS.
The didomain construct NP45-365 was concentrated to 10 mg/ml with an Amicon Ultra concentrator (Millipore, MA). Data were collected on the BL13A beam line of the National Synchrotron Radiation Research Center at 25°C (Hsinchu, Taiwan). The first 10 points of the data were excluded from analysis due to possible aggregation effects. The GNOM program was used to analyze the scattering profile and to obtain the radius of gyration (Rg), the pairwise distribution function [P(r)], and the maximal distance (dmax) (31). The BUNCH program was used to add flexible linkers assuming P1 symmetry (27). Atomic coordinates of the NTD monomer and the CTD dimer served as input to a modified version of CRYSOL (M. Petoukhov, personal communication). A total of 252 modeling runs were obtained, and the interdomain distances were measured by calculating the coordinates of the center of gravity of the two domains using in-house software.
Secondary-structure prediction and sequence alignment.
Representative N protein sequences from all groups of Coronaviridae were obtained from the SwissProt server. The JPred metaserver was used to obtain consensus secondary-structure predictions for the central flexible linkers of the various sequences (5). These sequences were then manually aligned based on the predicted structural and physicochemical properties. The sequence length was arbitrarily fixed to that of the SARS-CoV N protein flexible linker for easier visualization.
RESULTS
The N protein contains multiple ssRNA binding sites.
Figure 3 shows that inclusion of either the first 44 residues (aa 1 to 44) or the central flexible linker (residues 182 to 247) of the SARS-CoV N protein increases the apparent binding affinity for 20-mer poly(U) ssRNA three- to fourfold over that of the NTD (residues 45 to 181) alone. Inclusion of the flexible linker not only increases the apparent affinity; it also has a large effect on the apparent Hill coefficient. Similar results are obtained when either the central flexible linker or the C-terminal 54 residues (residues 366 to 422) are included in the construct of the CTD (residues 248 to 365), as shown in Fig. 4. The increase in apparent binding affinity is even more pronounced (six- to eightfold), probably due to the dimeric nature of the CTD, which has two attached disordered regions, whereas the NTD has one. The apparent binding parameters of various constructs are listed in Table 1. Notice that in fitting the EMSA data, the binding constants of probable intermediate species are ignored and only the overall binding constant is obtained. Our results recapitulate some of the observations in the literature. The NP248-422 construct contains the CTD and a previously identified RNA-binding region (3, 21, 32), and our results show that the construct has higher apparent affinity and a greater apparent Hill coefficient than the CTD alone. The didomain construct NP45-365, which contains both structural domains and the flexible linker, has the highest apparent binding affinity, in agreement with our earlier observations. Taken together, a common trend is quickly apparent: inclusion of the disordered regions enhances the binding affinity of any particular construct. Of particular interest is the central flexible linker, which not only increases the binding affinity but also greatly enhances the Hill coefficients of the constructs, suggesting the presence of cooperativity. This is interesting, and the source of the cooperativity will be discussed later.
FIG. 3.

Effects of the ID regions (residues 1 to 44 and 182 to 247) on the RNA binding affinity of the NTD. (A through C) Fitting of the binding isotherms of NP45-181 (NTD) (A), NP1-181 (B), and NP45-247 (C), based on the EMSA results. Each binding isotherm represents the overall fitting against three independent experiments, taking into account the standard deviation of each data point. (D through F) Representative EMSA results for NP45-181 (D), NP1-181 (E), and NP45-247 (F).
FIG. 4.

Effects of the ID regions (residues 182 to 247 and 366 to 422) on the RNA binding activity of the CTD. (A through C) Fitting of the binding isotherms of NP248-365 (CTD) (A), NP248-422 (B), and NP182-365 (C), based on the EMSA results. Each binding isotherm represents the overall fitting against three independent experiments, taking into account the standard deviation of each data point. (D through F) Representative EMSA results for NP248-365 (D), NP248-422 (E), and NP182-365 (F).
TABLE 1.
Binding coefficients for U20 ssRNA to various regions of the SARS-CoV N proteina
| Buffera and region (aa) | Apparent Kd (μM)b | Hill coefficientb |
|---|---|---|
| Buffer A | ||
| 1-181 | 2.98 ± 0.19 | 1.6 ± 0.15 |
| 45-181 | 9.81 ± 0.82 | 1.1 ± 0.09 |
| 45-247 | 2.73 ± 0.05 | 3.2 ± 0.16 |
| 182-365 | 1.35 ± 0.06 | 10.6 ± 1.0 |
| 248-365 | 10.2 ± 0.89 | 0.9 ± 0.06 |
| 248-422 | 1.62 ± 0.05 | 2.9 ± 0.22 |
| 45-365 | 0.74 ± 0.04 | 2.3 ± 0.26 |
| Buffer B | ||
| 45-181 | 9.40 ± 1.1 | 0.53 ± 0.04 |
| 248-365 | 9.30 ± 0.89 | 0.65 ± 0.04 |
| Sumo-1-FL | 15.6 ± 1.1 | 1.5 ± 0.14 |
Buffer A consists of 10 mM NaPi, 50 mM NaCl, and 1 mM EDTA (pH 6.0). Buffer B consists of 50 mM NaPi, 150 mM NaCl, and 1 mM EDTA (pH 7.4).
Values are averages for three individual experiments ± standard deviations.
The central flexible linker interacts with ssRNA with high affinity.
Since the flexible linker appears to play an important role in the binding mechanism, we decided to focus our attention on this region. Unfortunately, the flexible linker is prone to degradation, and initial attempts at expression in E. coli failed. We utilized a Sumo-1-tagged expression system to increase expression levels and avoid premature cleavage. The purified Sumo-1-tagged construct (Sumo-1-FL) binds to ssRNA, while the pure Sumo-1 protein has no binding affinity toward nucleic acids (data not shown), indicating that residues 181 to 246 are able to bind to ssRNA directly. The apparent binding affinity of this region is comparable to those of the NTD and CTD constructs, as listed in Table 1, highlighting the functional importance of the flexible linker domain in RNA binding.
The flexible linker is ID.
A combination of techniques was used to ascertain the intrinsic disorder of the flexible linker. 15N-edited heteronuclear single-quantum coherence (HSQC) spectra have been widely used as a tool to monitor the order and disorder of proteins (8). Well-dispersed spectra are indicative of a structured protein, while congested spectra with resonances clustered around a small region of 8.3 ± 0.5 ppm in the proton dimension are often disordered. Comparing the HSQC spectrum of NP45-247 with that of the NTD (NP45-181) in Fig. 5A, we observed additional resonances in the spectrum of NP45-247 clustered in the 7.5- to 8.5-ppm range on the proton chemical shift. This strongly suggests that the additional residues from aa 182 to 247 of NP45-247 are disordered. The dispersed resonances are almost exact matches between the two constructs, indicating that residues 182 to 247 do not affect the structure of residues 45 to 181. Furthermore, size exclusion chromatography of NP45-247 shows that the protein elutes out of the column with a Stokes radius corresponding to a globular protein of 41 kDa (Fig. 5B). The theoretical molecular mass of the construct is 22.9 kDa, suggesting that the NP45-247 construct has an elongated shape. This is in contrast to the NTD, which is mainly globular (13). We attribute this to residues 182 to 247 forming an extraneous “tail” that affects the hydrodynamic properties of the molecule. An alternative interpretation of dimer formation is excluded, because no additional well-dispersed resonance was observed. Our data presented in the next paragraph for CTD constructs also preclude dimer formation for residues 182 to 247.
FIG. 5.

Residues 182 to 247 are ID when attached to the NTD. (A) 15N-edited HSQC spectra of NP45-181 (NTD) (left) and NP45-247 (right) show additional resonances clustered in the middle of the spectrum of NP45-247. Axis units are ppm. (B) Size exclusion chromatogram of NP45-247. The corresponding apparent molecular weight was calculated from the equation log(MW) = 6.5404 − 0.1802 EV, where MW is the molecular weight in thousands and EV is the elution volume in milliliters.
We observed the same phenomenon, shown in Fig. 6, when comparing NP182-365 to the CTD (NP248-365). Again, no additional well-dispersed resonance was observed in the CTD construct that included the linker region between residues 182 and 247. Thus, the extraneous “tail” formed by residues 182 to 247 does not affect the structure of the CTD (Fig. 6A), but it does change the Stokes radius of the construct as calculated from the fast-performance liquid chromatography elution profile shown in Fig. 6B. The calculated molecular mass of NP182-365 is 21 kDa, and the expected molecular mass of NP182-365 is 42 kDa, since the construct includes the CTD, which forms a dimer. The molecular mass calculated from the experimental Stokes radius is 69 kDa, indicating that NP182-365 does not form a dimer of dimers, which is what one would expect if residues 182 to 247 really act as dimerization motifs. Taken together, our results are compatible with previous reports from this lab where the didomain construct NP45-365 was shown to have resonances in the disordered region of the spectrum without affecting resonances belonging to either structural domain (2). We conclude that the flexible linker (residues 182 to 247) forms a bona fide ID domain not affected by either structural domain in the context of the whole protein.
FIG. 6.

Residues 182 to 247 are ID when attached to the CTD. (A) 15N-edited HSQC spectra of NP248-365 (CTD) (left) and NP182-365 (right) show additional resonances clustered in the middle of the spectrum of NP182-365. Axis units are ppm. (B) Size exclusion chromatogram of NP182-365. The corresponding apparent molecular weight was calculated from the equation log(MW) = 6.5404 − 0.1802 EV, where MW is the molecular weight in thousands and EV is the elution volume in milliliters.
The flexible linker is partially extended in solution.
The conformation of the didomain construct NP45-365 was further studied by the SAXS technique to provide information on its shape. The results are shown in Fig. 7A. Data analysis showed that the radius of gyration of the NP45-365 dimer is 61 Å, much larger than expected for a 72-kDa globular protein (Fig. 7B). This is consistent with the model that the NTD and CTD do not interact, and the two NTDs in the dimer are likely to float freely in solution. A representative structure of NP45-365 based on CRYSOL simulations is shown in Fig. 7C. It should be mentioned that due to the ID nature of the linker region, this structure represents only a model of the conformational ensemble and does not represent a structure per se. However, the model captures features of the conformational ensemble and allows for the qualitative analysis of gross structural features. The most prominent feature of the model is that the flexible linker does not adopt a fully extended conformation, suggesting the existence of residual structures within the linker. However, the interdomain distances are still long enough to allow all five domains (two NTDs, two flexible linkers, one CTD dimer) of the NP45-365 dimer to interact with nucleic acids. This partially explains the increase in binding affinity whenever the flexible linker is attached to either structural domain; one is simply attaching one additional RNA-binding site.
FIG. 7.

SAXS results for the didomain construct NP45-365. (A) Scattering profile of NP45-365 (crosses) and normalization fitting with GNOM (dashed lines). J, scattering intensity; s, scattering angle vector. (B) Normalized results from GNOM showing the pairwise distance distribution [P(r)] and the maximum distance. The radius of gyration is fitted to 61 Å. “r” represents the pairwise distances. (C) Representative model of NP45-365 structure based on CRYSOL simulations of SAXS data. Only the alpha carbons are shown. Notice the difference in distance between the two NTDs and the CTD core.
The physicochemical characteristics of the flexible linker are conserved across coronaviruses.
Our findings prompted us to examine the sequences of flexible linkers from other coronavirus N proteins. Shown in Fig. 8 is an alignment of representative flexible linker sequences of the N proteins from all three coronavirus groups. Because the flexible linkers of different coronavirus species share very low homology, current alignment tools based on sequence and/or structure do not work well in this case. However, these linker sequences share a number of sequence and physicochemical attributes. A very prominent commonality is the sequential arrangement of motifs. All flexible linker sequences start with an SR-rich region, followed by the predicted helix, and end with a region rich in basic residues (Fig. 8). Overall, the flexible linkers from all coronavirus N proteins have high theoretical isoelectric points, >10.5, which could explain their nonspecific affinity for RNA. This feature does not show up in ordinary sequence analyses, especially in cases where the sequence/structure homology is marginal or nonexistent. Our results show that conservation of physicochemical properties extends beyond simple sequence or structural homology and could have functional significance. Interestingly, all flexible linker sequences have been predicted to contain a helical region. However, the predicted helix was not observed in our study, although we cannot rule out the possibility of the presence of a transient helix.
FIG. 8.

Alignment of the flexible linker regions from different coronavirus N proteins. Residues that are predicted by JPred to form a helix are boxed. The arginines of the SR-rich regions are underlined. The names of the coronaviruses (with SwissProt accession numbers and phylogenetic groups in parentheses) are as follows: SARS-CoV (P59595; group 2b); NL63, human coronavirus NL63 (Q6Q1R8; group 1b); 229E, human coronavirus 229E (P15139; group 1b); TGEV, porcine transmissible gastroenteritis virus strain Purdue (P04134; group 1a); OC43, human coronavirus OC43 (P33469; group 2a); MHV-1, murine hepatitis virus 1 (P18446; group 2a); IBV, avian infectious bronchitis virus strain Beaudette (P69596; group 3).
DISCUSSION
At present the molecular basis of SARS-CoV N protein-RNA interaction is unclear, and there is a paucity of quantitative information on the strength of N protein-RNA interaction. Several factors are hindering progress. First, the solubility of the N protein-RNA complex is very poor, making it very difficult to study the binding with standard solution techniques, such as isothermal titration calorimetry or other solution spectroscopic methods. Second, the presence of multiple RNA-binding sites on the N protein, as revealed by our results reported here, and the lack of RNA sequence specificity complicated the measurement and data analysis for methods such as surface plasmon resonance. The current EMSA method, which measures the amount of free RNA in solution, provides the best alternative for determining the N protein-RNA interaction, subject to the following limitations. First, the current technique does not accurately measure the apparent binding constant when the affinity is high (e.g., submicromolar) due to the amount of ssRNA required to obtain a good signal on the gel. Second, in cases where a number of binding domains are present in a protein (e.g., NP45-365), the reaction will be composed of multiple species. A single N protein may bind to multiple RNA molecules, and a single RNA molecule may bind multiple protein molecules. Since these species cannot be identified with certainty, the data cannot be analyzed correctly. It should also be noted that the substrate RNA used for the EMSA studies [20-mer poly(U)] is nonspecific both in structure and in sequence and may not completely reflect how the N protein binds to viral RNA. However, the current data, when taken semiquantitatively, can still reveal insightful information on the nature of N protein-RNA interaction.
The major conclusions from the present studies are as follows. (i) The SARS-CoV N protein is a modular protein consisting of two structured domains flanked by three long stretches of ID segments. (ii) The ID regions account for almost half of the molecule, and the central ID region exists in an extended conformation. (iii) There are multiple RNA-binding sites in the N protein with comparable binding affinities in the micromolar region. The binding sites are distributed in several regions of the molecule. Apparently this property is shared by all coronaviruses and perhaps by many nucleic acid-binding proteins. A large number of nucleic acid-binding proteins, including those of viral origin, contain long stretches of ID regions (34). Paramyxoviruses and flaviviruses, for example, have N and core proteins that contain considerable amounts of disordered residues, respectively (14, 17). The advantages of these properties can be put in the context discussed below. Their relevance to RNA packaging and their functions are also discussed.
Enhanced RNA-binding affinity.
The presence of intrinsic disorder and multiple binding sites together can confer high RNA-binding affinity. First, the extended conformation of the N protein due to the presence of ID segments increases the collision radius with RNA, much like in the “fly-casting” model proposed by Shoemaker et al. (29). Second, transcription factors and other allosteric cell signaling proteins contain a disproportionate number of domains or segments that are ID under native conditions. Hilser and Thompson have proposed a quantitative mechanistic model to assess the importance of intrinsic disorder for intramolecular site-to-site communication in a multidomain regulatory protein, the so-called “coupled-allostery” effect (12). They showed that site-to-site allosteric coupling is maximized when intrinsic disorder is present in the domains or segments containing one or both of the coupled binding sites. Although regulatory proteins generally have much higher affinity for their respective RNA or DNA targets than that presented here for the N protein, the same principles can be applied to this system. The N protein contains multiple RNA-binding sites and showed a large Hill coefficient, as revealed by our EMSA results. Thus, like that of a multidomain regulatory protein, the RNA binding of the N protein is allosteric, i.e., binding of a segment to RNA facilitates the binding of other segments to RNA. The flexibility of the ID region in the N protein allows the optimal alignment of RNA-binding site-containing segments of the N protein and facilitates their binding to the RNA molecule already bound to other sites of the same N protein molecule, resulting in enhanced binding affinity. It should be realized that the “coupled-allostery” effect is more robust and effective in enhancing binding affinity than the multivalence effect in a rigid molecule, since the binding sites do not have to align perfectly for initial binding. Thus, even though the RNA-binding affinity of the individual sites of the N protein is not particularly strong, the RNA-binding affinity of the full-length protein can be very high due to the combined “fly-casting” (29) and “coupled-allostery” (12) effects conferred by the modular N protein with ID linkers.
ID regions as interaction hubs.
One of the surprises in this study is the involvement of the flexible linker in RNA binding (Table 1), which has never been reported for the SARS-CoV N protein. The SR-rich region of the flexible linker has been implicated in a number of protein-protein interactions, including those with host proteins such as human heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and the phosphoprotein B23 (22, 37). It also plays a role in self-association (10, 23) and could have implications for the formation of the nucleocapsid. The SR-rich region also contains the highest density of positive charges in the flexible linker but is also a site for multiple phosphorylation and thus is a prime target for regulating RNA-binding activity (30). In fact, electrostatic charges have been shown to play an important role in the nonspecific RNA binding of the structured regions, and all the putative disordered regions of coronavirus N proteins are positively charged (3, 13, 32). The multifarious activities revolving around the flexible linker of the SARS-CoV N protein strongly suggest that this region acts as a “flexible-net” interaction hub (24), where intrinsic disorder plays a key role.
The flexible linker might not be the only region that could act as an interaction hub. The C-terminal disordered region, for example, has been found to participate in the oligomerization of the N protein (21). However, a polylysine stretch within the oligomerization region has also been shown to bind to nucleic acids. Moreover, earlier reports have shown that this C-terminal region interacts with the membrane (M) protein of SARS-CoV (11). Although the function of the N-terminal disordered domain has not yet been identified, it has been speculated that this region is involved in protein-protein interactions (25). Taken together, we speculate that the three disordered regions of the SARS-CoV N protein represent three interaction hubs that bind to different partners of the N protein interactome. This is consistent with the observation for the regulatory proteins that ID regions are able to recognize multiple partners.
Coupled nucleic acid binding and self-association.
Similar mechanisms may link RNA binding with N protein self-association in the disordered regions. Both the flexible linker and the C-terminal disordered region have been implicated in oligomerization of the N protein (10, 21), and our current findings showed that they also bind to nucleic acids. The effect of RNA binding on oligomerization could be even more dramatic for the ID regions. The extensively charged nature of the flexible linker and the C-terminal disordered region represents a large barrier to N-N interaction. In fact, repulsive forces between the domains may cause the large Rg observed for the didomain construct NP45-365 in our SAXS studies. While charge repulsion between the domains confers the advantage of avoiding interdomain interactions and results in a larger electrostatic binding surface, it also impedes oligomerization (4, 9) and formation of the nucleocapsid. Binding to nucleic acids may neutralize the charges on the N protein and allows two protein molecules to approach and oligomerize. This simple concept would couple capsid formation, which is essentially a self-association process, with RNA binding and guarantee the formation of nucleocapsids containing genetic material. Multiple phosphorylation of the SR-rich region, on the other hand, could provide an additional level of regulation to the RNA-binding process or the self-association process (26, 30). However, the functions and levels of phosphorylation of SARS-CoV NP are still uncertain, and whether phosphorylation really plays a role in RNA binding and/or capsid formation remains to be determined.
Insights into the linkage between RNA binding and RNP packaging.
The modular structure and the presence of ID segments in the N protein offer considerable advantages for the packaging of the genomic RNP and the expression of genomic information. We envision that a single RNA molecule will bind to multiple N proteins at a given moment. Since the bindings are electrostatic and nonspecific, the RNA-bound N proteins presumably can “slide” along the RNA molecule and interact with other RNA-bound N proteins (16). The flexible linker allows more freedom for the different parts of the N protein molecule to interact with each other, resulting in specific packaging of the helical RNP molecule. We have previously shown that in crystal the CTD packs to form two parallel, basic helical grooves, which may be oligonucleotide attachment sites (3). Thus, the RNA molecule would wrap around the CTD core in forming the helical RNP molecule. In the model, both the N and the C terminus of the CTD protrude out of the helical core, potentially allowing the linker, NTD, and N-terminal residues to interact with other parts of the RNA molecule. The ID regions will play a pivotal role in optimizing the interaction of the RNA molecule with all the other segments of the N protein. The SARS-CoV NTD and the NTD and CTD of avian infectious bronchitis virus have also been found to form helical packing in crystal (7, 15, 28). In the absence of the structure of RNA-bound N protein, we cannot exclude the possibility of other forms of helical packaging. Nonetheless, the two characteristics of the N protein, i.e., intrinsic disorder and multiple RNA-binding sites, will be of fundamental importance in understanding the packaging of the RNP.
The modular structure and multiple sites of moderate RNA-binding affinity of the N protein not only allow the packaging of a stable RNP but also offer an energetically favorable condition for the expression of the viral genomic information. One can envision an unzipping mechanism for unwinding of the viral RNA molecule and dissociation of the RNA molecule from the N protein in a stepwise manner, one module at a time, without the need to overcome a high-energy barrier, since each module of the N protein interacts with the RNA molecule with only moderate affinity. Whether such a mechanism exists will not be known until the detailed atomic resolution structure of the SARS-CoV RNP complex is available.
In conclusion, we showed that the SARS-CoV N protein is a modular protein containing multiple RNA-binding sites. A hallmark of this protein is the presence of long segments of ID regions, accounting for almost half of the sequence. We have also determined the RNA-binding affinity of each module semiquantitatively. The RNA-binding sites reside throughout the entire sequence, including the ID regions of the protein. The flexible linkers of different coronavirus N proteins share low homology, yet they exhibit similar physicochemical properties, implying a universal code of RNA binding in this protein family. The presence of multiple RNA-binding sites of moderate affinity, coupled with the presence of the long stretches of ID regions in the N protein structure, is likely to have fundamental consequences not only for the RNA-packaging mechanism and viral genome expression but also for interaction with other viral and host proteins.
Acknowledgments
This work is supported, in part, by Academia Sinica (Taiwan) under grant AS 95-TP-A07 and by grants NSC 96-2112-M-008-021-MY3 (M.-C. Wu), 97-2627-B-008-004 (M.-C. Wu), 96-2911-M-001-003-MY3 (C.-K. Hu), NSC 95-2311-B-001-066-MY3 (T.-H. Huang), and NSC-95-2113-M-001-035-MY3 (T.-H. Huang) and the National Center for Theoretical Sciences in Taiwan from the National Science Council of the Republic of China. The NMR spectra were obtained at the High-Field Nuclear Magnetic Resonance Center, Taiwan, which is supported by the National Research Program for Genomic Medicine, the Republic of China.
We also thank Maxim Petoukhov (EMBL, Heidelberg, Germany) for providing the modified CRYSOL program and for helpful discussions about SAXS techniques and data processing.
Footnotes
Published ahead of print on 3 December 2008.
REFERENCES
- 1.Chang, C. K., S. C. Sue, T. H. Yu, C. M. Hsieh, C. K. Tsai, Y. C. Chiang, S. J. Lee, H. H. Hsiao, W. J. Wu, C. F. Chang, and T. H. Huang. 2005. The dimer interface of the SARS coronavirus nucleocapsid protein adapts a porcine respiratory and reproductive syndrome virus-like structure. FEBS Lett. 5795663-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang, C. K., S. C. Sue, T. H. Yu, C. M. Hsieh, C. K. Tsai, Y. C. Chiang, S. J. Lee, H. H. Hsiao, W. J. Wu, W. L. Chang, C. H. Lin, and T. H. Huang. 2006. Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 1359-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, C. Y., C. K. Chang, Y. W. Chang, S. C. Sue, H. I. Bai, L. Riang, C. D. Hsiao, and T. H. Huang. 2007. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 3681075-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiti, F., M. Stefani, N. Taddei, G. Ramponi, and C. M. Dobson. 2003. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424805-808. [DOI] [PubMed] [Google Scholar]
- 5.Cuff, J. A., and G. J. Barton. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40502-511. [DOI] [PubMed] [Google Scholar]
- 6.Drosten, C., S. Gunther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguiere, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, and H. W. Doerr. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 3481967-1976. [DOI] [PubMed] [Google Scholar]
- 7.Fan, H., A. Ooi, Y. W. Tan, S. Wang, S. Fang, D. X. Liu, and J. Lescar. 2005. The nucleocapsid protein of coronavirus infectious bronchitis virus: crystal structure of its N-terminal domain and multimerization properties. Structure 131859-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia, P., L. Serrano, M. Rico, and M. Bruix. 2002. An NMR view of the folding process of a CheY mutant at the residue level. Structure 101173-1185. [DOI] [PubMed] [Google Scholar]
- 9.Guo, M., P. M. Gorman, M. Rico, A. Chakrabartty, and D. V. Laurents. 2005. Charge substitution shows that repulsive electrostatic interactions impede the oligomerization of Alzheimer amyloid peptides. FEBS Lett. 5793574-3578. [DOI] [PubMed] [Google Scholar]
- 10.He, R., F. Dobie, M. Ballantine, A. Leeson, Y. Li, N. Bastien, T. Cutts, A. Andonov, J. Cao, T. F. Booth, F. A. Plummer, S. Tyler, L. Baker, and X. Li. 2004. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 316476-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He, R., A. Leeson, M. Ballantine, A. Andonov, L. Baker, F. Dobie, Y. Li, N. Bastien, H. Feldmann, U. Strocher, S. Theriault, T. Cutts, J. Cao, T. F. Booth, F. A. Plummer, S. Tyler, and X. Li. 2004. Characterization of protein-protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 105121-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilser, V. J., and E. B. Thompson. 2007. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 1048311-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang, Q., L. Yu, A. M. Petros, A. Gunasekera, Z. Liu, N. Xu, P. Hajduk, J. Mack, S. W. Fesik, and E. T. Olejniczak. 2004. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 436059-6063. [DOI] [PubMed] [Google Scholar]
- 14.Ivanyi-Nagy, R., J. P. Lavergne, C. Gabus, D. Ficheux, and J. L. Darlix. 2008. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 36712-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayaram, H., H. Fan, B. R. Bowman, A. Ooi, J. Jayaram, E. W. Collisson, J. Lescar, and B. V. Prasad. 2006. X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: implications for nucleocapsid formation. J. Virol. 806612-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalodimos, C. G., N. Biris, A. M. Bonvin, M. M. Levandoski, M. Guennuegues, R. Boelens, and R. Kaptein. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305386-389. [DOI] [PubMed] [Google Scholar]
- 17.Karlin, D., F. Ferron, B. Canard, and S. Longhi. 2003. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 843239-3252. [DOI] [PubMed] [Google Scholar]
- 18.Kuiken, T., R. A. Fouchier, M. Schutten, G. F. Rimmelzwaan, G. van Amerongen, D. van Riel, J. D. Laman, T. de Jong, G. van Doornum, W. Lim, A. E. Ling, P. K. Chan, J. S. Tam, M. C. Zambon, R. Gopal, C. Drosten, S. van der Werf, N. Escriou, J. C. Manuguerra, K. Stohr, J. S. Peiris, and A. D. Osterhaus. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai, M. M. 1990. Coronavirus: organization, replication and expression of genome. Annu. Rev. Microbiol. 44303-333. [DOI] [PubMed] [Google Scholar]
- 20.Luo, C., H. Luo, S. Zheng, C. Gui, L. Yue, C. Yu, T. Sun, P. He, J. Chen, J. Shen, X. Luo, Y. Li, H. Liu, D. Bai, J. Shen, Y. Yang, F. Li, J. Zuo, R. Hilgenfeld, G. Pei, K. Chen, X. Shen, and H. Jiang. 2004. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem. Biophys. Res. Commun. 321557-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo, H., J. Chen, K. Chen, X. Shen, and H. Jiang. 2006. Carboxyl terminus of severe acute respiratory syndrome coronavirus nucleocapsid protein: self-association analysis and nucleic acid binding characterization. Biochemistry 4511827-11835. [DOI] [PubMed] [Google Scholar]
- 22.Luo, H., Q. Chen, J. Chen, K. Chen, X. Shen, and H. Jiang. 2005. The nucleocapsid protein of SARS coronavirus has a high binding affinity to the human cellular heterogeneous nuclear ribonucleoprotein A1. FEBS Lett. 5792623-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo, H., F. Ye, K. Chen, X. Shen, and H. Jiang. 2005. SR-rich motif plays a pivotal role in recombinant SARS coronavirus nucleocapsid protein multimerization. Biochemistry 4415351-15358. [DOI] [PubMed] [Google Scholar]
- 24.Oldfield, C. J., J. Meng, J. Y. Yang, M. Q. Yang, V. N. Uversky, and A. K. Dunker. 2008. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9(Suppl. 1)S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker, M. M., and P. S. Masters. 1990. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology 179463-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng, T. Y., K. R. Lee, and W. Y. Tarn. 2008. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2754152-4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petoukhov, M. V., and D. I. Svergun. 2005. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 891237-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saikatendu, K. S., J. S. Joseph, V. Subramanian, B. W. Neuman, M. J. Buchmeier, R. C. Stevens, and P. Kuhn. 2007. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 813913-3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoemaker, B. A., J. J. Portman, and P. G. Wolynes. 2000. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA 978868-8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surjit, M., R. Kumar, R. N. Mishra, M. K. Reddy, V. T. Chow, and S. K. Lal. 2005. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 7911476-11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svergun, D. I. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25495-503. [Google Scholar]
- 32.Takeda, M., C. K. Chang, T. Ikeya, P. Guntert, Y. H. Chang, Y. L. Hsu, T. H. Huang, and M. Kainosho. 2008. Solution structure of the C-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method. J. Mol. Biol. 380608-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang, T. K., M. P. Wu, S. T. Chen, M. H. Hou, M. H. Hong, F. M. Pan, H. M. Yu, J. H. Chen, C. W. Yao, and A. H. Wang. 2005. Biochemical and immunological studies of nucleocapsid proteins of severe acute respiratory syndrome and 229E human coronaviruses. Proteomics 5925-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tompa, P., and P. Csermely. 2004. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 181169-1175. [DOI] [PubMed] [Google Scholar]
- 35.Xu, X., Y. Liu, S. Weiss, E. Arnold, S. G. Sarafianos, and J. Ding. 2003. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 317117-7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu, I. M., M. L. Oldham, J. Zhang, and J. Chen. 2006. Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between Corona- and Arteriviridae. J. Biol. Chem. 28117134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng, Y., L. Ye, S. Zhu, H. Zheng, P. Zhao, W. Cai, L. Su, Y. She, and Z. Wu. 2008. The nucleocapsid protein of SARS-associated coronavirus inhibits B23 phosphorylation. Biochem. Biophys. Res. Commun. 369287-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao, X., J. M. Nicholls, and Y. G. Chen. 2008. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-β signaling. J. Biol. Chem. 2833272-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zúñiga, S., I. Sola, J. L. Moreno, P. Sabella, J. Plana-Durán, and L. Enjuanes. 2007. Coronavirus nucleocapsid protein is an RNA chaperone. Virology 357215-227. [DOI] [PMC free article] [PubMed] [Google Scholar]