

Abstract
Many viruses have evolved mechanisms to evade the repression of translation mediated by protein kinase R (PKR). In the case of murine cytomegalovirus (MCMV), the protein products of two essential genes, m142 and m143, bind to double-stranded RNA (dsRNA) and block phosphorylation of PKR and eukaryotic initiation factor 2α. A distinctive feature of MCMV is that two proteins are required to block PKR activation whereas other viral dsRNA-binding proteins that prevent PKR activation contain all the necessary functions in a single protein. In order to better understand the mechanism by which MCMV evades the PKR response, we investigated the associations of pm142 and pm143 with each other and with PKR. Both pm142 and pm143 interact with PKR in infected and transfected cells. However, the ∼200-kDa pm142-pm143 complex that forms in these cells does not contain substantial amounts of PKR, suggesting that the interactions between pm142-pm143 and PKR are unstable or transient. The stable, soluble pm142-pm143 complex appears to be a heterotetramer consisting of two molecules of pm142 associated with each other, and each one binds to and stabilizes a monomer of pm143. MCMV infection also causes relocalization of PKR into the nucleus and to an insoluble cytoplasmic compartment. These results suggest a model in which the pm142-pm143 multimer interacts with PKR and causes its sequestration in cellular compartments where it is unable to shut off translation and repress viral replication.
As a consequence of the threat viruses pose to the survival and fitness of their hosts, an array of cellular defenses have evolved to repress viral infection. In mammals, the interferon system mediates multifaceted innate immune responses to viral infections (25). Among the important effectors of this system is the interferon-stimulated double-stranded RNA (dsRNA)-activated protein kinase R (PKR). dsRNA, which is produced during many viral infections (29) and binds to PKR, causing a conformational change, dimerization, and autophosphorylation, followed by phosphorylation of the alpha subunit of initiation factor 2 (eIF2α). Phosphorylated eIF2α inhibits the activity of the guanine nucleotide exchange factor eIF2B, resulting in repression of translation initiation (8).
Because viral replication depends on ongoing translation, viruses have evolved a variety of mechanisms for evading the PKR response (22). One such mechanism utilizes virally encoded dsRNA-binding proteins such as the E3L protein of vaccinia virus and the NS1 protein of influenza virus. The observation that vaccinia virus lacking E3L (VVΔE3L) has a very limited host range in cell culture and is avirulent in animals but does replicate in PKR-deficient cells illustrates the importance of blocking the PKR response (3, 30, 31). Many viral dsRNA-binding proteins form homodimers and bind to PKR, but the relative contributions of the various interactions to anti-PKR functions vary. For example, in the case of E3L, binding to and sequestering dsRNA may be sufficient to prevent PKR activation (5), while in the case of NS1 direct binding to PKR appears to be more critical than binding to dsRNA (20).
In previous studies, we identified dsRNA-binding proteins encoded by two betaherpesviruses, human cytomegalovirus (HCMV) and murine cytomegalovirus (MCMV) (6, 7). The TRS1 and IRS1 genes of HCMV individually encode proteins that block PKR activation by a mechanism that appears to depend on binding to both dsRNA and PKR (10, 11). These proteins also have the unusual property that they cause accumulation of PKR in the nucleus.
In the case of MCMV, the m142 and m143 genes function by blocking PKR activation. Valchanova and coworkers reported that the failure of mutant viruses lacking either m142 or m143 to replicate is linked to activation of PKR and inhibition of protein synthesis (27). We found that the protein products of these two genes (pm142 and pm143) coimmunoprecipitate in infected cells and function together to bind dsRNA and to enable replication of VVΔE3L (7). Thus, the MCMV system differs from other viral systems studied thus far in that two different proteins are required to block PKR activation.
The limited understanding of how dsRNA binding proteins actually block PKR function, coupled with the unusual features of the MCMV system, led us to undertake studies aiming to clarify the interactions among pm142, pm143, and PKR. We found that like other well-studied viral dsRNA binding proteins, both pm142 and pm143 can bind to PKR. pm142 and pm143 appear to form a stable soluble heterotetrameric complex, which, interestingly, does not contain substantial amounts of PKR. Similar to HCMV infection, MCMV infection also causes relocalization of PKR to the nucleus as well as to insoluble cytoplasmic complexes. These results suggest a model in which interaction with pm142 and pm143 results in PKR sequestration into compartments where it is unable to shut off protein synthesis.
MATERIALS AND METHODS
Cells, virus, and infections.
NIH 3T3 cells, PKR null mouse fibroblasts (provided by Bryan Williams, Cleveland Clinic Foundation), and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% NuSerum (Collaborative Biomedical), as previously described (6). MCMV strain K181 and MCMV MC.55, a recombinant of a strain derived from K181 that expresses green fluorescent protein (GFP) (provided by Jeff Vieira, University of Washington) (28) were propagated in NIH 3T3 cells. Infections were performed at a multiplicity of infection (MOI) of 3.
Plasmids.
The plasmid pCS2+BirA, which expresses Escherichia coli biotin ligase, was obtained from Bruce Clurman (Fred Hutchinson Cancer Research Center). All other expression constructs used in these experiments were cloned into the pcDNA3.1/V5-His-TOPO vector (Invitrogen).
Plasmid pEQ1100, which expresses enhanced GFP (EGFP) with a C-terminal biotinylation signal and adjacent six-His tag (indicated in designations as B and H, respectively) was previously described (7). The plasmid pEQ1068 (TRS1[1-738]-B-H) was constructed by inserting primers encoding a biotinlylation signal (7) into the XhoI and XbaI sites of pEQ979 (10).
Construction of pEQ1073 (m142-B-H) was previously described (7). pEQ1163(m142-B) was derived from pEQ1073 by digestion with PmeI and AgeI, blunting with Klenow, and religating to remove the His tag. pEQ1063 (m142-H) was made by amplifying pEQ985 (7) using the oligonucleotides 461 (ACCATGGACGCCCTGTGCGC) and 548 (GTCGTCATCGTCGGCGTCCGC) and cloning the product into the TOPO vector.
pEQ1109 (m143-H) was made by amplifying pEQ939 (7) using the oligonucleotides 410 (ACCATGTCTTGGGTGACCGGAGAT) and 596 (CGCGTCGGTCGCTCTCTCGT) and cloning the product into the TOPO vector. The m143 open reading frame was removed from pEQ1109 by digestion with HindIII and EcoRV and cloned into pEQ1068 after digestion with the same enzymes to construct pEQ1126 (m143-B-H). pEQ1164 (m143-B) was derived from pEQ1126 by digestion with PmeI and AgeI, blunting, and religation.
Transient transfection.
Subconfluent HeLa cells in 12-well plates were transfected with plasmid DNA using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Biotin (25 μM) was added to the medium following transfection. At 24 to 48 h posttransfection the cells were washed with phosphate-buffered saline (PBS) at 4°C then lysed in 300 μl of radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, 0.2% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate) containing 250 mM NaCl. After a portion was reserved for subsequent analysis, the resulting lysates were used for binding to avidin as described below.
Immunoprecipitation, avidin agarose pull-down, and immunoblot analyses.
For coimmunoprecipitation analyses, NIH 3T3 cells in six-well plates were either mock infected or infected with MCMV MC.55. At 48 h postinfection the cells were washed twice with PBS (4°C) and then lysed in 300 μl of NP-40 lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% NP-40). The cells were incubated on a rotator for 20 min and then clarified by centrifugation at 16,000 × g for 10 min at 4°C. Three percent of the sample was saved for assessing protein expression in the lysates. The remaining supernatants were transferred to new tubes, and 5 μl of the indicated rabbit polyclonal antiserum (preimmune, anti-m142, or anti-m143; provided by Laura Hanson and Ann Campbell, Eastern Virginia Medical School) (12) was added. After 1 to 2 h on the rotator, protein A-Sepharose was added to each tube, and rotation was continued for ∼4 h. The samples were washed three times with NP-40 lysis buffer, followed by separation on 10% polyacrylamide gels and transfer to polyvinylidene difluoride (PVDF) membranes by electroblotting. The membranes were probed with a mixture of m142 and m143 antisera or with mouse monoclonal antibody PKR (B-10) (sc-6282; Santa Cruz Biotechnology).
For avidin-agarose binding assays, transfected cell lysates were incubated with immobilized avidin (Pierce) for 2 to 4 h, washed four times, and then immunoblotted as described above. Blots were probed with either Penta-His antibody (Qiagen) or avidin-alkaline phosphatase (AP) (Avidx-AP conjugate; Applied Biosystems) according to the manufacturer's recommendations.
For all immunoblot analyses, proteins were detected using the Western-Star chemiluminescent detection system (Applied Biosystems) according to the manufacturer's recommendations. Silver staining was performed using a SilverQuest silver staining kit (Invitrogen) according to the manufacturer's recommendations.
Metabolic labeling.
For pulse-chase analysis, HeLa cells were transfected as described above with pCS2+BirA and either m143-B-H alone or m143-B-H and m142-H. After 24 h, the cells were labeled for 1 h with 100 μCi/ml [35S]methionine (Easytag Express protein labeling mix; PerkinElmer) and, after two washes, chased by addition of medium containing 1 mM cold methionine. At the indicated times postchase, the cells were lysed with 300 μl of radioimmunoprecipitation assay buffer containing 250 mM NaCl, and the resulting lysates were incubated in the presence of avidin-agarose as described above. The resulting samples were separated on 10% polyacrylamide gels, dried, and subjected to phosphorimager analysis on a Typhoon Trio multimode imager.
Cell fractionation.
For glycerol gradient fractionation of infected cells, 100-mm dishes of NIH 3T3 cells were infected with MCMV MC.55. At 48 h postinfection the cells were scraped into PBS, pelleted, and then resuspended in 250 μl of cell fractionation buffer (20 mM HEPES [pH 8.0], 150 mM NaCl, 1.5 mM MgCl2, 1 mM dithiothreitol, 1% NP-40, 1 mM benzamidine, 10% glycerol). The samples were incubated for 30 min at 4°C on a rotator and then clarified by centrifugation (16,000 × g). Supernatants were separated on 5-ml, 15 to 35% glycerol gradients at 237,000 × g for 17 h at 4°C. Fractions were collected from the bottom of the gradients and separated on 10% polyacrylamide gels, transferred to PVDF membranes, probed with a mixture of m142 and m143 antisera, and then stripped and reprobed with anti-PKR antiserum. Molecular weight standards were centrifuged on parallel gradients, and their distribution was determined by polyacrylamide gel electrophoresis and Coomassie blue staining.
A similar glycerol gradient fractionation of transfected cells was performed after transfection of plasmids expressing m142-B-H and m143-B-H into HeLa cells. Lysates prepared at 24 h posttransfection were analyzed as described for infected cells, except that the PVDF membrane was probed with avidin-AP for protein detection.
Nuclear- and cytoplasmic-enriched fractions were prepared from 100-mm dishes of NIH 3T3 cells after mock infection or infection with MCMV MC.55. At 24, 48, and 72 h postinfection, the cells were washed with PBS and lysed with 1 ml of cell fractionation buffer (see above). The lysates were transferred to 1.5-ml microcentrifuge tubes and placed on a rotator at 4°C for 30 min. The samples were then centrifuged at 1,000 × g for 5 min at 4°C, and the supernatant (cytoplasmic-enriched fraction) was removed carefully without disturbing the pellet. The pellet fraction was then gently resuspended in 0.5 ml of fractionation buffer and microcentrifuged as before, and the remaining pellet (nuclear-enriched fraction) was resuspended in 60 μl of 2% SDS and sonicated to shear DNA. Equal amounts of protein were separated on 10% polyacrylamide gels, transferred to PVDF membranes, and immunoblotted with the following antisera according to the manufacturer's recommendations: PKR (B-10), lamin A/C (2032; Cell Signaling), calnexin (610523; BD Transduction Laboratories), ß-actin (A2066; Sigma), and a mixture of m142 and m143 antisera.
Immunofluorescence.
NIH 3T3 cells or PKR null mouse fibroblasts grown on glass coverslips were mock infected or infected with MCMV K181. At 48 h postinfection, the cells were washed three times with PBS (4°C), fixed in 100% MeOH at −20°C for 5 min, and then washed again with PBS. The coverslips were blocked with 5% normal goat serum for 1 h and then incubated for 1 h with PKR (B-10) antibody or an anti-ß-galactosidase antibody (Promega) that served as an immunoglobulin G2a isotype control. The coverslips were washed three times with PBS and then incubated with fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (Sigma) for 1 h in the dark. After cells were washed three times with PBS, they were incubated with Hoechst 33342 (5 μg/ml; Invitrogen) for 5 min, washed twice with PBS, and then mounted on slides using ProLong Gold antifade mounting medium (Invitrogen). All incubations were performed at room temperature. The samples were analyzed using a Deltavision RT Wide-field Deconvolution Microscope (Applied Precision, Inc.).
RESULTS
pm142 and pm143 form a complex that binds to PKR in MCMV-infected cells.
Several viral proteins that block PKR activation bind to both dsRNA and PKR (4, 5, 10, 11, 16, 20, 24). We have shown that MCMV pm142 and pm143 together bind to dsRNA and block PKR activation (7). Therefore, as a next step in elucidating the mechanism by which pm142 and pm143 function, we investigated whether they also physically associate with PKR.
First, we infected NIH 3T3 cells with MCMV and at 48 h postinfection prepared lysates. After immunoprecipitation with preimmune serum or antiserum specific for either pm142 or pm143, we analyzed the precipitated proteins by immunoblot analyses using either a mixture of rabbit anti-m142 and anti-m143 antisera or with a mouse monoclonal antibody directed against PKR (Fig. 1). Although the immunoglobulin heavy chain partially obscured the pm142 band following precipitation of MCMV-infected lysates with anti-m142 or anti-m143 antiserum, these experiments confirmed that pm142 and pm143 form a complex, as previously reported (7). Also, as noted previously, anti-m143 appeared to precipitate more pm142 than the amount of pm143 precipitated by anti-m142 (Fig. 1A, compare lanes 7 and 8). Importantly, both pm142 and pm143 antisera precipitated PKR while preimmune serum did not (Fig. 1B). The amount of PKR that coimmunoprecipitated with pm142 or pm143 was relatively small, suggesting the possibility that only a minority of PKR in the cell is associated with these two proteins (see below). As expected, pm142, pm143, and PKR were absent from precipitates from mock-infected cell lysates after precipitation with the same antisera. Thus, PKR associates with pm142 and pm143 in MCMV-infected cells.
FIG. 1.

The pm142 and pm143 proteins bind PKR in MCMV-infected cells. NIH 3T3 cells were mock infected (lanes 1, 3, 4, and 5) or infected with MCMV (lanes 2, 6, 7 and 8) at an MOI of 3, and at 48 h postinfection lysates were prepared and analyzed by immunoblot assay directly (lanes 1 and 2) or after immunoprecipitation (IP) with the preimmune serum (lanes 3 and 6), pm142 antiserum (lanes 4 and 7), or pm143 antiserum (lanes 5 and 8). (A) Unbound mock-infected and infected cell lysates (lanes 1 and 2; 3% of the amount immunoprecipitated) and 20% of each bound sample (lanes 3 to 8) were probed using a mixture of m142 and m143 antisera. The migration of pm142, pm143, and the immunoglobulin G heavy chain (HC) are indicated on the right. (B) Lysates and the remaining 80% of each bound sample were probed with the anti-PKR antibody. Ab, antibody.
pm142 and pm143 each bind to PKR in transfected HeLa cells.
In order to determine which of the MCMV proteins (pm142, pm143, or both) associates with PKR, we used an avidin-agarose pull-down system, as described in Materials and Methods. This system takes advantage of the high affinity of the biotin-avidin interaction and eliminates background resulting from the immunoglobulin heavy chain. The plasmids used in these assays expressed proteins that were either His-tagged (designated pm142-H, for example), or contained a 14-amino-acid biotinylation signal in addition to the His tag (e.g., pm142-B-H). When cotransfected into cells along with a plasmid containing the E. coli biotin ligase (BirA) gene and incubated in the presence of 25 μM biotin, biotin is efficiently added to a lysine residue in the biotinylation signal of signal-tagged proteins (2).
For these analyses, we transfected HeLa cells with various combinations of expression plasmids, prepared lysates at 48 h posttransfection, and detected proteins present in the total lysates and in the fractions that bound to avidin-agarose by assays using anti-His or avidin-AP. When probed with anti-His antibody, all of the His-tagged proteins were detected in the lysates (Fig. 2A, top panel). Consistent with the known interactions of pm142 and pm143, both proteins were detected in the avidin bead-bound fractions when either one was biotinylated (Fig. 2A, bottom panel, lanes 2 and 3), while neither protein bound to EGFP-B-H (lanes 7 and 8), even on a longer exposure (data not shown). Omitting the BirA plasmid from the transfection eliminated the binding of both pm142 and pm143 to the avidin beads (lane 4). Note that the biotinylation signal caused a slight but detectable increase in the size of the pm142 and pm143 proteins (compare lanes 2 and 3).
FIG. 2.
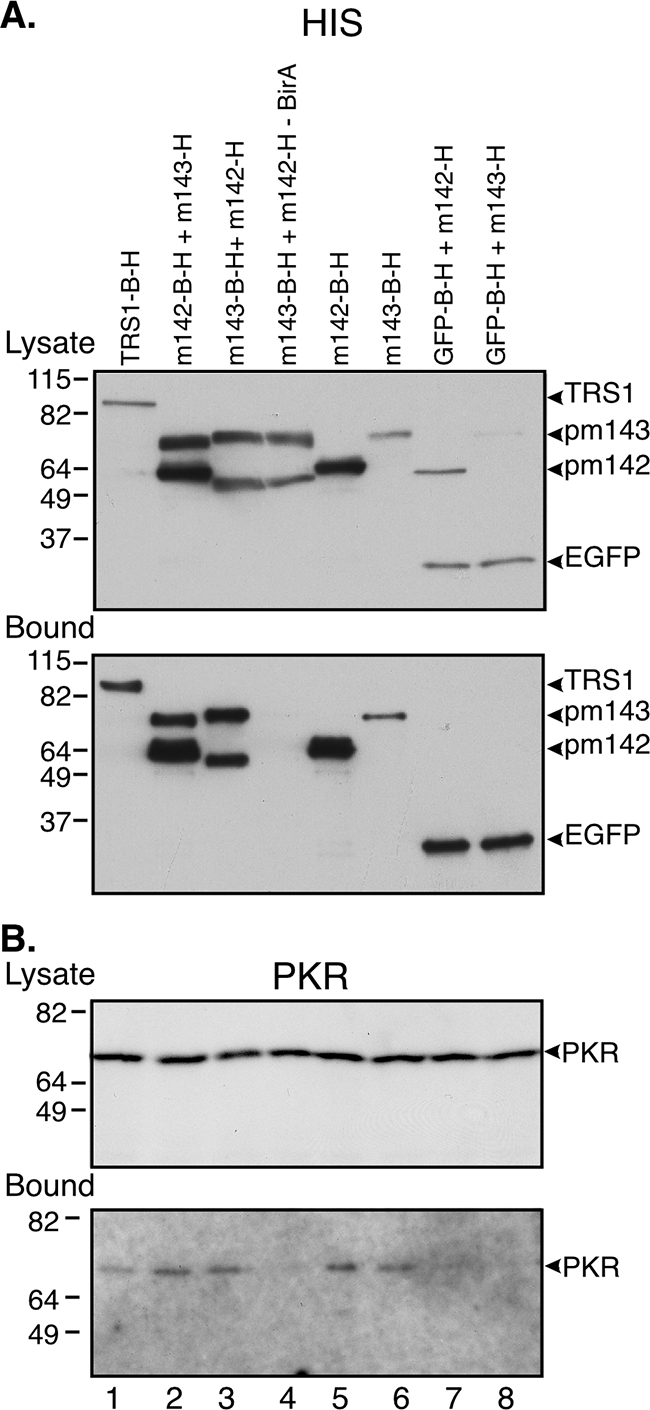
Both pm142 and pm143 bind to PKR in transfected cells. HeLa cells were transfected with the indicated plasmids, all of which express proteins containing His tags alone or with a biotinylation signal. All transfections, except that in lane 4, also included the plasmid encoding biotin ligase (pCS2+BirA). At 48 h posttransfection cell lysates were prepared and incubated with avidin-agarose and analyzed by immunoblot assay as described in Materials and Methods. (A) Membranes with unbound lysates (top panel) or 20% of the bound samples (bottom panel) were probed with the anti-His antibody. (B) Membranes with unbound lysate (top panel) and 80% of the bound samples (lower panel) were subjected to immunoblot analysis with the PKR antibody.
Immunoblot assays of the same samples using PKR antiserum revealed that at least a small amount of PKR was precipitated by the pm142-pm143 complex and by pm142 and pm143 individually (Fig. 2B, lanes 2, 3, 5, and 6). pTRS1-B-H also precipitated PKR (lane 1), consistent with our previous finding that pTRS1 binds to PKR (11). As expected, the negative controls (samples lacking BirA or pulled down with EGFP-B-H) did not precipitate PKR (Fig. 2B, lanes 4, 7, and 8). Treatment of the immunoprecipitated material with dsRNA-specific RNase (RNase III) under conditions that completely digested reovirus genomic dsRNA did not disrupt the association of pm143 with pm142 or with PKR (data not shown). However, dsRNA that is embedded in the complex might not be accessible to the RNase III, so these results do not exclude the possibility that dsRNA is a key component of the complex. However, these data do reveal that pm142 and pm143 each appear to bind to PKR independently.
Expression of pm142 stabilizes pm143.
We noticed that the abundance of pm143 in lysates was consistently much lower when it was expressed by itself or with EGFP than when expressed with pm142 (Fig. 2A, top, compare lanes 6 and 8 to lanes 2, 3, and 4). To evaluate the possibility that pm143 might be stabilized by its association with pm142, we measured the half-life of pm143 in cells transfected with pm143-B-H in the presence or absence of pm142-H (Fig. 3). At 24 h posttransfection, we pulse labeled cells with [35S]methionine for 1 h and then chased with excess cold methionine and analyzed proteins bound to avidin-agarose by electrophoresis and phosphorimager analysis. As is evident in Fig. 3, even by 2 h postchase there was substantially less pm143 in the absence of pm142 than when the proteins were coexpressed. The calculated half-life of pm143 was approximately 8 h in the absence of pm142 and 25 h when it was expressed in combination with pm142. Thus, pm143 is stabilized by coexpression of pm142.
FIG. 3.

The pm143 protein is stabilized by coexpression of pm142. HeLa cells were transiently transfected with m143-B-H alone or in combination with m142-H. At 24 h posttransfection the cells were pulse labeled with [35S]methionine for 1 h and then refed with medium containing excess cold methionine. Cell lysates were prepared at the indicated times postchase, and protein binding to avidin-agarose was analyzed by electrophoresis and phosphorimager analysis.
The pm142-pm143 complex is larger than a heterodimer but lacks substantial amounts of PKR.
The observation that pm142 and pm143 bind to each other and that both also bind to PKR (Fig. 1 and 2) suggested that these three proteins might form a heterotrimeric complex in infected cells. To test this hypothesis, we fractionated infected cell lysates over glycerol gradients and analyzed the distribution of these three proteins by SDS-polyacrylamide gel electrophoresis and immunoblot assays (Fig. 4). In parallel gradients, we fractionated molecular weight standards in an identical manner and determined their distribution by polyacrylamide gel electrophoresis and Coomassie blue staining.
FIG. 4.

Size fractionation of pm142-pm143 complexes and PKR on glycerol density gradients. NIH 3T3 cells were infected with MCMV (MOI of 3), and at 48 h postinfection cell lysates were prepared and centrifuged through 15 to 35% glycerol density gradients as described in Materials and Methods. Following centrifugation, fractions were collected from the bottom of the gradients, and the fractions were separated on polyacrylamide gels, transferred to PVDF membranes, and probed with a mixture of m142 and m143 antisera (A), after which the blot was stripped and reprobed with an anti-PKR antibody (B). Molecular size standards were size fractionated on a parallel gradient and then analyzed by polyacrylamide gel electrophoresis and Coomassie blue staining. The migration of the molecular size standards is indicated below panel B. Note that fractions 5 and 7 were lost during processing and that aliquots of the lysates (lys) prior to fractionation were included in the first and last lanes.
Most of the pm143 migrated with a peak of pm142 at an apparent size of ∼200 kDa (Fig. 4A, fractions 12 to 17), which is close to the calculated size of a heterotrimer (178 kDa) consisting of one molecule each of pm142 (49 kDa), pm143 (61 kDa), and PKR (68 kDa). Surprisingly, we detected little or no PKR in the fractions containing the pm142-pm143 complex (Fig. 4B). Instead, PKR migrated mostly in the range of ∼70 kDa to ∼160 kDa (fractions 17 to 24). The smaller size within this range corresponds to that of the PKR monomer (68 kDa), while the larger species could represent PKR dimers or PKR bound to other factors. Although the small amount of overlap between pm142, pm143, and PKR (fractions 16 to 18) might represent PKR-pm142-pm143 heterotrimers, these results indicate that the majority of the pm142-pm143 complex in infected cells is not bound to PKR. Conversely, most of the PKR in the cell appears not to be bound to the pm142-pm143 complex, which may explain the small amount of PKR associating with pm142 and pm143 in our immunoprecipitation experiments (Fig. 1 and 2). However, these data do not exclude the possibility that there is a PKR-pm142-pm143 trimeric complex in infected cells but that it dissociates under the conditions of these experiments.
In addition to its presence in the pm142-pm143 complexes, pm142 migrates as a smaller species, possibly representing pm142 monomers and homodimers (see below), as well as in larger complexes of unknown composition. Perhaps due to its labile nature in the absence of pm142 (Fig. 3), pm143 is present only in fractions containing pm142.
These results, suggesting that there is a stable ∼200-kDa complex consisting of pm142 and pm143 that lacks substantial amounts of PKR, raise two questions. First, what is the composition of the ∼200-kDa complex? Second, can we reconcile the observations that pm142 and pm143 can each bind to PKR and prevent its activation, yet most of the PKR in the cells appears not to be stably associated with pm142 and pm143? The subsequent studies were designed to evaluate these issues.
The pm142-pm143 complex lacks other proteins.
Since the pm142-pm143 complex migrated at a size of ∼200 kDa (Fig. 4) and a heterodimer of the two proteins would be expected to be only ∼110 kDa, we investigated the possibility that the complex contains additional proteins. First, we transfected cells with plasmids expressing pm142-B-H and pm143-B-H and at 24 h posttransfection prepared cytoplasmic lysates and analyzed the distribution of the proteins by glycerol gradient centrifugation. Detection of the two transfected proteins using avidin-AP demonstrated that, similar to the complex present in infected cells, the pm142-pm143 multimer sedimented as a ∼200-kDa complex in these transfected, uninfected cells (Fig. 5). This result demonstrates that no other MCMV proteins are required for formation of the major pm142-pm143 complex.
FIG. 5.

The pm142-pm143 complex does not contain additional MCMV proteins. HeLa cells were transfected with plasmids expressing BirA, m142-B-H, and m143-B-H, and at 24 h posttransfection lysates were prepared and fractionated on 15 to 35% glycerol density gradients as described in Materials and Methods. The indicated subset of fractions was analyzed by gel electrophoresis, transfer to PVDF membranes, and detection of the biotin-tagged pm142 and pm143 proteins using avidin-AP. The migration of molecular size standards analyzed on a parallel gradient is indicated below the panel.
To evaluate whether any cellular proteins were present in the pm142-pm143 multimer, we transfected cells with pm142-H and pm143-B-H and pulled down pm143-B-H and any associated proteins by binding to avidin-agarose. We then detected these proteins by gel electrophoresis followed by silver staining. We analyzed lysates from cells transfected with pm142-H and pm143-H, both lacking the biotinylation signal, as a negative control. For comparison, we also analyzed HCMV TRS1-B-H. Consistent with prior evaluations using immunoblot assays, pm142 was detectable in the pm143-bound material (Fig. 6). Although there were several other minor protein bands, some of which also were detected in the negative control sample, none approached the abundance of the bands corresponding to pm142 and pm143. Thus, the relatively large size of the pm142-pm143 multimer cannot easily be explained by the presence of either additional MCMV or cellular proteins.
FIG. 6.

The pm142-pm143 complex does not include cellular proteins. HeLa cells were transfected with plasmids expressing m143-B-H and m142-H, TRS1-B-H, or m143-H plus m142-H. All samples also included pCS2+BirA. At 48 h posttransfection lysates were prepared and incubated with avidin-agarose as described in Materials and Methods, and bound proteins were analyzed by gel electrophoresis and silver staining.
The pm142-pm143 complex may be a heterotetramer.
The absence of other proteins in the pm142-pm143 complex might be explained by its being a higher-order multimer of just these two proteins. To investigate this possibility, we again transfected various plasmids expressing proteins tagged with either His or the biotinylation signal into HeLa cells and assayed total and avidin-bound proteins using either anti-His antibody or avidin-AP.
When expressed by itself, pm142-B was detectable in lysates and the avidin-bound samples using avidin-AP, but not with anti-His (Fig. 7, lanes 1). On the other hand, pm142-H was expressed in lysates (Fig. 7A, lane 2), but it lacks the biotin tag, and so, as expected, did not bind to avidin-agarose (Fig. 7B, lane 2); nor was it detected by avidin-AP (Fig. 7C and D, lanes 2). However, when pm142-B and pm142-H were cotransfected, pm142-H was pulled down by avidin beads (Fig. 7B, lane 3), demonstrating that pm142-H binds to pm142-B, thus suggesting that pm142 can homodimerize.
FIG. 7.

The pm142 and pm143 proteins appear to form a heterotetrameric complex. HeLa cells were transfected with pCS2+BirA in addition to the indicated plasmids that express either His- or biotin-tagged forms of pm142 and pm143. At 48 h posttransfection lysates were collected, incubated with avidin-agarose, and subjected to immunoblot analysis as previously described. Membranes containing unbound lysates (3% of total) (A) and 50% of the bound samples (B) were probed with anti-His antibody. Membranes with unbound (3% of total) (C) and bound (50% of total) (D) lysates were probed with avidin-AP to detect biotin-tagged proteins.
Consistent with its relative instability in the absence of pm142 (Fig. 3), pm143-B transfection by itself resulted in only a faint band in both the lysate and bound fractions (Fig. 7C and D, lanes 4). On an exposure comparable to that shown in Fig. 7C and D, pm143-H was almost undetectable (data not shown), but with a longer exposure it was clearly present (Fig. 7A, lane 5). In lysates prepared after transfection of both pm143-B and pm143-H, we could detect both proteins in the lysates but only pm143-B in the bound fraction. The observation that all of the other proteins shown in Fig. 7 that bound directly or indirectly to avidin beads produced bands that appeared at least as intense in bound samples as in the lysates, coupled with the detection of pm143-H in the lysates but not the bound samples, suggests that pm143 does not self-associate.
Interestingly, when pm142-H was expressed along with pm143-B and pm143-H, pm143-H was present in the bound fraction along with pm142-H (Fig. 7B, lane 7). This result indicates that pm143-B and pm143-H were tethered together by one or more molecules of pm142. The absence of detectable pm142-H or pm143-H binding to the EGFP-B-H control (Fig. 7B, lane 8) supports the specificity of the binding reactions.
Collectively, the results shown in Fig. 3 to 6 suggest that the major form of the pm142-pm143 complex is a heterotetramer in which two pm142 molecules bind to each other in addition to each binding to separate pm143 monomers.
MCMV causes relocalization of PKR.
We previously reported that pTRS1 and pIRS1 not only bind to PKR but also cause accumulation of PKR in the nucleus (11). Because of the similarities between HCMV and MCMV, we hypothesized that MCMV infection might also cause PKR relocalization. To test this idea, we prepared nuclear- and cytoplasmic-enriched fractions of mock- or MCMV-infected NIH 3T3 cells at 24, 48, and 72 h postinfection and evaluated the distribution of PKR by immunoblot analysis (Fig. 8). PKR remained relatively constant in abundance and remained predominantly cytoplasmic during this time interval in mock-infected cells. In contrast, PKR abundance increased, and it redistributed to the nuclear fraction in MCMV-infected cells. We performed control immunoblotting on the same cytoplasmic and nuclear extracts to evaluate the efficacy of the fractionation procedure. Antibodies directed against the nuclear marker lamin A/C revealed its presence exclusively in the nuclear fraction. The cytoplasmic marker calnexin was upregulated by MCMV infection but remained predominantly cytoplasmic in the infected cells. An anti-ß-actin immunoblot showed that the abundance of ß-actin was similar in mock-infected and infected cells although it appeared to be slightly more concentrated in the nuclear than the cytoplasmic fractions in both cases. The same extracts were also probed with a mixture of anti-m142 and anti-m143 antisera, and as reported by Hanson et al. (12), pm142 and pm143 were present in both the cytoplasmic and nuclear fractions throughout infection.
FIG. 8.

MCMV infection causes nuclear relocalization of PKR. NIH 3T3 cells were mock infected or infected with MCMV (MOI of 3). Cell lysates were prepared at 24, 48, and 72 h postinfection and separated into cytoplasmic (cyt)- and nuclear (nuc)-enriched fractions as described in Materials and Methods. Equal amounts of each fraction were subjected to gel electrophoresis, transferred to PVDF membranes, and probed with the indicated antiserum including the nuclear marker lamin A/C and the cytoplasmic marker calnexin.
As a complementary method for assessing the effects of MCMV on PKR, we examined the distribution of PKR in mock- and MCMV-infected cells by indirect immunofluorescence using an Olympus Deltavision microscope, as described in Materials and Methods. In mock-infected NIH 3T3 cells (Fig. 9A), incubation with anti-PKR antibody showed the predominantly cytoplasmic localization of PKR, with a small amount of nuclear PKR appearing to localize in the nucleoli, as has been reported by others (14, 15) and consistent with our immunoblotting data (Fig. 8). At 48 h postinfection, PKR expression was elevated in MCMV-infected cells, and a substantially higher amount was detectable in the nucleus. Surprisingly, contrary to the results based on cell fractionation methods (Fig. 8), the majority of the PKR in the infected cells appeared to remain localized within the cytoplasm. The PKR signal in infected cells also seemed distributed in a coarser punctate pattern in the cytoplasm of infected cells than in mock-infected cells. Staining with an isotype-matched control antibody showed minimal background staining in these cells. As an additional control, we incubated mock- or MCMV-infected PKR-null murine embryonic fibroblasts with anti-PKR antiserum or the isotype-matched control antibody (Fig. 9B). In these cells, both the anti-PKR and control antibodies revealed only a low level of background fluorescence.
FIG. 9.

MCMV infection appears to cause PKR relocalization to both the nucleus and insoluble cytoplasmic complexes. NIH 3T3 and PKR-null (PKR−/−) cells grown on coverslips were mock infected or infected with MCMV (MOI of 3). At 48 h postinfection the cells were fixed, incubated with anti-PKR or an isotype control antibody, and prepared for immunofluorescence as described in Materials and Methods. Samples were analyzed by deconvolution microscopy. FITC, fluorescein isothiocyanate.
Thus, the results of both the immunoblot assays and immunofluorescence assays revealed an increase in the amount of nuclear PKR after MCMV infection. Compared to the immunoblot assays, the immunofluorescence studies indicate that there is more abundant PKR in the cytoplasm at late times postinfection. These results suggest that MCMV infection results in relocalization of PKR to both the nucleus and to insoluble cytoplasmic complexes.
DISCUSSION
The importance of PKR as a mediator of intracellular innate defenses against viral infections is highlighted by the remarkable number and diversity of viral factors and mechanisms that have evolved to counteract its inhibitory effects on host cell translation (22). In several viral systems, mutants lacking anti-PKR factors are severely attenuated for growth in cell culture and have extremely reduced virulence in animal models. Moreover, the replication defect of some of these viruses has been shown to be reversed at least in part in PKR-deficient cells and, in the case of herpes simplex virus type 1, even in animals, thus providing genetic support for the functional importance of the interactions between PKR and viral antagonists (19, 30, 31).
Many viruses encode a dsRNA-binding protein that can inhibit the PKR pathway. Some viruses, including vaccinia virus, herpes simplex virus type 1, and HCMV, have been shown to contain a second gene that also inhibits the PKR pathway (4, 6, 16, 22, 26). In these examples, the two genes act independently and, in the case of vaccinia virus and herpes simplex virus type 1, at different steps in the PKR activation pathway. The two HCMV genes are very closely related and likely act at the same step. MCMV also has two anti-PKR genes but is unusual in that the two genes act together as a single functional unit. Both m142 and m143 are required for effective dsRNA binding and for rescue of VVΔE3L replication (7). Furthermore, deletion of either gene results in phosphorylation of PKR and eIF2α and repression of protein synthesis in infected cells and eliminates MCMV replication (27). The genomic proximity of m142 and m143 combined with their functional similarities suggests that evolution of MCMV may have resulted in separation of the functions needed for blocking PKR into two proteins, while in other viruses these functions are combined in a single protein.
Many well-studied viral dsRNA-binding proteins that prevent PKR activation bind to PKR (4, 11, 20, 24). We found that the pm142-pm143 complex does so as well (Fig. 1). The observation that pm142 and pm143 can each bind to PKR in transfection assays (Fig. 2) argues against the possibility that the separation of dsRNA-binding protein functions in MCMV led to one protein's becoming specialized in binding to PKR. We do not know whether the interactions between pm142-pm143 and PKR are direct ones. Data showing that pm142 and pm143 are each able to bind to PKR yet that the combination of both is required for strong binding to dsRNA (7) suggest that a dsRNA tether is not required for PKR binding to pm142 or pm143, but it remains possible that there are other proteins or nucleic acids contributing to the interaction.
We also do not yet know whether binding to PKR is required for pm142-pm143 function although, based on studies of other viral dsRNA-binding proteins, we suspect that it is. HCMV pTRS1 (and pIRS1) and influenza NS1 bind to PKR, and these interactions appear to be essential for its inhibition (12, 20). In the case of vaccinia virus E3L, the C-terminal dsRNA binding domain, in the absence of the N-terminal PKR-binding domain, is sufficient for viral replication in many cell types, but it is required for preventing PKR activation late in infection (18), for blocking the inhibitory effects of PKR in a Saccharomyces cerevisiae growth assay (24), and for virulence in animal models (3). Evaluating whether E3L function requires it to bind to PKR is complicated by the presence of other functions, including an N-terminal Z-DNA binding domain that may be responsible for some of the observed phenotypes (17). Adding to the complexity, binding to PKR does not always inhibit PKR function, as is clearly illustrated by the activation of PKR resulting from its interaction with the cellular regulator PACT (21). Nonetheless, the fact that both pm142 and pm143 associate with and inhibit PKR does suggest that this is a functionally important physical interaction.
The observed binding of pm142, pm143, and PKR to each other in each binary combination suggested that these three proteins might form a heterotrimeric complex. Although we found clear evidence for a complex of pm142 and pm143 in infected or transfected cells, little, if any, PKR comigrated with the pm142-pm143 complex. One possible explanation for the observed interaction between PKR and pm142-pm143 by coimmunoprecipitation but not by glycerol gradient fractionation is that the interaction is unstable, and therefore the complex dissociated under the conditions used in the glycerol gradient experiments. Alternatively, the interaction might be a transient one, possibly resulting in inactivation of PKR by a mechanism that does not require continued association with the MCMV proteins.
We also discovered that MCMV alters the distribution of PKR in infected cells. Our analyses of uninfected cells are consistent with reports indicating that approximately 20% of the cellular PKR is present in the nucleus, usually in association with the nucleolus (14, 15). Fractionating cells with detergent revealed accumulation of PKR in the nuclear-enriched pellet fraction by 24 h postinfection (Fig. 8). Since such pellet fractions may also contain insoluble cytoplasmic material, complementary methods are important for evaluating protein distribution. Using immunofluorescence assays, we found that PKR did, indeed, appear to accumulate in the nucleus during infection, but a large fraction of it remained in the cytoplasm. PKR accumulates in the nucleus after HCMV infection but not after vaccinia infection (11). Using recombinant vaccinia viruses, we showed that pTRS1 or pIRS1 could mediate this relocalization, suggesting that pm142 and pm143 are plausible candidates for instigating the redistribution of PKR during MCMV infection.
Altered distribution of PKR has been reported in other settings. Endoplasmic reticulum stress can cause PKR to accumulate in the nucleus (23). Activated PKR was recently shown to be enriched in the nucleus of high-risk myelodysplastic syndrome lymphoblasts (9) and in neurons expressing the human immunodeficiency virus type I gp120, where it may contribute to neurodegeneration (1). Human papilloma virus causes PKR to relocalize both to the nucleus and to cytoplasmic P bodies (13). At present, the role of nuclear accumulation of PKR in infected cells is unknown, but it might serve to keep PKR away from its cytoplasmic targets or, alternatively, to enable interaction with a nuclear target and enhance viral replication. The relocalization of PKR into insoluble cytoplasmic compartments might represent another mechanism for ensuring that PKR is sequestered away from translational machinery. Note that our glycerol gradient experiments would not have detected nuclear or insoluble cytoplasmic complexes since these were removed during processing, so it remains possible that pm142 and pm143 are associated with PKR in these sites. Consistent with this possibility, Hanson et al. found that pm142 and pm143 appear to be primarily cytoplasmic by immunofluorescence but were present in the “nuclear” fractions as well when analyzed by cell fractionation and immunoblot assay (12).
Our data suggest that pm142 and pm143 form a stable complex, likely a heterotetramer, consisting of two molecules of pm142 associated with each other and each one associating with a molecule of pm143 (Fig. 10). Data supporting this proposed structure include the observations that in infected cells, pm142 and pm143 (i) colocalize by immunofluorescence (12), (ii) coimmunoprecipitate (Fig. 1 and 7), and (iii) migrate through glycerol gradients as a complex having a size consistent with a heterotetramer (Fig. 4). Further evidence comes from transfection assays showing that (iv) pm143 stability requires pm142, (v) the molarity of the two proteins in the complex is similar (based on experiments in which their relative amounts can be compared, as in Fig. 2A, 5, and 6), and (vi) pm142 can self-associate while pm143 appears not to do so (Fig. 7).
FIG. 10.

Model depicting putative mechanisms of PKR inactivation and relocalization by pm142 and pm143. In the absence of viral antagonists, PKR is activated by dsRNA, which leads to dimerization (1), auto-phosphorylation, and subsequent phosphorylation of the alpha subunit of the translation initiation factor eIF2, thereby blocking translation and inhibiting viral replication. During MCMV infection, a stable heterotetrameric pm142-pm143 complex forms that can interact with PKR and dsRNA. Whether or not pm142 and pm143 remain associated with PKR is not yet clear, but we hypothesize that these viral proteins cause sequestration of PKR in the nucleus and in insoluble cytoplasmic complexes (gray area), where it is unlikely to be able to shut off translation and block viral replication (2).
Based on our results, we propose a model in which pm142 and pm143 form a stable soluble heterotetrameric cytoplasmic complex during infection (Fig. 10). This complex serves as a reservoir, poised to interact with PKR and dsRNA. As has been proposed for the mechanism by which E3L functions, the pm142-pm143 complex may associate with PKR and prevent its homodimerization (24). The absence of a stable soluble pm142-pm143-PKR complex may result from its relocalization to the nucleus and insoluble cytoplasmic fractions where PKR would be unable to shut off translation, although it is also possible that a transient interaction with pm142 and pm143 is sufficient to cause PKR to relocalize. Although we do not yet understand the significance of the separation of the PKR-inhibitory function into two proteins in MCMV, this organization is unique among viruses studied thus far, and we expect that further exploration of this system will help uncover fundamentals of the mechanisms by which dsRNA-binding proteins interfere with PKR function and enable viral replication.
Acknowledgments
We thank Bryan Williams (Cleveland Clinic Foundation) for providing PKR-null murine embryonic fibroblasts, Laura Hanson and Ann Campbell (Eastern Virginia Medical School) for providing antisera, Bruce Clurman (Fred Hutchinson Cancer Research Center) for the BirA expression plasmid, and the Fred Hutchinson Cancer Research Center Scientific Imaging and Genomics Cores for technical assistance.
This work was supported by NIH grant AI26672.
Footnotes
Published ahead of print on 10 December 2008.
REFERENCES
- 1.Alirezaei, M., D. D. Watry, C. F. Flynn, W. B. Kiosses, E. Masliah, B. R. Williams, M. Kaul, S. A. Lipton, and H. S. Fox. 2007. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J. Neurosci. 2711047-11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckett, D., E. Kovaleva, and P. J. Schatz. 1999. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8921-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt, T. A., and B. L. Jacobs. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 75850-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassady, K. A., M. Gross, and B. Roizman. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 728620-8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, H. W., L. H. Uribe, and B. L. Jacobs. 1995. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 696605-6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Child, S. J., M. Hakki, K. L. De Niro, and A. P. Geballe. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78197-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Child, S. J., L. K. Hanson, C. E. Brown, D. M. Janzen, and A. P. Geballe. 2006. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J. Virol. 8010173-10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dever, T. E., A. C. Dar, and F. Sicheri. 2007. The eIF2α kinases, p. 319-344. In M. B. Matthews, N. Sonenberg, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 9.Follo, M. Y., C. Finelli, S. Mongiorgi, C. Clissa, C. Bosi, G. Martinelli, W. L. Blalock, L. Cocco, and A. M. Martelli. 22 May 2008. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia. doi: 10.1038/leu.2008.122. [Epub ahead of print.] [DOI] [PubMed]
- 10.Hakki, M., and A. P. Geballe. 2005. Double-stranded RNA binding by human cytomegalovirus pTRS1. J. Virol. 797311-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hakki, M., E. E. Marshall, K. L. De Niro, and A. P. Geballe. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 8011817-11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanson, L. K., B. L. Dalton, L. F. Cageao, R. E. Brock, J. S. Slater, J. A. Kerry, and A. E. Campbell. 2005. Characterization and regulation of essential murine cytomegalovirus genes m142 and m143. Virology 334166-177. [DOI] [PubMed] [Google Scholar]
- 13.Hebner, C. M., R. Wilson, J. Rader, M. Bidder, and L. A. Laimins. 2006. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 873183-3193. [DOI] [PubMed] [Google Scholar]
- 14.Jeffrey, I. W., S. Kadereit, E. F. Meurs, T. Metzger, M. Bachmann, M. Schwemmle, A. G. Hovanessian, and M. J. Clemens. 1995. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp. Cell Res. 21817-27. [DOI] [PubMed] [Google Scholar]
- 15.Jimenez-Garcia, L. F., S. R. Green, M. B. Mathews, and D. L. Spector. 1993. Organization of the double-stranded RNA-activated protein kinase DAI and virus-associated VA RNAI in adenovirus-2-infected HeLa cells. J. Cell Sci. 10611-22. [DOI] [PubMed] [Google Scholar]
- 16.Khoo, D., C. Perez, and I. Mohr. 2002. Characterization of RNA determinants recognized by the arginine- and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. J. Virol. 7611971-11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, Y. G., M. Muralinath, T. Brandt, M. Pearcy, K. Hauns, K. Lowenhaupt, B. L. Jacobs, and A. Rich. 2003. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 1006974-6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langland, J. O., and B. L. Jacobs. 2004. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology 324419-429. [DOI] [PubMed] [Google Scholar]
- 19.Leib, D. A., M. A. Machalek, B. R. Williams, R. H. Silverman, and H. W. Virgin. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 976097-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, S., J. Y. Min, R. M. Krug, and G. C. Sen. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 34913-21. [DOI] [PubMed] [Google Scholar]
- 21.Li, S., G. A. Peters, K. Ding, X. Zhang, J. Qin, and G. C. Sen. 2006. Molecular basis for PKR activation by PACT or dsRNA. Proc. Natl. Acad. Sci. USA 10310005-10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohr, I. J., T. Pe'ery, and M. B. Mathews. 2007. Protein Synthesis and Translational Control during Viral Infection, p. 545-599. In M. B. Matthews, N. Sonenberg, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Onuki, R., Y. Bando, E. Suyama, T. Katayama, H. Kawasaki, T. Baba, M. Tohyama, and K. Taira. 2004. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer's disease. EMBO J. 23959-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romano, P. R., F. Zhang, S. L. Tan, M. T. Garcia-Barrio, M. G. Katze, T. E. Dever, and A. G. Hinnebusch. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol. Cell. Biol. 187304-7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadler, A. J., and B. R. Williams. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8559-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shors, S. T., E. Beattie, E. Paoletti, J. Tartaglia, and B. L. Jacobs. 1998. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J. Interferon Cytokine Res. 18721-729. [DOI] [PubMed] [Google Scholar]
- 27.Valchanova, R. S., M. Picard-Maureau, M. Budt, and W. Brune. 2006. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 8010181-10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Den Pol, A. N., E. Mocarski, N. Saederup, J. Vieira, and T. J. Meier. 1999. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J. Neurosci. 1910948-10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber, F., V. Wagner, S. B. Rasmussen, R. Hartmann, and S. R. Paludan. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 805059-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang, Y., R. C. Condit, S. Vijaysri, B. Jacobs, B. R. Williams, and R. H. Silverman. 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 765251-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang, P., B. L. Jacobs, and C. E. Samuel. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J. Virol. 82840-848. [DOI] [PMC free article] [PubMed] [Google Scholar]