

Abstract
Degradation of mRNA is one of the key processes that control the steady-state level of gene expression. However, the rate of mRNA decay for the majority of genes is not known. We successfully obtained the rate of mRNA decay for 19 977 non-redundant genes by microarray analysis of RNA samples obtained from mouse embryonic stem (ES) cells. Median estimated half-life was 7.1 h and only <100 genes, including Prdm1, Myc, Gadd45 g, Foxa2, Hes5 and Trib1, showed half-life less than 1 h. In general, mRNA species with short half-life were enriched among genes with regulatory functions (transcription factors), whereas mRNA species with long half-life were enriched among genes related to metabolism and structure (extracellular matrix, cytoskeleton). The stability of mRNAs correlated more significantly with the structural features of genes than the function of genes: mRNA stability showed the most significant positive correlation with the number of exon junctions per open reading frame length, and negative correlation with the presence of PUF-binding motifs and AU-rich elements in 3′-untranslated region (UTR) and CpG di-nucleotides in the 5′-UTR. The mRNA decay rates presented in this report are the largest data set for mammals and the first for ES cells.
Key words: mRNA decay, mRNA degradation, microarray, transcript, exon junction, AU-rich elements (ARE), embryonic stem cells, leukemia inhibitory factor (LIF), retinoic acid (RA), cell differentiation, mouse strain
1. Introduction
Post-transcriptional regulation of gene expression includes mRNA processing, splicing, editing, transport, stability and translation. Degradation of mRNA is an important component of gene function that controls the steady-state concentration of functional transcript levels in the cell.1 For example, in human mRNA, stability is estimated to control the mRNA level of about 5–10% of all genes.2 Quantification of this process is important for interpretation of gene-expression data obtained with microarrays as well as data from gene manipulation experiments. Changes in gene expression may be delayed by several days if the corresponding transcript is stable. This may cause serious errors in reconstruction of gene regulatory networks.
Studies in several organisms revealed that majority of transcripts are stable.3–5 It appeared that the specific half-life of each mRNA is precisely related to its physiological role.4–8 For example, most housekeeping genes have long mRNA half-lives. In contrast, proteins that are required only for limited time in the cell, e.g. at a certain stage of the cell cycle, during development, growth or differentiation or in response to external stimuli, often have mRNAs with short half-lives.9 Transcriptionally inducible genes are disproportionately represented in the class of genes with rapid mRNA turnover.5
RNA stability can be altered in response to external stimuli such as hormones10 or various types of stress. For example, response to UV-B exposure leads to stabilization of many short-lived mRNAs in mammalian cells.11 Conversely, in yeast, mild heat shock promotes rapid degradation of mRNAs encoding the ribosomal proteins,12 while glucose starvation, amino acid starvation, or sugar-induced osmotic stress causes stabilization of at least some yeast mRNAs.13–15 Changes in RNA stability can also play an important role during embryonic development or cell differentiation. In plasmodium it has been shown that a major determinant of mRNA decay rate appears to be tightly linked to intra-erythrocytic development cycle, and to a lesser extent the functional category of the mRNAs themselves.16 During primordial germ cells specification of Oryzias latipes (Japanese medaka) some specific RNA molecules are protected from degradation, thus establishing a specific mRNA expression pattern controlled by post-transcriptional mechanisms.17 Expression of a wide variety of transcripts is controlled by changes in mRNA stability during neuronal development.18,19 During muscle differentiation mRNA half-lives for muscle-specific genes—myogenin and myoD—have been shown to be the highest during differentiation, but declines when differentiation is completed.20 Abnormal changes in RNA stability can be a cause of cell malfunction leading to cancer21,22 and other diseases like diabetic nephropathy,22 muscular atrophy,23 neurological disorders such as Alzheimer disease.9
Decay of mRNA is controlled by complex mechanisms that are not fully understood. This mechanism is integrated with other mRNA-related molecular processes including transcript elongation, splicing, polyadenylation, transport and translation.6,9 RNA decay mechanisms include interaction between cis-acting elements and trans-acting factors. Major cis-factors affecting decay rates include AU-rich elements (ARE), iron-responsive elements, histone stem-loops, coding region determinants, Jun-kinase response elements, retained introns, and exon junctions.24–26 Trans-factors that control mRNA decay include RNAses that are often assembled into exosomes, RNA-binding proteins (poly-A binding, WD40 repeats, ARE-binding), siRNA, miRNA, piRNA. P-bodies in the cytoplasm can facilitate miRNA-mediated mRNA degradation induced by stress, and ribosomes control nonsense-mediated decay (NMD).27 Recent studies have shown that PUF-domain proteins can promote mRNA degradation of genes involved in development via Pop2-mediated deadenylation.28 Binding motif of PUF protein PUM2 has been identified in mouse.29
Estimates of mRNA decay rates may depend on the method of measurement. Functional degradation, defined as decrease of the numbers of functional mRNA capable of producing correct protein, should be distinguished from physical degradation (or mass decay), which represents the decrease of mRNA numbers measured by PCR amplification or hybridization intensity.30 Analysis of functional degradation is appropriate for individual genes, but it is not an option for high-throughput analysis targeted at the whole transcriptome. Thus, we will consider only physical degradation of mRNA with understanding that this measurement may not capture some aspects of mRNA function. The most widely used methods for high-throughput analysis of mRNA degradation is based on transcriptional inhibitors, for example by actinomycin D, which inhibits transcription via binding to single-strand DNA at the transcription initiation complex and preventing elongation by RNA polymerase II.31
Multiple studies attempted to measure mRNA decay rates in human,3,5 but only one study has relatively large data of 5245 genes in hepatoma cells.4 There are no data for mouse or any other mammals. Among eukaryotes, comparable data have been generated for plasmodium,16 yeast7 and Arabidopsis.32 To obtain baseline data for the mRNA half-life in mouse, we have selected mouse embryonic stem (ES) cells. ES cells are often used as experimental models for cell differentiation and for reconstruction of gene regulatory networks. Genes that control mRNA decay rates may also affect the differentiation of ES cells.33 Despite increasing interest in the study of ES cells, there have been no attempts to measure mRNA decay rates in these cells. Here, we have described mRNA half-life of essentially all mouse genes in mouse ES cells.
2. Materials and methods
2.1. Cell culture
We used two mouse ESC lines: MC1 derived from mouse strain 129S6/SvEvTac and MC2-B6 derived from strain C57BL/6J. These lines were purchased from The Transgenic Core Laboratory of the Johns Hopkins University School of Medicine (Baltimore, MD, USA). To remove contaminating feeder cells, ES cells were first cultured for two passages on gelatin-coated culture dish in complete ES medium [DMEM, 15% FBS; Leukemia Inhibitory Factor (LIF) (ESGRO) 1000 U/ml; 1 mM sodium pyruvate; 0.1 mM NEAA, 2 mM glutamate, 0.1 mM β-mercaptoethanol and penicillin/streptomycin (50 U/50 µg per ml)]. Cells were then seeded in gelatin-coated 6-well plates at the density of 1–2 × 105 cells/well (1–2 × 104/cm2) and cultured for 3 days before the test for mRNA stability. Cells were incubated at 37°C and 5% CO2. Medium was changed daily. Differentiated ES cells were obtained from MC1 cells by LIF withdrawal (complete medium without LIF, below referenced as LIF−) or by retinoic acid (RA) treatment (complete medium with 1 µM RA, below referenced as RA) for 7 days before the test. To measure mRNA stability, transcription was blocked by adding actinomycin D (Sigma, cat # A-9415) to the medium at the concentration of 5 µg/ml. Cells were harvested at 0, 1, 2, 4 and 8 h after addition of actinomycin D.
2.2. Microarray experiments
RNA was extracted using Phase lock gel™ (Eppendorf/Brinkman) columns according to the manufacturer’s protocol after adding 1 ml of Trizol™ (Invitrogen) per well. RNA was precipitated with isopropanol, washed with 70% ethanol and dissolved in DEPC dH2O. RNA was hybridized to the NIA Mouse 44K Microarray v2.3 (whole genome 60-mer oligonuleotide probe; manufactured by Agilent Technologies, #014951)34 in duplicate for each cell type and time point. Fluorescently labeled microarray targets were prepared from 2.5 µg aliquots of total RNA samples using a Low RNA Input Fluorescent Linear Amplification Kit (Agilent). A reference target (Cy5-CTP-labeled) was produced from Stratagene Universal Mouse Reference RNA (UMR), and all other targets were labeled with Cy3-CTP. Targets were purified using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol, and quantified on a NanoDrop scanning spectrophotometer (NanoDrop Technologies). All hybridizations were carried out by combining a Cy3-CTP-labeled experimental target and a Cy5-CTP-labeled UMR target. Microarrays were hybridized and washed according to Agilent protocol (G4140-90030; Agilent 60-mer oligonucleotide microarray processing protocol—SSC Wash, v1.0). Slides were scanned on an Agilent DNA Microarray Scanner, using standard settings, including automatic photo-multiplier tube adjustment.
2.3. Estimation of mRNA decay rates
Outliers in microarray data were detected using criterion of z > 7 for log-transformed Cy3 and Cy5 data separately and removed (0.17% of the data). Then, Cy3 signals were normalized by Cy5 signals for the same probe in log scale, except for 4.34% of probes in which strong correlation between log Cy5 and Cy3 signals (slope of log10(Cy5/Cy3) > 0.25) was likely a hybridization artifact (termed ‘chain effect’), because of low Cy5/Cy3 signal ratio (>3-fold difference) and high variance (>0.05) of log10(Cy3) (chain effect is described in Section 2.4). Degradation rate of mRNA was estimated using linear regression of log-transformed (base 10) signal intensity values y versus time t:
| 1 |
where t is time, b is the slope, a is intercept and d = b*ln(10) is instantaneous decay rate. The decrease of mRNA amount measured by microarray usually matched well with this model at all time points (Fig. 1A). However, for some mRNA species, the decay leveled off at some time point, which may be attributed to either heterogeneity in degradation rates (i.e. short-lived fraction degraded first) or non-specific hybridization to probes on the array (Fig. 1B). For these mRNA species we estimated decay rates after omitting the later time points to obtain the most accurate estimate of degradation rate. Time points were removed sequentially from the end of the time course till the lower confidence limit for b was maximized and the first time point (t = 0) was within the confidence interval of the regression. Out of 32 601 probes on the arrays, for which mRNA decay rates were estimated, 95.5% matched well to the exponent and all five time points were used for analysis; in 3.4, 0.9 and 0.2% cases decay rates were estimated using four, three and two time points, respectively. The same number of time points was used for different cell types and culture conditions (MC2-B6-LIF+, MC1-LIF+, MC1-LIF− and MC1-RA) to ensure proper comparison of mRNA degradation rates. Because the same amount of RNA was used for array hybridization, degradation of some mRNA species after block of transcription resulted in the increase of relative abundance of other stable mRNA species. Thus, additional correction was needed to account for global mRNA degradation. Yang et al.4 used β-actin for normalization assuming that it is very stable. However, Actb appeared not very stable in our experiments (half-life = 7.9 h). Thus, for global normalization, we used 200 most stable non-redundant genes with average log intensity of >2.5 and for which decay rates were successfully measured for all four types of cells. Average mRNA degradation rate for these genes was estimated for each cell type using Equation (1) (e.g. d = −0.1012 for undifferentiated MC1 cells) and then was subtracted from all estimated mRNA degradation rate for this cell type. Half-life of each mRNA species was estimated as H = min [24, ln(2)/d] for positive d, and H = 24 h for negative d. We truncated half-life estimates by 24 h, because it is difficult to project mRNA half-life beyond this time point based on the experiment that lasted for 8 h. We also estimated the average decay rate of each gene in all examined cell types and the corresponding half-life. Statistical significance of the differential mRNA decay rates in various cell types was determined using false discovery rate (FDR < 0.05), estimated from z-values: 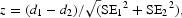 where d1 and d2 are decay rates in cell types 1 and 2, respectively, and SE1 and SE2 are standard errors of these decay rates estimated from regression. FDR, which is necessary for multiple hypothesis testing, was estimated using the method of Benjamini and Hochberg.35 In addition to FDR, we used the criterion of >2-fold difference between half-lives to be significant.
where d1 and d2 are decay rates in cell types 1 and 2, respectively, and SE1 and SE2 are standard errors of these decay rates estimated from regression. FDR, which is necessary for multiple hypothesis testing, was estimated using the method of Benjamini and Hochberg.35 In addition to FDR, we used the criterion of >2-fold difference between half-lives to be significant.
Figure 1.

Stability of mRNA in mouse ES cells. (A and B) Regression of gene expression level (microarray signal intensity in log scale) versus time after suppressing transcription by actinomycin D (y = a + bx) is used to estimate the rate of mRNA decay, d = b*ln(10). All 10 data points are used for an example (A) and only six data points are used for and example (B). (C) Comparison between mRNA decay rates estimated for MC1 ES cells and those estimated for MC2-B6 ES cells. Rates are log-transformed, log10(d + 0.1). (D) Comparison between mRNA decay rates estimated using different probes (#1 and #2) on the microarray, which match to the main transcript of the same gene. Only the data for MC1 cells are shown. (E) Comparison of mRNA decay rates identified with the best probe (#1) from the main transcript of each gene with decay rates estimated using other probe (#2) that matched the same gene; pairs of probes were first classified into groups according to the difference in estimated mRNA decay rate (group size is shown below bars, n), and then the proportion of probes #2 that matched different types of transcripts and/or introns was estimated and plotted for each group. (F) Frequency distribution of mRNA half-life among all genes. (G) Frequency distribution of mRNA half-life in different functional categories of genes; color circles indicate that the functional group was previously identified as being over-represented among stable or unstable mRNA species.
2.4. Calibration of microarray signal intensities
Reliable estimation of mRNA degradation rates requires accurate scaling of expression values to ensure that the ratio of hybridization signal intensities is equal to the ratio of mRNA abundance uniformly across the entire range of values. We tested the scaling using a series of dilutions (5×, 1×, 0.2× and 0.04×) of the mRNA pool (obtained from newborn mice of C57BL/6J strain) labeled with Cy3 and hybridized together with the constant amount of the UMR sample labeled with Cy5. Regression of log-transformed Cy3 signals versus log RNA input was close to 1 for most probes with log signal >1.5 (Supplementary Fig. S1), which indicates proper scaling. The results confirmed accurate calibration of intensity signals across the entire range of values.
Unexpectedly, Cy5 signals of some probes on the microarray also showed strong positive dependence on RNA input despite the fact that the amount of Cy5-labeled UMR was constant in all the arrays (Supplementary Fig. S2). This effect was observed only when Cy3 signal was much stronger than Cy5 signal. The mechanism of this effect is unknown, but we hypothesize that it stems from the chain hybridization when free ends of Cy3-labeled mRNA bound to probes on the array can hybridize with Cy5-labeled mRNA either non-specifically or via rare antisense segments (termed ‘chain effect’). This effect may cause underestimation of gene expression difference between cell types if values are normalized by UMR signal. To avoid distortion, we did not use UMR for normalization for a small portion of probes where we expected chain effect.
Another challenge in interpretation of microarray measurements is that expression values often do not fall below some ‘background’ level, which most likely results from non-specific hybridization. If a gene has low expression and is measured close to probe’s background level, it may show little change in expression value after actinomycin D treatment, which can be erroneously viewed as evidence of high mRNA stability. To avoid these errors, we estimated mRNA decay rates only for sets of data with expression values that were either high, >2.5 in log10 scale, or substantially above the background, >2 fold.
2.5. Microarray data analysis by gene ontology terms
Analysis of gene ontology (GO) terms in a selected list of genes was done using the hypergeometric distribution and FDR ≤ 0.05 criterion using NIA Mouse Gene Index (ver. mm8) software.36 Only non-redundant genes with gene symbols were used for analysis. Because many GO categories are redundant (contain similar lists of genes), we adjusted the calculation of FDR in the following way. First, we identified all redundant pairs of GO terms that had a correlation of gene presence ≥0.7. Second, in the list of GO terms ordered by increasing P-values, pi, estimated from the hypergeometric distribution, GO term i was considered redundant, if it was redundant to at least one preceding term.
2.6. Real-time quantitative reverse transcriptase–polymerase chain reaction
Total RNAs were extracted from ES cells using Trizol™ (Invitrogen) and Phase lock gel™ (Eppendorf/Brinkman) columns according to the manufacturers’ protocols. RNAs were precipitated with isopropanol, washed with 70% ethanol and dissolved in DEPC dH2O. Primers for quantitative reverse transcriptase–PCR (qRT–PCR) were designed using the Vector NTI Advance 9.1 software (Invitrogen) and tested for SYBR Green chemistry using an established in-house protocol.37 Reactions were run on the ABI 7500 Sequence Detection Systems using the default cycling program, and data were processed using SDS 2.2 software (Applied Biosystems). Gene expression levels were normalized to a Cyclin D3 as an internal control and to total RNA amount.
3. Results and discussion
3.1. Measuring mRNA decay rates in mouse ES cells
To quantify mRNA decay rates in mouse ES cells we measured changes in gene expression with whole-genome microarrays in a time course (0, 1, 2, 4 and 8 h) after treating cells with actinomycin D. Experiments were done for two ES cell lines—MC1 (129S6/SvEvTac) and MC2-B6 (C57BL/6J) cultured in the standard condition in the presence of LIF. To increase the number of genes that can be assayed, we also carried out the analysis of MC1 ES cells undergoing differentiation into two different culture conditions—in the absence of LIF and in the presence of RA for 7 days. Expression data for all genes are available in Supplementary Table S1. Proper calibration of signal intensities in the full range of gene expression values was confirmed using hybridization of different RNA amounts to microarrays (see details in Section 2.2). Expression values for most genes were normalized using UMR, except for 1824 probes (4.3%) where normalization was not possible because UMR signal was low and unstable (see details in Section 2.4). Decay rates were estimated using linear regression of log-transformed data (Fig. 1A and B) and normalized by average expression change of 200 most stable genes (see details in Section 2.4; Supplementary Table S1).
Because the earlier work did not fully address the data reproducibility issue, where analysis was limited to estimating statistical (technical) error and validation of expression measurements by PCR for a small number of genes, we felt that it was important to evaluate the reproducibility of the measurements. To this end, we used the following two methods: (i) comparing mRNA decay rates in two independent experiments with different ES cell lines (MC1 and MC2-B6) cultured in the standard conditions and (ii) comparing mRNA decay rates estimated using different oligonucleotide probes on the microarray for the main transcript of the same gene. The first method yielded very high consistency between estimates of mRNA degradation rate (r = 0.925), indicating biological reproducibility of our database (Fig. 1C). The second method gave somewhat lower match (r = 0.744), but the correlation was still within the range expected from the comparison of gene expression data obtained with different array platforms (Fig. 1D).
Discrepancy between mRNA half-lives estimated using different oligonucleotide probes for the same gene was likely related to the biological complexity of mRNA processing rather than to the error in measurement. For example, probes located in 3′-UTR often showed shorter mRNA half-lives than probes located in the open reading frame (ORF) (Supplementary Fig. S3A). This may be caused by either alternative termination of transcription coupled with a differential degradation rate or active mRNA truncation at some early step of mRNA processing. Interestingly, some genes of histone H1 family had the opposite pattern of degradation: probes located in the short 3′-UTR showed longer half-lives than probes located in the ORF (Supplementary Fig. S3B). However, because of a high sequence similarity between histone genes and a partial cross-hybridization of probes, this effect may be an artifact.
To validate the microarray data, we carried out qRT–PCR analysis and found that qRT–PCR results matched well to those obtained by DNA microarrays for six out of seven selected genes (Supplementary Fig. S4). An exception was Sox17, whose degradation rates measured by qRT-PCR were much lower than those measured by microarrays. The closer inspection of probe locations revealed that all microarray probes for Sox17 were located in the 3′-UTR of the gene, whereas qRT-PCR primers were located in the ORF. This result was, therefore, consistent with differential degradation rates of Sox21 measured with probes located in 3′-UTR and ORF (Supplementary Fig. S3A).
3.2. Effect of alternative transcription and splicing on estimation of RNA stability
It is conceivable that the influence of probe locations on the estimation of mRNA stability goes beyond the aforementioned simple difference between 3′-UTR and ORF. For example, oligonucleotide probes recognized different transcripts, which are produced by alternative transcription start sites (TSSs), splicing and polyA signals, may lead to the estimation of different half-lives for the same gene. To determine the effect of alternative transcription and splicing on mRNA stability, we compared decay rates identified with the best probe from the main transcript of each gene with decay rates estimated using other probes that matched the same gene. Probes from alternatively spliced transcripts often showed shorter mRNA half-lives compared with the best probe in the main transcript (Fig. 1E). This effect was strongest for probes located in the retained introns of alternative transcripts. Similar effect of alternative splicing was reported in Arabidopsis; however, it was not statistically significant because of the small number of alternatively spliced forms captured by the array.32 Thus, we, for the first time, statistically confirm that alternative transcripts, especially those with retained introns, have shorter mRNA half-life compared with main transcripts.
This phenomenon can be simply explained by the possibility that alternative transcripts have a higher degradation rate due to specific sequences located in retained introns or alternatively spliced exons. Alternatively, it is also possible that the degradation of alternative transcripts appears to be accelerated by the splicing that may continue even after the blocking of transcription by actinomycin D. Because RNAs were extracted from whole cells in our experiments, it may contain heterogeneous nuclear RNA with some retained introns, which are able to hybridize with intron-matching probes in the array. If splicing continues for a while after the blocking of transcription by actinomycin D, the number of heterogeneous nuclear RNA would decrease. Further experiments are needed to distinguish between the two possible mechanisms. All subsequent analysis was done only for main transcripts of each gene. We would like to note that for 1391 genes, mRNA decay rates were not consistent between different microarray probes (>2-fold difference). Therefore, mRNA half-lives for these genes estimated with the best oligonucleotide should be considered with caution (marked by an asterisk in Supplementary Table S2).
3.3. General trends in stability of mRNAs in ES cells
We were able to estimate mRNA half-lives of 19 977 non-redundant genes (Supplementary Table S2). For 14 815 genes, estimates are available for two ES cell lines and three culture conditions. We were not able to estimate mRNA half-lives of 7260 genes due to the low level of expression in ES cells and those of 541 genes due to the lack of acceptable probes in the main transcript. Most of the genes had long half-lives, which was in concordance with published data.3–5 In our analysis, median estimated half-life for all genes was 7.1 h (Fig. 1F), which was less than reported for human hepatoma cells (10 h);4 however, this difference may be related to normalization method, as discussed below (see Section 3.9). In our analysis, only 54 genes (including Prdm1, Myc, Gadd45 g, Foxa2, Hes5 and Trib1) showed a half-life shorter than 1 h. Published data indicate that more genes showed RNA half-life shorter than 30 min.3 Therefore, it is possible that the number of genes with very short mRNA life may be underestimated in our study, because we did not collect samples at the time points earlier than 1 h.
3.4. Functional annotation of stable and unstable transcripts
We analyzed over representation of GO terms in the 610 genes with half-life shorter than 2 h and in 4310 genes with half-life longer than 12 h to identify functional categories of genes with unstable and stable mRNA, respectively (Supplementary Tables S3 and S4). Using GO database and additional information on protein domains, we manually assembled lists of genes with functions that were characteristic to stable and unstable mRNA species (Supplementary Table S2). The major functional categories of genes with unstable mRNA were ‘Regulation of transcription’, ‘Cell cycle’, ‘Apoptosis’, ‘Signal transduction’ and ‘Development’, whereas the major functional categories of genes with stable mRNA were ‘Metabolism’, ‘Protein biosynthesis’, ‘Extracellular matrix’ and ‘Cytoskeleton’ (Fig. 1G). Most of these results confirm findings published earlier;4,5,32 however, overrepresentation of ‘Extracellular matrix’ and ‘Cytoskeleton’ groups among stable mRNA species is reported here for the first time. Many transcription factors showed very short mRNA half-life, which may be important for the quick expression changes of target genes. Genes with half-life from 1 to 2 h (n = 557) included early response and cell cycle regulation and also some genes important for pluripotency and early development: for example, Sox2, Klf4, Foxd3, Sox7, Nodal, Ctgf, Id1, Id2, Id3 and Wnt signaling.
3.5. Strain-dependent stability of mRNA
In the previous study, we have shown that there are genes differentially expressed between ES cells derived from 129 mouse stain and ES cells derived from C57BL/6 mouse strain (strain-specific signature genes).38 We, therefore, asked if the observed differences were related to differences in mRNA half-life of corresponding genes. We found that a small number of genes (n = 92) had statistically significant differences in mRNA decay rates between MC1 (129S6/SvEvTac) and MC2-B6 (C57BL/6J) ES cells (FDR <0.05 and >2-fold change of half-life) (Supplementary Table S5) and only 10 of these genes overlapped with strain-specific signature genes. Although differential gene expression of at least some strain-specific genes can be explained partially by differences in mRNA stability, we were not able to identify correlation between differential gene expression and mRNA stability between strains on a global scale.
3.6. Alteration of mRNA half-life in differentiating ES cells
Differentiation of ES cells in the absence of LIF or in the presence of RA caused changes in mRNA stability in a large set of genes (n = 3332 in LIF− and n = 2257 in RA) (Fig. 2A and B, Supplementary Tables S6 and S7). These changes were much stronger than the strain difference in mRNA stability between MC1 and MC2-B6 ES cells (Fig. 1C). In general, decay rates of mRNA increased after LIF withdrawal: the median half-life declined from 7.07 to 5.48 h; and out of 3332 genes with significant change of mRNA stability, 77.9% had increased decay rates compared with undifferentiated ES cells (Fig. 2A and C). In contrast, differentiation of ES cells in RA conditions caused a general increase of mRNA stability: the median half-life increased from 7.07 to 8.60 h; and out of 2257 genes with significant change of mRNA stability, 87.1% had decreased decay rates compared with undifferentiated ES cells (Fig. 2B and C). This indicates a substantial difference between two protocols of ES cell differentiation. But despite these global differences, gene-specific changes in mRNA stability in these differentiating ES cells showed a good correlation: the change in decay rates of mRNA (in log scale) in RA conditions was positively correlated with the change of decay rates (in log scale) in LIF− conditions (r = 0.593) (Supplementary Fig. S5A). This means that changes in mRNA decay rates can be viewed as a superposition of two effects: (i) gene-specific effect that is consistent between LIF− and RA conditions and (ii) non-specific effect associated with the culture medium that caused general decrease in stability in LIF− conditions and general increase in mRNA stability in RA conditions.
Figure 2.

Change in mRNA decay rates in ES cells after differentiation. (A) Change of mRNA decay rates after 7 days of differentiation in LIF− conditions; significant changes (FDR <0.05 and >2-fold change of half-life) is shown by red and green dots; rates are log-transformed, log10(d + 0.1). (B) Change in mRNA decay rates after 7 days of differentiation in RA conditions. (C) Frequency distribution of mRNA half-life in undifferentiated MC1 cells (LIF+) compared with cells differentiated after LIF withdrawal (LIF−) and retinoic acid (RA) treatment. (D) Half-life of mRNA of genes associated with pluripotent state of ES cells in undifferentiated cells (LIF+) and upon differentiation (LIF− and RA).
To select genes whose mRNA were either destabilized or stabilized in differentiated cells in both LIF− and RA conditions, we used one standard deviation cut-off of log decay rates (Supplementary Fig. S5A). Genes with increased rate of mRNA decay upon differentiation were enriched in GO terms ‘transcription regulation’, ‘nervous system development’, ‘apoptosis’, ‘ubiquitin cycle’, ‘ribosome’ (mostly mitochondrial ribosomal proteins), ‘chromatin binding’ and ‘methyltransferase activity’ (Supplementary Table S8). Genes with decreased rate of mRNA decay upon differentiation were enriched in GO terms ‘extracellular space’, ‘extracellular matrix’, ‘calcium ion binding’, ‘actin cytoskeleton’ and ‘lysosome’ (Supplementary Table S9). During the ES cell differentiation, unstable mRNA species became even less stable, whereas stable mRNA species became more stable (Supplementary Fig. S5B).
It is believed that in general the differentiation of ES cells is associated with destabilization of pluripotency-related genes and stabilization of lineage-specific genes. Indeed, our data showed that mRNAs of some pluripotency-related genes (Sall4, Eed, Esrrb, Notch4, Mras, Tbx3, Btbd14b and Sap30) were destabilized upon differentiation (Fig. 2D). Interestingly, the strongest increase in mRNA decay rate was observed for Btbd14b (Nac1), which is a known cofactor of NANOG and POU5F1 proteins.39,40 Similarly, mRNAs of some differentiation-related genes (e.g. Bmp1, Acta1, Igfbp1, Igfbp4, Igfbp6, Frzb, Cited1, Flt1, Slit2, Krt8, Krt14, Cnn1, Mest, Wnt3 and Amot) were stabilized. On the other hand, the change in mRNA stability for all genes had no correlation with the change in gene expression levels (Supplementary Fig. S5C and D), indicating that mRNA stability has no global role in controlling gene expression changes during ES cells differentiation.
One way to control mRNA degradation is to modulate the expression levels of particular ribonucleases. In metazoans, it is known that the modulation of the expression of ribonucleases and/or associated factors play an important role in the control of mRNA stability during development.6 It has also been shown that many ribonucleases and associated factors are differentially regulated during development. For example, in Drosophila, the expression of ribonucleases pacman/XRN1, twister/SKI2 and tazman/DIS3/RRP44, varies during development at both RNA and protein levels.41–43 We, therefore, focused on genes that are involved in the mRNA degradation or binding and found that 103 genes changed their expression at >2-fold between undifferentiated and differentiated ES cells (Supplementary Table S10). The multiplicity of mRNA degradation pathways makes it difficult to relate expression of these genes with observed patterns of mRNA degradation. However, we noted that the expression levels of Lsm3, Lsm5, Lsm6, Lsm7, Lsm12, Lsmd1, Cnot4, Cnot10, Dcp2, Exosc1, which are known to promote mRNA degradation, decreased in the differentiated ES cells, especially in the RA condition. This may explain increased stability of mRNA in cells differentiated in the RA condition. In contrast, degradation-promoting genes—Zfp36l1 and Cnot6l—showed increased expression in differentiated ES cells, which may have contributed to lower mRNA stability in the LIF− condition, but it does not explain increased mRNA stability in the RA condition.
3.7. Multivariate analysis of gene structural features that affect mRNA stability
Decay rates of mRNA vary substantially between genes (Fig. 1F); however, little is known about factors and mechanisms responsible for these differences. Two major structural factors that affect mRNA stability are known: (i) the presence of introns in genes (i.e. multi-exon genes), which makes mRNA more stable;44 and (ii) the presence of AREs in the 3′-UTR of genes, which makes mRNA less stable.45,46 However, all the previous results have been obtained using single-factor analysis, which may easily lead to erroneous conclusions if factors are correlated. We, therefore, used multivariate regression analysis, which helps to distinguish direct causal effects from mere correlations. We incorporated as many structural characteristics of genes as possible, including the length of ORF, the length of 5′-UTR, the length of 3′-UTR, the number of exons, the occurrence of different kinds of ARE, the presence of CpG di-nucleotides and the presence of PUF binding motifs in all parts of the transcript (Supplementary Table S11). AREs were detected by using three types of reported consensus sequences: AUUUA (ARE1), UUAUUUAWW (ARE2), WWWUAUUUAUWWW (ARE3)45,47 and non-specific A/U 12-mers with no more than one mismatch (ARE4). We used UGUANAUA as PUF consensus motifs.29,48,49 CpG elements were added to the list after we unexpectedly found that they can affect mRNA stability. We did not analyze the effects of miRNA on the mRNA stability, because it would require extensive data mining and integration with the existing miRNA databases.
Multiple regression analysis showed that mRNA stability was correlated positively with the number of exons and negatively with the ORF length, number of ARE2, ARE4 and PUF in 3′-UTR, number of ARE4 and PUF in ORF, and number of CpG di-nucleotides in 5′-UTR (Supplementary Table S11). The effect of ORF length was nearly equal to the effect of the number of exons with the opposite sign, which indicated that the number of exon junctions per unit ORF length may be a better predictor of mRNA stability than the number of exons per gene. This is also supported by the fact that most exon junctions are located in the ORF region of genes. Indeed, log-transformed rate of mRNA decay correlated stronger with log-transformed number of exon junctions per unit ORF length (r = −0.326) than with log-transformed number of exons (r = −0.111). Therefore, below we always used the ratio of exon junctions per 1 kb of ORF length as a predictor of mRNA stability.
The final form of regression after removing non-significant terms for MC1 cells in the standard culture conditions (LIF+) was
| 2 |
where y is log-transformed rate of mRNA degradation, log10(b+0.1); x1 is log-transformed number of exon junctions per 1 kb ORF length; x2 is log-transformed ORF length; x3 is log-transformed number of ARE4 in 3′-UTR; x4 is log-transformed number of ARE4 in ORF; and x5 is log-transformed number of ARE2 in 3′-UTR; x6 is log-transformed number of CpG di-nucleotides in 5′-UTR; x7 is log-transformed number of PUF motifs in 3′-UTR; and x8 is log-transformed PUF motifs in ORF. Log-transformation was used because it increased the predictive power of the regression and made all regression coefficients dimensionless and comparable with each other; log10(n + 1) was used to handle zero counts as arguments. This regression accounted for 19.8% of variation in mRNA degradation rates, which was only slightly lower than the regression with all 19 factors (20.0%). Equation (2) was not precise enough to predict mRNA degradation rates for individual genes, but it worked well to estimate the average degradation rate in groups of genes identified in microarray experiments.
According to the regression equation [Equation (2)], we conclude that the number of exon junctions per unit ORF length shows the strongest and the most consistent effect on mRNA stability between cells types. This conclusion is further supported by the alternative data presentation shown in Fig. 3A. The proportion of genes with ≥5 exon junctions per unit ORF length was higher in genes with longer mRNA half-life, whereas the proportion of genes associated with the destabilizing factors (CpG di-nucleotides in 5′-UTR, and ARE4 and PUF in 3′-UTR) was higher in genes with shorter mRNA half-life (Fig. 3A). Although the role of introns in stability of mRNA has been reported both in experimental and statistical studies,44 here we present the first evidence that the number of exon junctions per unit ORF length is the most important known structural factor that increases mRNA stability. After intron is excised by spliceosome, the exon junction location is marked by binding of the exon junction complex (EJC), which consists of at least 10 proteins.50 EJC remains bound to mRNA after transport to cytoplasm, can stimulate translation, and mediate NMD of mRNA during the pioneer round of translation.50 It is indeed possible that some components of EJC can remain bound to mRNA after the first round of translation and decrease the rate of mRNA degradation.44 But the mechanism of this process remains unknown.
Figure 3.

Effect of structural and functional characteristics of genes on the rate of mRNA decay. (A) Proportion of genes with increased number of exon junctions, CpG di-nucleotides in 5′-UTR, and ARE4 and PUF motifs in 3′-UTR in groups of genes with different range of mRNA half-life. (B) Regression coefficients for the effect of structural elements (cis-factors) and gene function on the mRNA decay rate; abundance of structural elements and mRNA decay rates are log-transformed. (C) Correlation between functional characteristics of genes and abundance of structural elements (log-transformed).
According to the regression equation [Equation (2)], the second strongest factor affecting the mRNA stability was the number of PUF motifs in 3′-UTR, which decreased the mRNA stability. The third strongest factor affecting the mRNA stability was the presence of AREs. Unexpectedly, the sequence non-specific ARE4 in 3′-UTR (to a smaller extent, in ORF) decreased mRNA half-life more strongly than the consensus-matching AREs. Among different consensus matching AREs, only ARE2 in 3′-UTR showed a significant effect on mRNA stability. Several mechanisms are known to be involved in ARE-mediated mRNA degradation. For example, ARE-binding proteins can recruit PARN or exosomes.51 It is also known that the stability of mRNA can be modified by immune factors via AREs in 3′-UTR.52
The fourth strongest factor that affects mRNA stability was the number of CpG di-nucleotides in 5′-UTR. Because this was not known before, this is indeed the first report about the effect of CpG di-nucleotides on the mRNA stability. We suspected that the occurrence of CpG di-nucleotides in 5′-UTR may be correlated with the presence of CpG islands at TSS. We, therefore, estimated the number of CpG di-nucleotides near TSS (from −400 to +600 bp) and included it into the regression analysis. We found that the numbers of CpG di-nucleotides in 5′-UTR appeared to have a 5-fold stronger effect on mRNA stability than their numbers in the CpG island adjacent to TSS (Supplementary Table S12), which indicates that CpG di-nucleotides need to be located within 5′-UTR to affect mRNA decay. The mechanism of this effect is unknown, but considering a strong effect of splicing on mRNA stability we can hypothesize that it may be mediated by methyl CpG-binding proteins that regulate splicing.53
We have, thus far, shown that both functional categories of genes (Section 3.4) and the structural features of genes (this section) affect the mRNA stability. It will be, thus, interesting to see which factors affect mRNA stability more strongly. To this end, we carried out the regression analysis after including both structural features and functional categories (Fig. 3B, Supplementary Table S13). Among functional groups of genes, the strongest destabilizing effect was shown in the category of ‘Regulation of transcription’ followed by ‘Signal transduction’ and ‘Cell cycle’. In contrast, ‘Metabolism’, ‘Extracellular matrix’ and ‘Cytoskeleton’ functional categories increased mRNA stability, which is consistent with our previous results (Fig. 1G). However, we found that all the effects of structural factors listed in Equation (2) remained strong and statistically significant, and the sign of regression coefficients did not change after adding functional gene categories. This indicates that the stability of mRNAs is more significantly correlated with the structural features of genes than the function of genes.
Furthermore, the differential mRNA stability in various functional groups of genes can be partially explained by structural features of genes. For example, low stability of mRNA among genes in the functional category of ‘Regulation of transcription’ and ‘Development’ can be explained by the low number of exon junctions per ORF length and high number of ARE4 and PUF in 3′-UTR and CpG in 5′-UTR (Fig. 3C). Similarly, high stability of mRNA among genes in the category of ‘Metabolism’ can be explained by the high number of exon junctions per ORF length and low number of ARE4 and PUF in 3′-UTR and CpG in 5′-UTR (Fig. 3C). However, several functional categories of genes (e.g. ‘Regulation of transcription’ or ‘Extracellular matrix’) had a significant effect on mRNA stability based on regression (Fig. 3B). This indicates that the effect of gene function cannot fully be explained by structural characteristics of transcripts included in the regression.
As mentioned above, differentiation of ES cells in both LIF− and RA conditions caused changes in mRNA stability in a large set of genes (Fig. 3A and B, Supplementary Tables S6 and S7). To identify structural and functional factors that affected these global changes in mRNA stability, we compared the coefficients of multiple regression that related decay rates (log scale) with various factors in undifferentiated and differentiated ES cells in LIF− and RA conditions (Fig. 3B, Supplementary Table S13). Degradation-promoting effects of CpG di-nucleotides in 5′-UTR and ARE in 3′-UTR appeared much stronger in cells differentiated in the LIF− condition compared with undifferentiated cells and cells differentiated in the RA condition. Cells differentiated in the RA condition had weaker degradation-promoting effects of ARE4 in 3′-UTR than in undifferentiated cells. These effects may explain the increased mRNA decay rates in ES cells differentiated in LIF− and the decreased decay rates in ES cells differentiated in the RA condition. Protection of mRNA from degradation by exon junctions was stronger in both types of differentiated ES cells than in undifferentiated cells.
3.9. Comparison with human data
Interspecies comparison of mRNA decay rates has been done only among distant evolutionary lineages (plant, yeast and human). The new mouse data set provides an opportunity to compare the two mammalian species: mouse and human. Using Homologene database54 we identified 4153 pairs of orthologous genes for which mRNA rates were estimated both in mouse (this study) and human (Supplementary Table S3). For compatibility with our data, we converted estimates of mRNA decay rates in human from log2 to loge. Because human data are normalized using β-actin, whose mRNA is not highly stable (based on our data), a large set of human genes (11.6%) shows negative degradation rates.4 We, therefore, re-normalized the estimates of human mRNA decay rates using the method we used for mouse (by average decay rate of 200 genes with most stable mRNA). Median rate of mRNA decay for all orthologous genes in mouse (d = 0.098 h−1) was lower than in human (d = 0.137 h−1). Because these experiments differed not just in species (i.e. mouse versus human), but also in cell types (ES cells versus hepatoma), and were done in different laboratories using different microarray platforms, it was impossible to attribute this difference to any specific factor. However, correlation between mRNA decay rates in mouse and human was high (r = 0.610, Fig. 4), which was only slightly lower than correlation between estimates of mRNA decay rates obtained with different probes that matched the same mouse transcript (r = 0.744, see Section 3.1). This finding indicates that pathways of mRNA degradation are mostly conserved between mammalian species. Interestingly, mRNA decay rates in human hepatoma were more similar to decay rates in differentiated ES cells (r = 0.605 for LIF− and r = 0.616 for RA) than to decay rates in undifferentiated ES cells (r = 0.534, SE = 0.013, P = 0.0001). This may be explained by the fact that hepatoma cells are differentiated and, thus, the profile of mRNA decay rate in hepatoma matched better with that in differentiated ES cells. However, this similarity needs to be confirmed with further experiments that use the same cell types in both species.
Figure 4.

Comparison of mRNA decay rates for orthologous genes in mouse ES cells and in human hepatocytes (from the reference4) Rates are log-transformed, log10(d + 0.1). Human mRNA decay rates were re-normalized using average decay rate of 200 most unstable mRNA species for proper comparison.
3.10. Conclusion
In this paper we present a whole-genome analysis of mRNA decay rates in mouse ES cells. Decay rates were reliably measured for 19 977 non-redundant genes from time course of microarray data in undifferentiated ES cells originated from different mouse strains and in differentiated ES cells obtained by LIF withdrawal (LIF−) or RA treatment. The novelty of this study can be summarized as follows: (i) this is the first study of mRNA decay rates in mouse. (ii) This is the first study of mRNA decay rates in ES cells of any mammalian species. (iii) We have analyzed the difference in mRNA decay rates between cells derived from two different mouse strains and between undifferentiated and differentiated cells. (iv) The set of genes for which mRNA decay rates estimated is 3.8 times larger than in the most extensive earlier study for human. (v) Close match between data from different cell lines has confirmed high reproducibility of mRNA decay rate estimates. (vi) The microarrays have been calibrated. (vii) We have applied multivariate statistics for the analysis of factors affecting mRNA decay rates, which identifies direct effects and distinguishes them from mere correlations. (viii) We have explored the effect of various structural features of transcripts and gene function on mRNA stability in both general and during ES cell differentiation. The major result is that the number of exon junctions per ORF is the strongest factor promoting mRNA stability, whereas decay is promoted by CpG di-nucleotides in 5′-UTR region as well as by non-canonical ARE (without specific consensus sequence) in 3′-UTR and in ORF. We believe that the presented data on mRNA decay rates provides a valuable resource for future experiments in interpreting various gene manipulation and gene expression studies as well as the computational modeling and simulation on gene regulatory networks.
Supplementary data
Supplementary data are available online at www.dna.research.oxfordjournals.org.
Funding
The work was supported by the Intramural Research Program of the National Institute on Aging, NIH (Z01 AG000656).
Supplementary Material
Footnotes
Edited by Osamu Ohara
References
- 1.Raghavan A., Bohjanen P. R. Microarray-based analyses of mRNA decay in the regulation of mammalian gene expression. Brief Funct. Genomic Proteomic. 2004;3:112–124. doi: 10.1093/bfgp/3.2.112. [DOI] [PubMed] [Google Scholar]
- 2.Bolognani F., Perrone-Bizzozero N. I. RNA–protein interactions and control of mRNA stability in neurons. J. Neurosci. Res. 2008;86:481–489. doi: 10.1002/jnr.21473. [DOI] [PubMed] [Google Scholar]
- 3.Raghavan A., Ogilvie R. L., Reilly C., et al. Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res. 2002;30:5529–5538. doi: 10.1093/nar/gkf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang E., van Nimwegen E., Zavolan M., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam L. T., Pickeral O. K., Peng A. C., et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:research0041.1–research0041.11. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newbury S. F. Control of mRNA stability in eukaryotes. Biochem Soc. Trans. 2006;34:30–34. doi: 10.1042/BST20060030. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Martinez J., Aranda A., Perez-Ortin J. E. Genomic run-on evaluates transcription rates for all yeast genes and identifies gene regulatory mechanisms. Mol. Cell. 2004;15:303–313. doi: 10.1016/j.molcel.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Wang J., Chang Y. F., Hamilton J. I., Wilkinson M. F. Nonsense-associated altered splicing: a frame-dependent response distinct from nonsense-mediated decay. Mol. Cell. 2002;160:951–957. doi: 10.1016/s1097-2765(02)00635-4. [DOI] [PubMed] [Google Scholar]
- 9.Hollams E. M., Giles K. M., Thomson A. M., Leedman P. J. MRNA stability and the control of gene expression: implications for human disease. Neurochem. Res. 2002;27:957–980. doi: 10.1023/a:1020992418511. [DOI] [PubMed] [Google Scholar]
- 10.Sahmi M., Nicola E. S., Price C. A. Hormonal regulation of cytochrome P450 aromatase mRNA stability in non-luteinizing bovine granulosa cells in vitro. J. Endocrinol. 2006;190:107–115. doi: 10.1677/joe.1.06827. [DOI] [PubMed] [Google Scholar]
- 11.Gowrishankar G., Winzen R., Bollig F., et al. Inhibition of mRNA deadenylation and degradation by ultraviolet light. Biol. Chem. 2005;386:1287–1293. doi: 10.1515/BC.2005.146. [DOI] [PubMed] [Google Scholar]
- 12.Herruer M. H., Mager W. H., Raue H. A., Vreken P., Wilms E., Planta R. J. Mild temperature shock affects transcription of yeast ribosomal protein genes as well as the stability of their mRNAs. Nucleic Acids Res. 1988;16:7917–7929. doi: 10.1093/nar/16.16.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greatrix B. W., van Vuuren H. J. Expression of the HXT13, HXT15 and HXT17 genes in Saccharomyces cerevisiae and stabilization of the HXT1 gene transcript by sugar-induced osmotic stress. Curr. Genet. 2006;49:205–217. doi: 10.1007/s00294-005-0046-x. [DOI] [PubMed] [Google Scholar]
- 14.Benard L. Inhibition of 5′ to 3′ mRNA degradation under stress conditions in Saccharomyces cerevisiae: from GCN4 to MET16. RNA. 2004;10:458–468. doi: 10.1261/rna.5183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jona G., Choder M., Gileadi O. Glucose starvation induces a drastic reduction in the rates of both transcription and degradation of mRNA in yeast. Biochim. Biophys. Acta. 2000;1491:37–48. doi: 10.1016/s0167-4781(00)00016-6. [DOI] [PubMed] [Google Scholar]
- 16.Shock J. L., Fischer K. F., DeRisi J. L. Whole-genome analysis of mRNA decay in Plasmodium falciparum reveals a global lengthening of mRNA half-life during the intra-erythrocytic development cycle. Genome Biol. 2007;8:R134. doi: 10.1186/gb-2007-8-7-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herpin A., Rohr S., Riedel D., Kluever N., Raz E., Schartl M. Specification of primordial germ cells in medaka (Oryzias latipes) BMC Dev. Biol. 2007;7:3. doi: 10.1186/1471-213X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deschenes-Furry J., Perrone-Bizzozero N., Jasmin B. J. The RNA-binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticity. Bioessays. 2006;28:822–833. doi: 10.1002/bies.20449. [DOI] [PubMed] [Google Scholar]
- 19.Ratti A., Fallini C., Cova L., et al. A role for the ELAV RNA-binding proteins in neural stem cells: stabilization of Msi1 mRNA. J. Cell Sci. 2006;119:1442–1452. doi: 10.1242/jcs.02852. [DOI] [PubMed] [Google Scholar]
- 20.Figueroa C., Vojtek A. B. Akt negatively regulates translation of the ternary complex factor Elk-1. Oncogene. 2003;22:5554–5561. doi: 10.1038/sj.onc.1206496. [DOI] [PubMed] [Google Scholar]
- 21.Champelovier P., Pautre V., Elatifi M., et al. Resistance to phorbol ester-induced differentiation in human myeloid leukemia cells: a hypothetic role for the mRNA stabilization process. Leuk. Res. 2006;30:1407–1416. doi: 10.1016/j.leukres.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Benjamin D., Moroni C. mRNA stability and cancer: an emerging link?, Expert Opin. Biol. Ther. 2007;7:1515–1529. doi: 10.1517/14712598.7.10.1515. [DOI] [PubMed] [Google Scholar]
- 23.Heier C. R., Gogliotti R. G., DiDonato C. J. SMN transcript stability: could modulation of messenger RNA degradation provide a novel therapy for spinal muscular atrophy? J. Child Neurol. 2007;22:1013–1018. doi: 10.1177/0883073807305669. [DOI] [PubMed] [Google Scholar]
- 24.Stoecklin G., Mayo T., Anderson P. ARE-mRNA degradation requires the 5′–3′ decay pathway. EMBO Rep. 2006;7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Hir H., Gatfield D., Izaurralde E., Moore M. J. The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Hir H., Izaurralde E., Maquat L. E., Moore M. J. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon–exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamasaki S., Stoecklin G., Kedersha N., Simarro M., Anderson P. T-cell intracellular antigen-1 (TIA-1)-induced translational silencing promotes the decay of selected mRNAs. J. Biol. Chem. 2007;282:30070–30077. doi: 10.1074/jbc.M706273200. [DOI] [PubMed] [Google Scholar]
- 28.Goldstrohm A. C., Hook B. A., Seay D. J., Wickens M. PUF proteins bind Pop2p to regulate messenger RNAs. Nat. Struct. Mol. Biol. 2006;13:533–539. doi: 10.1038/nsmb1100. [DOI] [PubMed] [Google Scholar]
- 29.White E. K., Moore-Jarrett T., Ruley H. E. PUM2, a novel murine puf protein, and its consensus RNA-binding site. RNA. 2001;7:1855–1866. [PMC free article] [PubMed] [Google Scholar]
- 30.Kennell D. Processing endoribonucleases and mRNA degradation in bacteria. J. Bacteriol. 2002;184:4645–4657. doi: 10.1128/JB.184.17.4645-4657.2002. Discussion 4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobell H. M. Actinomycin and DNA transcription. Proc. Natl Acad. Sci. USA. 1985;82:5328–5331. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narsai R., Howell K. A., Millar A. H., O’Toole N., Small I., Whelan J. Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana. Plant Cell. 2007;19:3418–3436. doi: 10.1105/tpc.107.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegmuller D., Raineri I., Gross B., Oakeley E. J., Moroni C. A cassette system to study embryonic stem cell differentiation by inducible RNA interference. Stem Cells. 2007;25:1178–1185. doi: 10.1634/stemcells.2006-0106. [DOI] [PubMed] [Google Scholar]
- 34.Carter M. G., Sharov A. A., VanBuren V., et al. Transcript copy number estimation using a mouse whole-genome oligonucleotide microarray. Genome Biol. 2005;6:R61. doi: 10.1186/gb-2005-6-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamini Y., Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 36.Sharov A. A., Dudekula D. B., Ko M. S. Genome-wide assembly and analysis of alternative transcripts in mouse. Genome Res. 2005;15:748–754. doi: 10.1101/gr.3269805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter M. G., Hamatani T., Sharov A. A., et al. In situ-synthesized novel microarray optimized for mouse stem cell and early developmental expression profiling. Genome Res. 2003;13:1011–1021. doi: 10.1101/gr.878903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharova L. V., Sharov A. A., Piao Y., et al. Global gene expression profiling reveals similarities and differences among mouse pluripotent stem cells of different origins and strains. Dev. Biol. 2007;307:446–459. doi: 10.1016/j.ydbio.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J., Rao S., Chu J., et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 40.Wang J., Levasseur D. N., Orkin S. H. Requirement of Nanog dimerization for stem cell self-renewal and pluripotency. Proc. Natl Acad. Sci. USA. 2008;105:6326–6331. doi: 10.1073/pnas.0802288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cairrao F., Arraiano C., Newbury S. Drosophila gene tazman, an orthologue of the yeast exosome component Rrp44p/Dis3, is differentially expressed during development. Dev. Dyn. 2005;232:733–737. doi: 10.1002/dvdy.20269. [DOI] [PubMed] [Google Scholar]
- 42.Seago J. E., Chernukhin I. V., Newbury S. F. The Drosophila gene twister, an orthologue of the yeast helicase SKI2, is differentially expressed during development. Mech. Dev. 2001;106:137–141. doi: 10.1016/s0925-4773(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 43.Till D. D., Linz B., Seago J. E., et al. Identification and developmental expression of a 5′–3′ exoribonuclease from Drosophila melanogaster. Mech. Dev. 1998;79:51–55. doi: 10.1016/s0925-4773(98)00173-7. [DOI] [PubMed] [Google Scholar]
- 44.Zhao C., Hamilton T. Introns regulate the rate of unstable mRNA decay. J. Biol. Chem. 2007;282:20230–20237. doi: 10.1074/jbc.M700180200. [DOI] [PubMed] [Google Scholar]
- 45.Chen C. Y., Shyu A. B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 46.Shaw G., Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- 47.Bakheet T., Frevel M., Williams B. R., Greer W., Khabar K. S. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie X., Lu J., Kulbokas E. J., et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cora D., Di Cunto F., Caselle M., Provero P. Identification of candidate regulatory sequences in mammalian 3′ UTRs by statistical analysis of oligonucleotide distributions. BMC Bioinformatics. 2007;8:174. doi: 10.1186/1471-2105-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang Y. F., Imam J. S., Wilkinson M. F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 51.Garneau N. L., Wilusz J., Wilusz C. J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 52.Tebo J., Der S., Frevel M., Khabar K. S., Williams B. R., Hamilton T. A. Heterogeneity in control of mRNA stability by AU-rich elements. J. Biol. Chem. 2003;278:12085–12093. doi: 10.1074/jbc.M212992200. [DOI] [PubMed] [Google Scholar]
- 53.Young J. I., Hong E. P., Castle J. C., et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl Acad. Sci. USA. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wheeler D. L., Barrett T., Benson D. A., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.