

Abstract
Renal hypertrophy and deposition of extracellular matrix proteins are consistent findings in diabetic nephropathy and these processes can be halted or reversed by euglycemic control. Using DNA microarray analysis of glomerular RNA from control and diabetic rats we found that the expression levels of insulin-like growth factor 1 receptor (IGF-1R) were increased while those of suppressor of cytokine signaling 2 (SOCS2) and STAT5 were decreased. All of these changes were normalized by islet cell transplantation. Overexpression of SOCS2 in rat mesangial cells inhibited IGF-1-induced activation of extracellular signal-regulated kinase, which subsequently reduced type IV collagen and DNA synthesis, an effect due to interaction of SOCS2 with IGF-1R. Inhibition of SOCS2 overexpression by small interfering RNA suppressed IGF-1R-mediated actions by preventing phosphorylation of tyrosine 317 in the p66Shc adaptor protein; however, overexpression of either SOCS1 or SOCS3 did not affect IGF-1R signaling. Insulin directly increased STAT5 and SOCS2 expression in mesangial cells. This study shows that insulin can inhibit the mitogenic action of IGF-1 in mesangial cells by regulating STAT5/SOCS2 expression. Insulin deficiency may contribute to the mesangial expansion found in diabetes through reduced STAT5/SOCS2 expression.
Keywords: diabetes, insulin, SOCS2, STAT, signaling, nephropathy
Diabetic nephropathy is a leading cause of end-stage renal failure,1 and the initial phases are characterized by the activation and proliferation of glomerular and renal tubular cells, resulting in enlarged kidneys. These pathological changes involve renal cellular proliferation and increased expression of extracellular matrix (ECM) proteins. A combination of nephromegaly and mesangial expansion leads to glomerulosclerosis and tubulointerstitial fibrosis, contributing to renal failure.2,3 Many growth factors, cytokines and vasoactive hormones can affect mesangial cell proliferation and synthesis of ECM proteins, and abnormal regulation of the expression of these molecules has been demonstrated in the early stages of diabetic nephropathy.4 Amongst these, growth hormone and insulin-like growth factor-1 (IGF-1) have been implicated due to their mitogenic actions on glomerular and tubular cells. However, the potential mechanisms used by these growth factors to induce the renal phenotype are not clear, as these growth factors do not display consistent elevated levels of expression in renal tissues.5 Thus, the exact mechanism by which hyperglycemia or diabetes can increase IGF-1's action on glomerular cells and upregulate the expression of extracellular proteins in glomerular tissues is unclear.
We have thus used a DNA microarray to identify changes in the expression of various genes in the glomeruli from control rats, untreated diabetic rats and diabetic rats receiving islet cell transplantation (ICT). ICT was chosen to be the treatment because it is the most effective method to restore normal physiology in the diabetic state and has been reported to partially reverse the glomerular pathology in diabetic patients.6 From the panel of changes in gene expression that can be induced by diabetes and subsequently normalized by ICT, we have identified that the mRNA expression of IGF-1R and suppressor of cytokine signaling 2 (SOCS2) were consistently altered. Increased expression of IGF-1R has been reported in diabetes and can induce ECM protein synthesis in mesangial cells. However, the plasma levels and the expression levels of IGF-1 in the renal parenchyma may be decreased in diabetes with poor glycemic control.7 However, it was the novel finding that the SOCS2 mRNA expression was also decreased that suggested a potential regulatory mechanism by which the action of IGF-1 can be enhanced to cause glomerular abnormalities.
SOCS2 is a member of SOCS family, a group of related proteins implicated in the negative regulation of cytokine action through inhibition of the Janus kinase/signal transducers and activators of transcription (STAT) signal-transduction pathway.8 Several SOCS isoforms have been reported to alter tyrosine kinase activity of growth factor receptors.9-12 SOCS2 may have a negative regulatory role in IGF-1 signaling because it has been reported to associate with IGF-1R and decrease its biological actions. SOCS2-deficient mice exhibit accelerated postnatal growth resulting in gigantism.13,14 However, there have been no reports about metabolic regulation of SOCS2 on IGF-1 signaling through IGF-1R in pathological states such as diabetic nephropathy. In addition, the possibility of insulin's role in the regulation of SOCS2 and inhibition of the mitogenic effects of IGF-1 has also not been shown.
In this study, we have identified a decrease in the expression of SOCS2 in both renal glomeruli commonly involved in diabetic microvascular complications. Furthermore, we have characterized in detail the interaction between SOCS2 and IGF-1R as well as the potential cellular signaling pathways that these two molecules utilize to the expression of ECM proteins and DNA synthesis in mesangial cells.
RESULTS
Physiological phenotypes of control, diabetic, and islet cell transplant-treated diabetic rats
Three groups of rats were used for this study including nondiabetic control rats, diabetic rats, and diabetic rats that received ICT 4 weeks after the onset of diabetes.15 All rats were sacrificed 8 weeks after the onset of diabetes for analysis. Diabetic rats displayed hyperglycemia (339±88 mg/100 ml versus 95±6 mg/100 ml for control rats) that was completely normalized by ICT (105±9 mg/100 ml; Supplementary Figure 1). Rats with 8 weeks of diabetes have increased kidney/body weight (100 gram) ratio (0.99±0.12 versus 0.81±0.08 of controls, P<0.05, n=20) that was rectified following euglycemic control by 4 weeks of ICT (0.84±0.07, P<0.05 versus diabetic group; Figure 1a). The presence of nephromegaly is associated with alterations of kidney function such as increased glomerular filtration rate (GFR; 3.15±0.74 ml/min in diabetic rats versus 1.75±0.42 ml/min in control rats, P<0.05) and calculated filtration fraction (0.34±0.07 in diabetic rats versus 0.22±0.05 in control rats, P<0.05; Figure 1b–d). These mimic the abnormal renal functions in diabetic patients with poor glycemic control.16 All of these changes were reversed after 4 weeks of euglycemic controls by ICT (GFR of 2.29±0.64 ml/min and filtration fraction of 0.22±0.06; Figure 1b–d), suggesting that they are specific to diabetes.
Figure 1. Effects of islet cell transplantation (ICT) on the physiological parameters in diabetic rats (STZ).
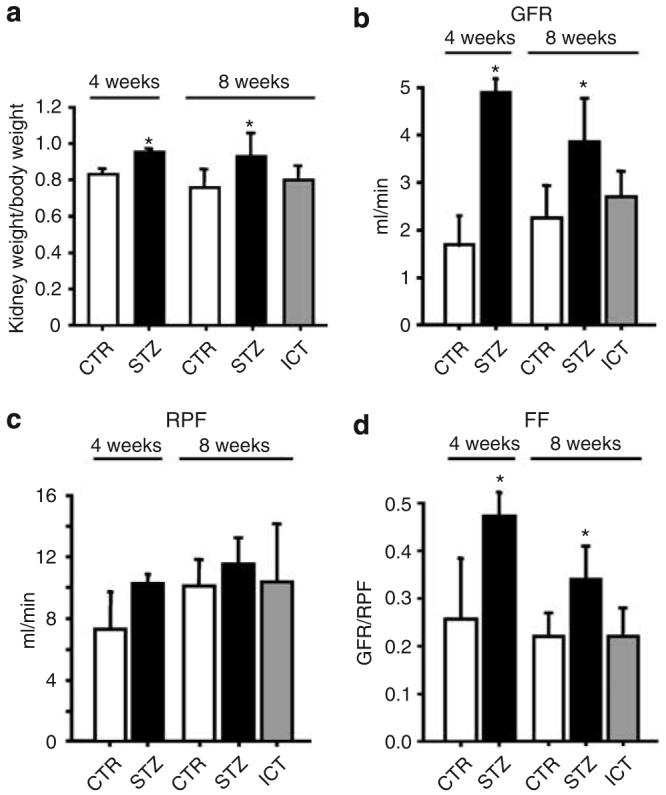
(a) Kidney weight/100 g body weight was increased in STZ-treated group and can be reversed by ICT (n=20, mean±s.d., *P<0.05 as compared to control (CTR) rats). (b) Glomerular filtration rate (GFR) was significantly increased in rats with diabetes for 4 and 8 weeks (P<0.05, n=5–7 rats per group), respectively, that was normalized by ICT. (c) Renal plasma flow (RPF). (d). Calculated filtration fraction (FF) was increased in rats with diabetes for 4 or 8 weeks (P<0.05, n=5–7 rats per group), respectively, and was normalized by ICT.
Expression profiling of renal glomeruli
Following the demonstration of renal dysfunction and enlargement in this diabetic rat model, we profiled the gene expression pattern in the glomerular tissues from the three groups of rats.17 Using a fivefold change as the cutoff criterion, 148 genes were identified to have increased expression in diabetes whereas the mRNA 223 other genes was downregulated. From these genes, the expressions of 32 upregulated and 69 downregulated genes were normalized by ICT (Supplementary database). Among the genes with altered expressions, SOCS2 were selected for further investigation because it had not been associated with diabetic renal complications or any other vascular pathologies, and it is associated with the action of IGF-1 and cellular growth which could be important for renal hypertrophy and mesangial expansion in early diabetic nephropathy.
The results of the DNA microarray analysis also identified a consistent 1.5-fold increase of IGF-1R in the renal glomeruli of STZ rats, which remained elevated with ICT treatment (Figure 2a). The mRNA expression of IGF-1 was not altered by diabetes. The changes in IGF-1R and SOCS2 mRNA expression detected by microarray analysis were confirmed by reverse transcription (RT)–PCR using total RNA from glomeruli of all three groups of rats (Figure 2b). The protein expressions of IGF-1R and SOCS2 in the glomeruli of three groups were confirmed by immunoblot analysis. IGF-1Rβ protein expression was elevated in glomeruli of STZ by 48% (n=11, P<0.05) that was completely normalized to basal level after 4 weeks of ICT (Figure 2c). Similarly, SOCS2 protein expression in the glomeruli of STZ rats was decreased by 30% (P<0.05, n=4; Figure 2d), in parallel to changes in mRNA levels as shown by microarray study. Islet cell transplant also revised the decrease in SOCS2 protein expression (Supplementary Figure 2). Immunohistochemical studies were performed and presence of SOCS2 was observed in the glomeruli (data not shown). The possible expression of SOCS2 by the mesangial cells was further studied. The effects of insulin and hyperglycemia on the expressions of IGF-1Rβ and SOCS2 protein levels in cultured rat mesangial cells were also determined (Figure 2e). Increasing extracellular glucose concentrations from 5.6 to 27.8 mm enhanced IGF-1R protein expression by 69% (n=6, P<0.05). Addition of 100 nm of insulin for 3 days in media containing 27.8 mm glucose decreased IGF-1R to that of 5.6 mm. Unlike IGF-1R, the expression of SOCS2 protein was increased significantly by co-incubation with 100 nm of insulin and 27.8 mm of glucose in mesangial cells. Elevating glucose levels alone from 5.6 to 27.8 mm did not have any effect on SOCS2 protein expression in the mesangial cells.
Figure 2. Diabetes altered the glomerular expression of IGF-1R and SOCS2 in vivo and in vitro.

(a) Affymatrix analysis revealed increased IGF-1R expression in glomeruli from diabetic rats (STZ) and diabetic rats treated with ICT. By contrast, SOCS2 expression was reduced in diabetic glomerular tissues but was normalized in rats treated with ICT. (b) RT–PCR confirmed the mRNA expression changes as detected by microarray analysis. (c) Immunoblotting using antibodies against IGF-1R and SOCS2 confirmed the changes in the expression of IGF-1Rβ and SOCS2 in diabetic glomerular tissues (n=11, mean±s.d., *P<0.05). (d) Expression of SOCS2 protein in glomerular tissues isolated from rats with (DM) or without (Ctl) 8 weeks of strepozotocine-induced diabetes (n=4, P<0.05). (e). Protein expression of IGF-1R and SOCS2 in isolated mesangial cells cultured in media containing either 5.5 or 27.8 mm glucose with or without 100 nm insulin (n=3–6, mean±s.d., *P<0.05).
The effect of diabetes on decreasing SOCS2 expression in the renal glomeruli appears to be specific because the protein expression of both SOCS1 and SOCS3 was not altered in diabetes (data not shown).
Effects of SOCS2 on IGF-1-induced DNA synthesis and type IV collagen expression in mesangial cells
The proliferation of mesangial cells and the excessive accumulation of ECM proteins such as type IV collagen contribute to glomerular or mesangial expansion and nephromegaly.4 We tested the possibility that changes in SOCS2 expression can alter IGF-1's biological actions. SOCS2 was overexpressed using adenoviral vectors, and its effects on IGF-1-induced collagen production and cell proliferation were studied. Infection of cultured mesangial cells by adenoviral vector containing SOCS2 (multiplicity of infection of 100–400) resulted in a dose-dependent increase in SOCS2 protein expression (Figure 3a). To differentiate this induction is due to exogenously expressed SOCS2 rather than upregulation of endogenous SOCS2, recombinant SOCS2 protein was fused to a hemagglutinin (HA) tag which subsequently demonstrated that the increase in SOCS2 was derived from adenoviral vector infection (Figure 3a). Infection with adenoviral vector containing green fluorescent protein (GFP) was used as a control. As shown in Figure 3, IGF-1 (50 nm) significantly increased the 5-bromo-2-deoxyuridine (BrdU) incorporation, a surrogate marker for DNA synthesis in mesangial cells by approximately twofold (n=6, P<0.05). The overexpression of SOCS2 protein reduced IGF-1's action on BrdU incorporation by 30% (n=6, P<0.05 versus GFP+IGF-1; Figure 3b). Induction of SOCS2 protein in mesangial cells by adenoviral infection did not change DNA synthesis in mesangial cells without the addition of IGF-1 (Figure 3b).
Figure 3. Overexpression of SOCS2 in cultured mesangial cells suppressed IGF-1-induced collagen IV synthesis and cellular proliferation.
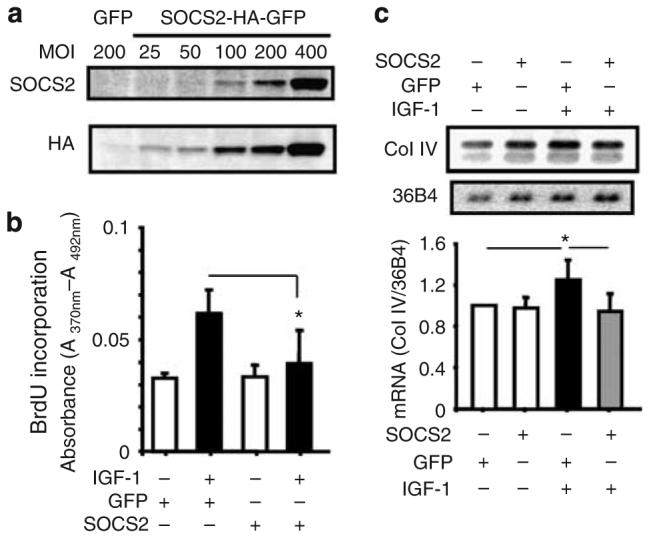
(a) Exogenous SOCS2 was introduced into mesangial cells using adenoviral vectors containing SOCS2 cDNA-tagged with HA. Immunoblotting displayed the presence of SOCS2 and HA protein in mesangial cells. (b) Transduction of adenoviral vector containing SOCS2 (MOI=400) inhibited IGF-1-induced BrdU incorporation (n=3–6, mean±s.d., *P<0.05 versus IGF-1/GFP 400). Mesangial cells were infected with SOCS2 or GFP expressing adenovirus, incubated with IGF-1 (50 nm) for 16 hrs, and assayed for incorporation of BrdU. (c) mRNA expression of collagen IV detected by northern blotting analysis in mesangial cells infected with SOCS2 adenoviral vectors with or without 50 nm IGF-1 for 6 h. Expression of collagen IV was normalized to 36B4 mRNA and quantified in the lower panel (n=4, mean±s.d., *P<0.05).
Furthermore, IGF-1 increased the mRNA expression of collagen IV in cultured mesangial cells by 1.3-fold as detected by northern blotting following the normalization to the expression of housekeeping gene 36B4 (n=4, P<0.05 versus unstimulated control cells; Figure 3c). Overexpression of SOCS2 totally suppressed IGF-1's induction on collagen IV mRNA expression (Figure 3c).
The inhibitory effect of SOCS2 on IGF-1's action is apparently isoform specific because overexpressions of SOCS1 and SOCS3, respectively, to a similar level in mesangial cells, did not block IGF-1-induced BrdU incorporation in mesangial cells (data not shown).
Effects of gene silencing of endogenous SOCS2 on IGF-1's action
As overexpression of SOCS2 suppressed the effects of IGF-1 on mesangial cell proliferation and ECM protein production, we further investigated whether downregulation of SOCS2, similar to that in diabetic glomerular tissues, can enhance IGF-1's action. RNAi that selectively targets to SOCS2 but not other isoforms was designed. As shown in Figure 4, transfection with SOCS2 RNAi decreased the protein expression of exogenously overexpressed SOCS2 by 80% whereas the expression of SOCS1 and three isoforms were not changed (Figure 4a). Downregulation of SOCS2 increased IGF-1-induced extracellular signal-regulated kinase (ERK)1/2 phosphorylation by 40%. Interestingly, the reduction of SOCS2 did not alter the expression of ERK at the basal states (Figure 4b). In contrast to SOCS2 overexpression, gene silencing of SOCS2 significantly enhanced IGF-1-induced mesangial cell proliferation, as measured by BrdU incorporation compared to control cells infected with adv-GFP by 40% (n=6, P<0.05; Figure 4c).
Figure 4. Effects of SOCS2 knockdown by RNAi on IGF-1 signaling in mesangial cells.
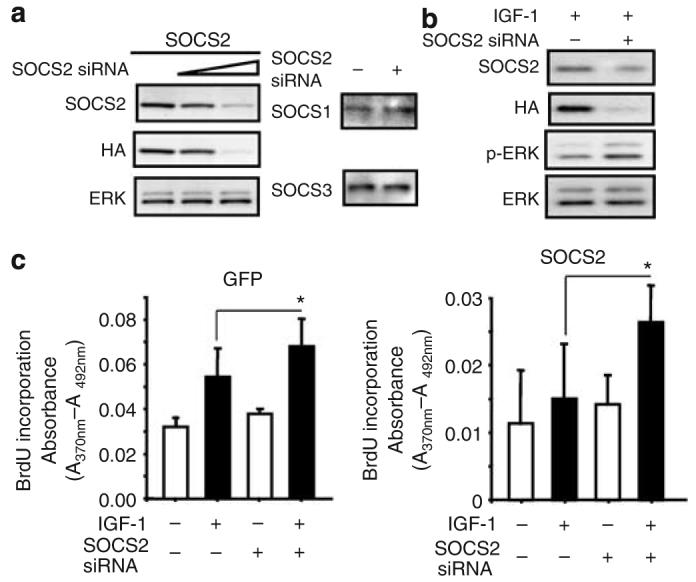
(a) SOCS2 RNAi decreased SOCS2 protein expression in cultured mesangial cells but did not changed the expression of SOCS1 and SOCS3 isoforms. Cells were transfected with SOCS2 RNAi and infected with SOCS2-adenovirus as indicated in the methods section. The levels of SOCS2, HA, and ERK1 (control) were determined by immunoblotting with anti-SOCS2, anti-HA, and anti-ERK1 antibodies, respectively. (b) Downregulation of SOCS2 enhanced IGF-1-stimulated phosphorylation of ERK1/2. (c) SOCS2 RNAi increased IGF-1-induced BrdU incorporation in GFP (left) or SOCS2 (right) adenoviral infected cells (n=6, P<0.05 comparing IGF-1-treated cells with or without the transfection of SOCS2siRNA).
Effect of SOCS isoforms on the intracellular signaling pathway of IGF-1
Although SOCS2 overexpression completely inhibited IGF-1-induced phosphorylation of ERK1/2 (Figure 5), this action is specific to SOCS2 because overexpression of SOCS1/3 isoforms respectively failed to inhibit ERK1/2 phosphorylation in mesangial cells induced by IGF-1 (Figure 5). IGF-1 can also activate phosphatidylinositol 3 (PI3) kinase/Akt pathway, however this pathway is not regulated by SOCS because overexpression of SOCS1–3 isoforms did not change the phosphorylation of Akt, a surrogate marker for the PI3 kinase/Akt pathway, with or without IGF-1 (data not shown).
Figure 5. Effects of SOCS2 protein overexpression on IGF-1-induced ERK1/2 phosphorylation.
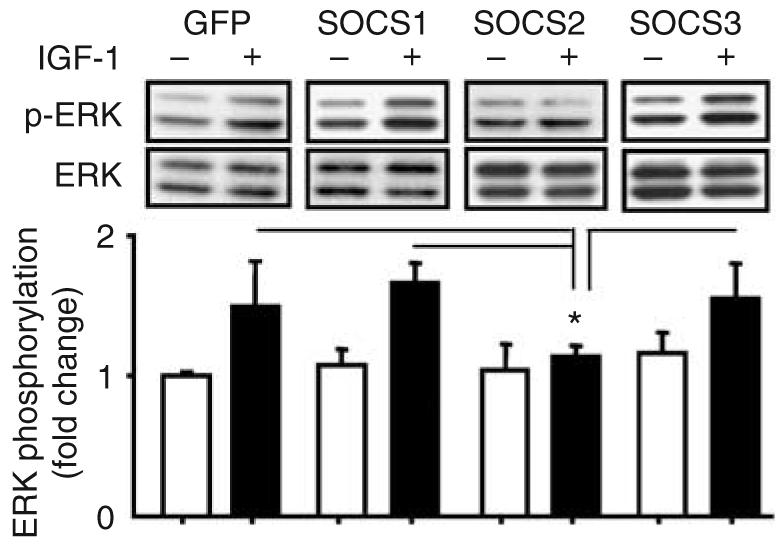
SOCS2 protein overexpression inhibited IGF-1-induced ERK1/2 phosphorylation whereas SOCS1 or 3 protein overexpression did not have this inhibitory effects (n=3, mean±s.d., *P<0.05 versus others with IGF-1 stimulation).
Shc has been reported as one of IGF-1R's substrates and is upstream to ERK phosphorylation and activation.18 In mesangial cells, Shc phosphorylation at Tyr239/240/317 was not changed at basal states without the addition of IGF-1 to cells infected with ad-GFP or SOCS2-HA-GFP. However, addition of 50 nm IGF-1 for 5 min increased p66Shc phosphorylation at Tyr317 (n=5, P<0.05) in GFP adenoviral vector-infected mesangial cells but the phosphorylation of Tyr239/240 site was unchanged (n=5, nonstatistical significant; Figure 6a and b). Interestingly, overexpression of SOCS2 (multiplicity of infection=400) reduced IGF-1-induced phosphorylation of Tyr317 of p66Shc to basal states (n=4, P<0.05 versus IGF-1-stimulated GFP-infected cells, nonstatistical versus GFP or SOCS2-infected mesangial cells). We have also evaluated tyrosine phosphorylation at 239/240/317 sites of p52Shc. Western blotting revealed SOCS2 overexpression did not alter the basal expression of Tyr239/240/317 in mesangial cells, nor did it affect IGF-1's action on these residues (Figure 6a and b). This study suggests that phosphorylation of p66Shc at Tyr317, but not Tyr239/240 is relevant to the downstream effects of IGF-1 in mesangial cells. In addition, this conclusion is further supported by studies downregulating SOCS2 expression using RNAi. We have shown a significant induction of phosphorylation of p66Shc at Tyr317 that was partially inhibited by the overexpression of SOCS2 (Supplementary Figure 3).
Figure 6. Effects of SOCS2 protein overexpression on IGF-1-induced phosphorylation of Shc.
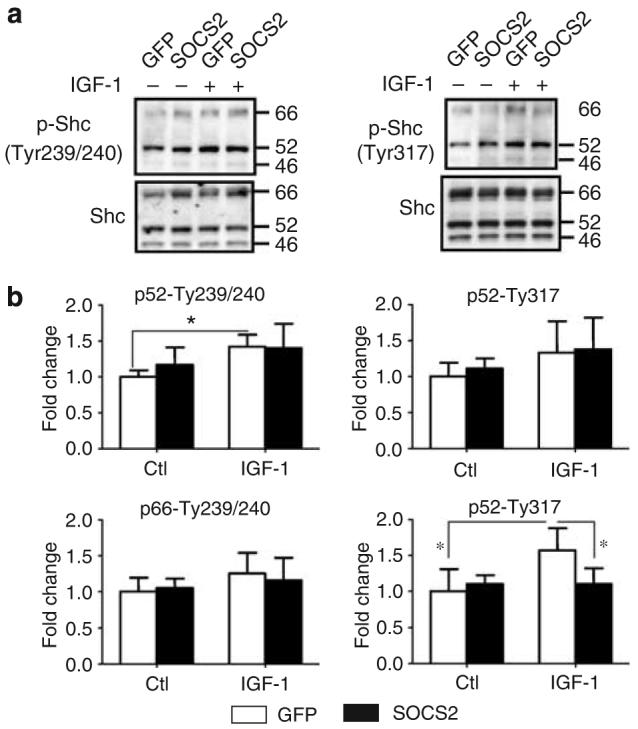
(a) Overexpression of SOCS2 protein inhibits IGF-1-induced phosphorylation of p66Shc at Tyr317 but not at Tyr239/240. Phosphorylation and protein levels were determined by immunoblotting with appropriate antibodies, as indicated. Cells were also stimulated with IGF-1 (50 nm) for 5 min. (b) Phosphorylation of Tyr317 as well as Tyr239/240 of both p66Shc and p52Shc were quantified and plotted as bar graph (n=4–11, P<0.05).
Direct interaction between SOCS2 and IGF-1 receptor
The potential association of SOCS2 with the IGF-1 receptor to mediate its actions was characterized in mesangial cells overexpressing SOCS2. Direct interaction between SOCS2 and IGF-1Rβ was determined by co-immunoprecipitation studies. Cell lysate of mesangial cells infected either with adenoviral GFP or adv-SOCS2-HA-GFP were immunoprecipitated using SOCS2 antibody. The resultant immunoprecipitates were analyzed further by immunoblot analysis using antibodies against HA, revealing the presence of exogenously expressed SOCS2 protein at 26 kDa in SOCS2-infected cells with or without IGF-1 but not in control cells. Interestingly, antibodies to IGF-1Rβ did not detect a significant signal in the immunoprecipitants without the addition of IGF-1. However, IGF-1Rβ subunit was clearly detected in SOCS2-infected cells following the addition of 50 nm IGF-1 (Figure 7). The interaction between SOCS2 and IGF-1Rβ was further confirmed by immunoprecipitation with antibody against IGF-1Rβ and then immunoblotted with HA antibody. Similarly, a strong signal was detected in the immunoprecipitant from SOCS2-overexpressing mesangial cells treated with IGF-1 whereas a much weaker signal in SOCS2-infected cells without IGF-1 (Figure 7). Control cells infected with viruses containing GFP did not show the presence of HA-tagged SOCS2 (Figure 7). These data clearly demonstrate that SOCS2 and IGF-1Rβ are part of the same protein complex in the presence of IGF-1 in mesangial cells.
Figure 7. SOCS2 interacted with IGF-1R as determined by co-immunoprecipitation.
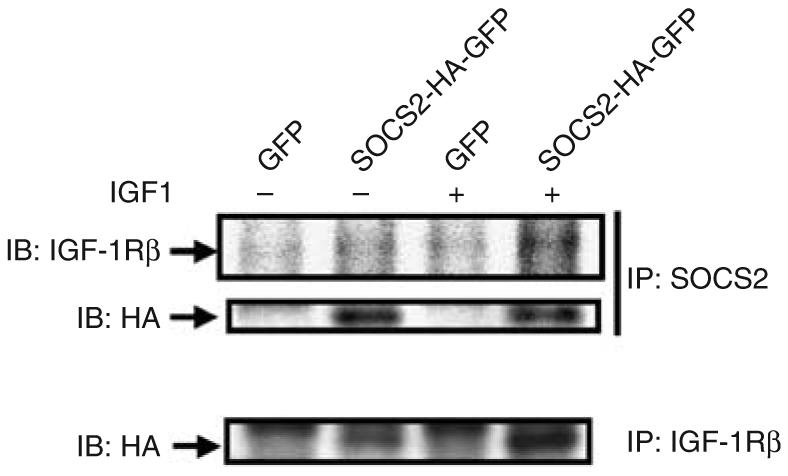
Protein lysates from mesangial cells infected with either GFP adenoviral control or SOCS2 were immunoprecipitated using antibodies against SOCS2 (top panel) or IGF-1Rβ (bottom panel) and then immunoblotted with antibodies against IGF-1Rβ or HA respectively as indicated.
Effect of SOCS1, 2, and 3 on insulin's signal and effects in mesangial cells
As insulin and IGF-1 share extensive similarities in cellular actions,19 and insulin has also been known to induce cellular DNA synthesis, we investigated the effects of SOCS proteins on insulin's action on mesangial cells. Insulin (100 nm) was also able to increase BrdU incorporation by 1.5-fold in mesangial cells (n=4–7, P<0.05 versus insulin treatment), but overexpression of SOCS1–3 isoforms failed to alter this effect of insulin on mesangial cells (n=4–7, P<0.05; Figure 8a). Similarly, overexpression of SOCS1–3 did not have effect on insulin-induced phosphorylation of ERK1/2 (Figure 8b).
Figure 8. Effects of overexpression of SOCS2 on insulin's action on mesangial cells.
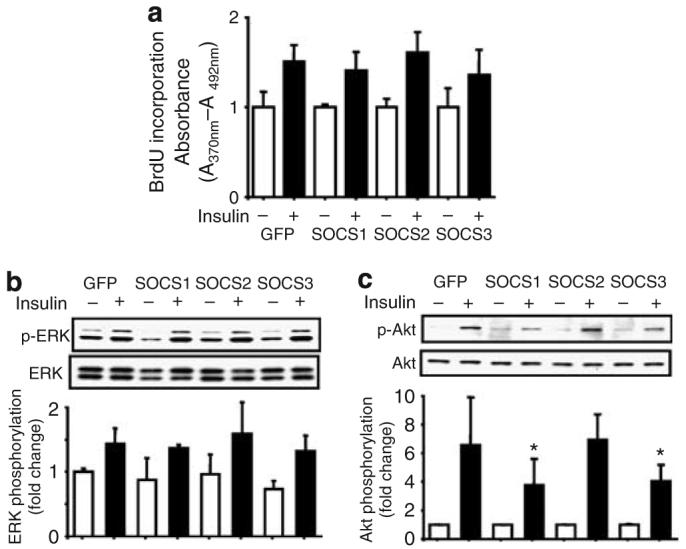
(a) Overexpression of all SOCS isoforms did not alter the effect of insulin on BrdU incorporation. (b) Overexpression of SOCS1–3 isoforms did not changed insulin-induced phosphorylation of ERK1/2. (c) Overexpression of SOCS1 and 3, but not SOCS2 isoforms inhibited Akt phosphorylation by insulin (n=3–6, mean±s.d., *P<0.05 versus GFP, SOCS2 with insulin stimulation).
However, insulin-induced Akt phosphorylations were significantly inhibited by approximately 50% following the overexpression of SOCS1 and 3 (n=3–6, P<0.05; Figure 8c). Overexpression of SOCS2 protein, on the contrary, did not have any effect on insulin's activation on PI3 kinase/Akt pathway (Figure 8c).
Insulin induced expression of SOCS2 through STAT5
Putative mechanisms underlying the reduced SOCS2 expression in the glomeruli in diabetic states were also explored. STAT5 has been reported to regulate SOCS2 expression in skeletal muscle cells.20 Microarray analysis revealed a concomitant reduction of mRNA expression of STAT5 by twofold in diabetic glomeruli that were normalized by ICT (Figure 9a). Immunoblot analysis revealed an approximately 30% reduction of STAT5a expression that can be normalized by insulin treatment, confirming the finding at mRNA level in the glomerular tissues (Figure 9b). Interestingly, cultured mesangial cells exposed to media containing 27.8 mm d-glucose for 3 days showed no change in the protein expression of STAT5 (Figure 9c). To determine whether insulin can directly regulate STAT5, expression of STAT5 proteins was studied in cultured mesangial cells exposed to 100 nm of insulin. STAT5 expression was increased by 49% (n=5, P<0.05) in the presence of insulin in media containing either 5.6 or 27.8 mm of glucose. (Figure 9c).
Figure 9. Downregulation of STAT5 expression in diabetic states.
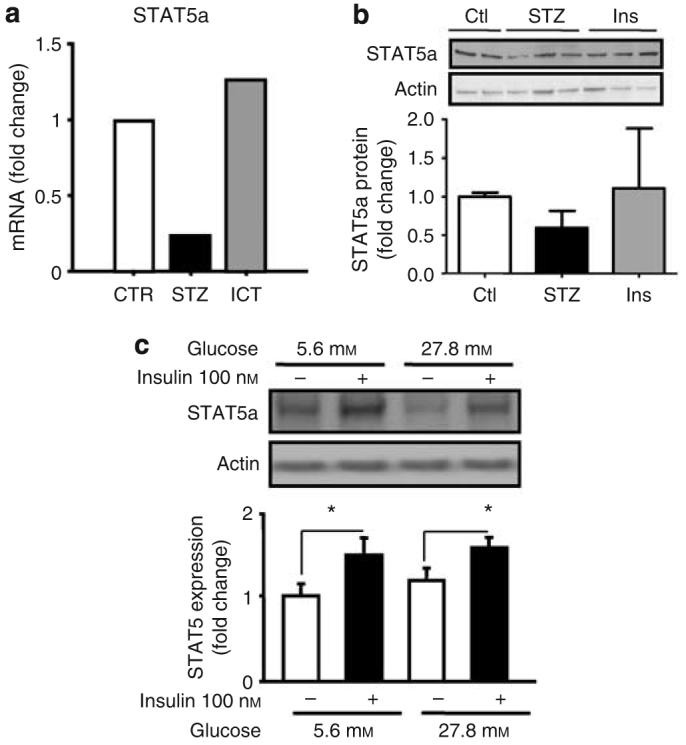
(a) Affymatrix gene array analysis revealed STAT5a mRNA expression was decreased in glomeruli from diabetic rats (STZ) that was normalized by islet transplantation (ICT). (b) Immunoblotting confirmed the reduced STAT5 protein expression in diabetic states that was reversed by insulin treatment. (c). Expression of STAT5 protein in cultured mesangial cells grown in media containing 5.6 or 27.8 mm of glucose with or without 100 nm of insulin for 3 days (n=5, P<0.05 versus without insulin).
DISCUSSION
Numerous studies have reported on the biochemical mechanisms by which elevated glucose levels cause molecular and functional abnormalities in renal and vascular cells in diabetes.21 However, very few studies have assessed the cellular consequences of insulin deficiency or inhibition of insulin's action on the metabolism, function, and proliferation of renal vascular and glomerular cells. Intervention with intensive insulin treatment or islet cell transplant can clearly prevent or may even reverse renal pathologies in diabetic states.6,22,23 Multiple lines of evidence supported potentially an important role for insulin in the regulation of renal functions such as on Na+/K+ ATPases in tubular cells.24 In this study, we have provided evidence showing that insulin has important direct effects on the alteration of expression profile in glomeruli and mesangial cells. Further, the current study has also identified a new pathway by which the IGF-1 signaling can be inhibited by insulin through its actions on SOCS2 expression and IGF-1R/Shc complex in the renal glomeruli.
These results showed for the first time that diabetic rats had decreased expression of SOCS2 in the glomerular tissues. This is unlikely due to a direct action of hyperglycemia alone because mesangial cells cultured in media containing 27.8 mm glucose did not change SOCS2 or STAT5 expression. Our data indicated that insulin has a direct effect on SOCS2 expression in mesangial cells. Previously, Sadowski et al.20 has reported that insulin increased the expression of both SOCS2 and 3 isoforms in C2C12 skeletal muscle cell line. However, our results indicated that insulin's regulatory effect on SOCS expression is isoform specific in mesangial cells because the expressions of SOCS1 and 3 isoforms were not changed by insulin. These results therefore suggested a specific action of SOCS2 in mediating insulin's biological responses in mesangial cells. Interestingly, normalization of glucose by ICT versus insulin pellet appears to have different effects on the expression of IGF-1R. The reason for this difference is not known but hyperinsulinemia is more likely associated with insulin pellet than those received ICT treatment in the renal capsule.
It has been previously shown that SOCS2 can interact with IGF-1 receptor in cultured cells overexpressing IGF-1 receptor following the addition of IGF-1 to the culture.13 However, this effect is not known in renal tissues such as mesangial cells and the downstream biological effects of this interaction were not reported.13 SOCS2 null mice exhibited gigantism phenotype with enlargement of most internal organs.14 They also showed a dramatic increase in the accumulation of collagen I in intestinal myofibroblasts following IGF-1 infusion as compared to wild-type control mice.25 We have clearly demonstrated that downregulation of SOCS2 in mesangial cells in diabetic states led to increased actions of IGF-1 on ECM protein production and DNA synthesis, phenotypes that are present in the progression of diabetic nephropathy. The effects of IGF-1 on DNA synthesis and type IV collagen are not large. However, their effects can be physiologically significant because changes in diabetic nephropathy require months or even years to develop. Thus, a change of 30% in the matrix protein levels, with such a slow turnover rate can be very significant. Furthermore, we have characterized a potential signaling pathway that confers this regulation. Following the activation of IGF-1R, transphosphorylation will recruit insulin receptor substrate proteins as well as other intracellular signaling molecules such as Grb2, Nck, and Shc.26 This will in turn activate PI3 kinase/Akt and Ras/Raf-1/MEK/ERK1/2 signaling cascades to elicit downstream biological actions such as gene expression, DNA synthesis and the blockage of cellular apoptosis.19 Activation of Ras/Raf-1/MEK/ERK1/2 pathway has been well documented to be essential for the regulation of gene transcription and DNA synthesis following the phosphorylation of IGF-1R.19 We have shown that SOCS2 protein can suppress the IGF-1-induced activation of ERK1/2 and therefore provide a molecular explanation for the inhibitory effect of SOCS2 on IGF-1. This inhibition is pathway-specific because SOCS2 did not inhibit IGF-1's action on PI3K/Akt pathway. In addition, the inhibitory effects of SOCS2 on IGF-1's actions were also isoform specific. Interestingly, although insulin and IGF-1 receptor share extensive homology and IGF-1 can bind to and activate insulin receptor at nonphysiological concentrations,19 SOCS2 did not have an effect on insulin's activation of MEK/ERK1/2 pathway, suggesting that SOCS2 action to inhibit IGF-1's action is ligand specific. Furthermore, SOCS2 overexpression can suppress the phosphorylation of Tyr317 of p66Shc by IGF-1 whereas this inhibition can be reversed by gene silencing of SOCS2, suggesting that the inhibition of ERK1/2 pathway is at the level close to IGF-1R because p66Shc is known to interact with IGF-1R directly.27 It is possible that SOCS2 can interrupt the binding of p66Shc to IGF-1R and block the downstream events, and this possibility requires further investigation. Further, SOCS2 can even enhance cytokine signaling because studies have shown that SOCS2 can bind to SOCS3 to enhance its degradation, thus by decreasing SOCS3 levels, SOCS2 can enhance the signaling of cytokines such as interleukin2 and interleukin3.28 Nevertheless, we have demonstrated a new inhibitory mechanism by which insulin can suppress IGF-1/IGF-1R/Shc/ERK1/2 pathway via SOCS2 protein and the subsequent reduced gene expression of type IV collagen and DNA synthesis. It is interesting to note that the overexpression of SOCS2 in mesangial cells did not significantly reduce basal DNA synthesis, but only during IGF-1 stimulations, supporting that the effects of SOCS2 is limited to IGF-1.
A great deal of previous studies have shown that lack of SOCS2's inhibitory action could lead to the uncontrolled cellular proliferation in multiple organs including bone, heart, liver, and kidney.14,29 We have reported here that the expression of SOCS2 but not SOCS1/3 is decreased in the glomeruli of diabetic rats, thus providing the very first evidence suggesting that the reduced SOCS2 protein expression may be responsible for nephromegaly in diabetic states. Further studies using diabetic SOCS2-null mice are required to establish a definitive role of SOCS2 in diabetic glomerulopathy. The mechanisms of SOCS2 downregulation by diabetes are likely due to either insulin deficiency or resistance. We have shown here for the first time that insulin can increase SOCS2 expression in the mesangial cells and in the glomeruli. In insulin deficient state, expression of both STAT5 and SOCS2 were decreased at mRNA and protein levels, which were normalized by ICT or insulin treatment. In cultured mesangial cells, insulin directly increased the expression of SOCS2 without affecting the expression of SOCS1 and 3. The concomitant downregulation of STAT5 induced by diabetes is likely the cause for the reduction of SOCS2 because this protein has been shown to regulate the expression and action of SOCS2.30 Further studies are needed to clarify how insulin regulates STAT5 level and whether this change will decrease SOCS2 expression. The isoform-specific effect of insulin to increase SOCS2 is important because a general action to increase SOCS1–5 will result in the inhibition of insulin's own actions. Our data have provided the interesting finding that insulin may have inhibitory effect on IGF-1's actions through a STAT5/SOCS2-dependent pathway. This is very unusual because IGF-1 can generally enhance the action of insulin. However, our observation provided evidence for the first time that insulin may also inhibit IGF-1's action at postreceptor levels. This could be highly significant because these data provide a mechanism to explain the enhanced actions of IGF-1 in mesangial cells observed in diabetic or obese states, even though most studies have not observed an increase in IGF-1 expression. In addition, these results have suggested a novel idea that insulin can inhibit IGF-1's cellular actions in cells at physiological conditions. The pathological significance of these findings for diabetic nephropathy needs to be evaluated directly because plasma IGF-1 levels are generally decreased in diabetic states associated with very poor glycemic control31 and probably not changed in the renal parenchyma. Our results suggest that IGF-1's action in the renal cells can be enhanced due to loss of insulin's inhibitory effects and the increased expression of IGF-1 receptor even without obvious increased levels of IGF-1. In addition, it is interesting to note that IGF-1's contribution on glomerular enlargement and nephropathy in obesity could be the results of decreases in the inhibitory actions of insulin on STAT5/SOCS2. Further studies on the changes in STAT5/SOCS2 in animal models of obesity are in progress. These data also showed that SOCS isoforms inhibited insulin and IGF-1's signaling very differently, with PI3K/Akt pathway being inhibited with insulin and Shc/ERK1/2 pathway affected when activated by IGF-1. This finding may also be extrapolated into other animal model of diabetes. It has been previously reported that action of IGF-1R's pathway was increased in mesangial cells isolated from NOD mice, possibly due to increases in autocrine expression of IGF-1.32 This may also be due to the reduced SOCS2 expression.
In summary, we have shown that insulin inhibited IGF-1's action by inducing the expression of STAT5 and SOCS2 to inhibit IGF-1 and IGF-1R activation of ERK pathway. This action apparently involves the tyrosine phosphorylation of Tyr317 of p66Sch to reduce the expression of type IV collagen and mesangial proliferation. In insulin deficiency or diabetic states, IGF-1's actions on glomerular cells are significantly enhanced by the loss of inhibitors actions due to reduction of STAT5 and SOCS2 and the reduced expression of IGF-1R. These data have provided new understanding on the regulatory effects of insulin on IGF-1's actions and a novel mechanism in addition to hyperglycemia, by which diabetes can cause mitogenic actions in glomerular mesangial cells and also glomerular dysfunctions and pathologies in diabetic rats.
MATERIALS AND METHODS
Materials
Anti-SOCS2, anti-IGF-1Rβ, anti-ERK1, anti-HA, anti-SOCS1, anti-SOCS3, anti-Stat5a, and anti-Shc were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). In part of the experiments, antibody against SOCS2 was from Abcam (Cambridge, MA). Anti-phospho MAPK p42/44, anti-phospho Akt, anti-Akt, anti-phospho Shc (Tyr 239/240), and anti-phospho Shc (Tyr 317) were from Cell Signaling Inc. (Beverly, MA, USA). Human SOCS2 construct and adenoviral vectors containing HA-tagged human SOCS2, SOCS1, and SOCS3 - GFP fusion genes were generously provided by Dr Jung-Song Lee and Dr Steve Shoelson in Joslin Diabetes Center. Recombinant human IGF-1 was purchased from Calbiochem (San Diego, CA, USA). LITMUS 28i vector was from New England BioLabs Inc. (Beverly, MA). Dicer siRNA Generation Kit was from Gene Therapy System Inc. (San Diego, CA, USA).
Animals and in vivo procedures
Diabetes was induced in male Lewis rats (100–120 g) by single intraperitoneal injection of STZ (75–90 mg/kg body weight) in 0.1 m citrate buffer (pH 4.5). The levels of blood glucose were measured 2 days after STZ injection and rats with blood glucose over 250 mg/100 ml) were used as diabetic rats. In diabetic rats, insulin (2–3 units) were injected everyday subcutaneously in order to maintain weight gain and prevent dehydration and ketoacidosis due to severe hyperglycemia and insulin deficiency. Four weeks after STZ injection, rats were randomly divided to three experimental groups: control rats (CTR), diabetic rats without treatment (STZ), diabetic rats treated with ICT. For ICT, the isolation of islet cells from donor rats were performed at the day before the transplantation. About 3000 islets isolated from the pancreas of three rats were transplanted under the capsule of the left kidney of a recipient rat.33 All rats were allowed free access to food and water. Four weeks after starting treatment and eight weeks after the initiation of the study, renal glomeruli were isolated from rats by sieving method as described previously.34 All procedures were done in accordance with guidelines set by Joslin Institutional Animal Care and Use Committee.
Physiological study
GFR and renal plasma flow were measured using a protocol we have previously established.35 Briefly, inulin (0.6%) and PAH (1.5%) in saline were infused via a catheter surgically placed in the left jugular vein of the anesthetized rats at a rate of 6.0 ml/h for 30 min, followed by a sustained infusion of 2.0 ml/h throughout the experiment. After 60 min, two clearance studies (each 30 min) were performed by measuring the concentrations of inulin and PAH in both the urine and blood samples collected from the rats. GFR and renal plasma flow were determined by inulin and PAH clearance, respectively, and the filtration fraction was calculated from the ratio of GFR to renal plasma flow.
Microarray
Total RNA was prepared from isolated glomeruli from four kidneys of two rats in same group using TRI reagent (Molecular Research Center Inc., Cincinnati, OH). Complementary DNA synthesis and cRNA labeling reaction were performed according to Affymetrix GeneChip Expression Technical Manual and hybridization to the Rat U34A GeneChip (Affymetrix, Santa Clara, CA). Hybridization and gene chip expression analysis were performed at Joslin Diabetes Center Affymetrix DNA microarray Core (DERC, Boston, MA, USA).
Cell culture
Mesangial cells were cultured from glomeruli isolated from male Sprague–Dawley rats (∼150 g). The cells were grown in Dulbecco's modified Eagle's medium (glucose 5.6 mm) containing 20% fetal bovine serum. Cultured cells were identified as mesangial cells using a previously established criteria.34 Cells were used between second to seventh passages.
Adenovirus infection
Determination of viral titer and multiplicity of infection for mesangial cells has been described in figure legends for each experiment.
RT–PCR
To verify the microarray results, RT–PCR using total RNA from glomeruli and immunoblot analysis in glomeruli and mesangial cells were performed. RT–PCR for SOCS2, IGF-1Rβ, and GAPDH as control were performed using standard protocols as previously described.36 The primer sequences for SOCS2 were sense 5′-GAGCTCAGTCAAACAGGATGGTACT-3′, antisense 5′-AGAATCCAATCTGAATTTCCC-3′. For IGF-1Rβ: sense 5′-CAACGACTATCAGCAGCTGA-3′, antisense 5′-TGGTGGAGAGGTAACAGA-3′. For GAPDH: sense 5′-GGTGAAGGTCGGTGTCAACGGATT-3′, antisense 5′-GATGCCAAAGTTGTCATGGATGACC-3′.
Protein isolation and immunoblotting
Homogenized glomeruli and cells were dissolved in lysis buffer (10 mm Tris-HCl at pH 7.5, 100 mm sodium chloride, 1% NP-40, 1% Triton X, 50 mm sodium fluoride, 2 mm EDTA, 1 mm pheyl methylsulphonyl fluoride, 10 μg/ml leupeptin and aprotinin). Some samples were immunoprecipitated with the indicated antibodies and protein A/G-Sepharose beads. SDS-polyacrylamide gel electrophoresis was performed on 10 or 12% Tris-glycine gel. Immunoblot analyses were identified with enhanced chemiluminscence (Amersham, Buckinghamshire, UK).
Northern blot analysis
Total RNA was prepared from mesangial cells using TRI reagent and northern analysis was performed as previously described.34
BrdU-incorporation assay
Cells were cultured in 96-well plates to ∼70% confluence infected with or without SOCS2-adenovirus or transfected with or without siRNA of SOCS2 and starved for 24 h. Recombinant IGF-1 was then added at the indicated concentration to cells for 16 h. The BrdU-incorporation assay was performed using the manufacturer's specifications (Roche, Nutley, NJ, USA) with an incorporation time of 2 h.
RNA interference
HA-tagged human SOCS2 cDNA were ligated to LITMUS 28i vector (New England BioLabs Inc.) and amplified by PCR using T7 primer. dsRNA synthesis and RNAi generation reaction were performed according manufacturer's specifications of Dicer RNAi Generation Kit (Gene Therapy System Inc.). Mesangial cells were plated at 1×105 cells per well in a 24-well plate for immunoblot analysis and at 1×104 cells per well in a 96-well plate for BrdU incorporation assay. Twelve hour after plating, mesangial cells were transfected with 100–500 ng of RNAi with or without indicated constructs using GeneSilencer siRNA Transfection Reagent (Gene Therapy System Inc.). One day after transfection, some cells were infected with HA-tagged SOCS2-adenovirus. The level of SOCS2 was determined by immunoblotting with anti-SOCS2 antibody and anti-HA antibody.
Statistical analysis
Data are expressed as mean±s.d. Comparisons between two groups were analyzed by Student's t-test. Comparisons among three or more groups were analyzed by one-way analysis of variance, followed by either Scheffe's test or Bonferroni/Dunn's multiple comparison test to evaluate statistical difference between two groups. P-values less than 0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by a grant from National Institute of Health (DK53105) to GLK, and DK36836 (Joslin's Diabetes and Endocrinology Research Center Grant). KI and YY are recipients of American Diabetes Association mentor-based fellowship. ZH is a recipient of Juvenile Diabetes Research Foundation fellowship.
Footnotes
DISCLOSURE
The authors state no conflict of interest.
REFERENCES
- 1.US Renal Data System . USRDS 2002 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease; Bethesda, MD: Jul, 2002. 2002. [Google Scholar]
- 2.Young BA, Johnson RJ, Alpers CE, et al. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 1995;47:935–944. doi: 10.1038/ki.1995.139. [DOI] [PubMed] [Google Scholar]
- 3.Mahadevan P, Larkins RG, Fraser JR, et al. Effect of prostaglandin E2 and hyaluronan on mesangial cell proliferation. A potential contribution to glomerular hypercellularity in diabetes. Diabetes. 1996;45:44–50. doi: 10.2337/diab.45.1.44. [DOI] [PubMed] [Google Scholar]
- 4.Wolf G, Ziyadeh FN. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 1999;56:393–405. doi: 10.1046/j.1523-1755.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- 5.Twigg SM, Cooper ME. The time has come to target connective tissue growth factor in diabetic complications. Diabetologia. 2004;47:965–968. doi: 10.1007/s00125-004-1423-6. [DOI] [PubMed] [Google Scholar]
- 6.Fioretto P, Steffes MW, Sutherland DE, et al. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- 7.Fujihara M, Uemasu J, Kawasaki H. Serum and urinary levels of insulin-like growth factor I in patients with chronic renal disease and diabetes mellitus: its clinical implication. Clin Nephrol. 1996;45:372–378. [PubMed] [Google Scholar]
- 8.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113(Pt 16):2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 9.Krebs DL, Hilton DJ. A new role for SOCS in insulin action. Suppressor of cytokine signaling. Sci STKE. 2003;2003:PE6. doi: 10.1126/stke.2003.169.pe6. [DOI] [PubMed] [Google Scholar]
- 10.Hortner M, Nielsch U, Mayr LM, et al. Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol. 2002;169:1219–1227. doi: 10.4049/jimmunol.169.3.1219. [DOI] [PubMed] [Google Scholar]
- 11.De Sepulveda P, Okkenhaug K, Rose JL, et al. Socs1 binds to multiple signalling proteins and suppresses steel factor-dependent proliferation. EMBO J. 1999;18:904–915. doi: 10.1093/emboj/18.4.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emanuelli B, Peraldi P, Filloux C, et al. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 13.Dey BR, Spence SL, Nissley P, et al. Interaction of human suppressor of cytokine signaling (SOCS)-2 with the insulin-like growth factor-I receptor. J Biol Chem. 1998;273:24095–24101. doi: 10.1074/jbc.273.37.24095. [DOI] [PubMed] [Google Scholar]
- 14.Metcalf D, Greenhalgh CJ, Viney E, et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405:1069–1073. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- 15.He Z, Way KJ, Arikawa E, et al. Differential regulation of angiotensin II-induced expression of connective tissue growth factor by protein kinase C isoforms in the myocardium. J Biol Chem. 2005;280:15719–15726. doi: 10.1074/jbc.M413493200. [DOI] [PubMed] [Google Scholar]
- 16.Parving HH. Initiation and progression of diabetic nephropathy. N Engl J Med. 1996;335:1682–1683. doi: 10.1056/NEJM199611283352212. [DOI] [PubMed] [Google Scholar]
- 17.Yechoor VK, Patti ME, Saccone R, et al. Coordinated patterns of gene expression for substrate and energy metabolism in skeletal muscle of diabetic mice. Proc Natl Acad Sci USA. 2002;99:10587–10592. doi: 10.1073/pnas.142301999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Natalicchio A, Laviola L, De Tullio C, et al. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J Biol Chem. 2004;279:43900–43909. doi: 10.1074/jbc.M403936200. [DOI] [PubMed] [Google Scholar]
- 19.Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818–835. doi: 10.1210/edrv.22.6.0452. [DOI] [PubMed] [Google Scholar]
- 20.Sadowski CL, Choi TS, Le M, et al. Insulin Induction of SOCS-2 and SOCS-3 mRNA expression in C2C12 Skeletal Muscle Cells Is Mediated by Stat5*. J Biol Chem. 2001;276:20703–20710. doi: 10.1074/jbc.M101014200. [DOI] [PubMed] [Google Scholar]
- 21.He Z, King GL. Microvascular complications of diabetes. Endocrinol Metab Clin North Am. 2004;33:215–238. doi: 10.1016/j.ecl.2003.12.003. xi-xii. [DOI] [PubMed] [Google Scholar]
- 22.DCCT Investigators The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 23.DCCT Investigators Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinciguerra M, Mordasini D, Vandewalle A, et al. Hormonal and nonhormonal mechanisms of regulation of the NA,K-pump in collecting duct principal cells. Semin Nephrol. 2005;25:312–321. doi: 10.1016/j.semnephrol.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Fruchtman S, Simmons JG, Michaylira CZ, et al. Suppressor of cytokine signaling-2 modulates the fibrogenic actions of GH and IGF-I in intestinal mesenchymal cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G342–G350. doi: 10.1152/ajpgi.00413.2004. [DOI] [PubMed] [Google Scholar]
- 26.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 27.Giorgetti S, Pelicci PG, Pelicci G, et al. Involvement of Src-homology/collagen (SHC) proteins in signaling through the insulin receptor and the insulin-like-growth-factor-I-receptor. Eur J Biochem. 1994;223:195–202. doi: 10.1111/j.1432-1033.1994.tb18983.x. [DOI] [PubMed] [Google Scholar]
- 28.Tannahill GM, Elliott J, Barry AC, et al. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol. 2005;25:9115–9126. doi: 10.1128/MCB.25.20.9115-9126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenhalgh CJ, Rico-Bautista E, Lorentzon M, et al. SOCS2 negatively regulates growth hormone action in vitro and in vivo. J Clin Invest. 2005;115:397–406. doi: 10.1172/JCI22710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenhalgh CJ, Bertolino P, Asa SL, et al. Growth enhancement in suppressor of cytokine signaling 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b) Mol Endocrinol. 2002;16:1394–1406. doi: 10.1210/mend.16.6.0845. [DOI] [PubMed] [Google Scholar]
- 31.Bideci A, Camurdan MO, Cinaz P, et al. Ghrelin, IGF-I and IGFBP-3 levels in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab. 2005;18:1433–1439. doi: 10.1515/jpem.2005.18.12.1433. [DOI] [PubMed] [Google Scholar]
- 32.Tack I, Elliot SJ, Potier M, et al. Autocrine activation of the IGF-I signaling pathway in mesangial cells isolated from diabetic NOD mice. Diabetes. 2002;51:182–188. doi: 10.2337/diabetes.51.1.182. [DOI] [PubMed] [Google Scholar]
- 33.Napoli R, Davalli AM, Hirshman MF, et al. Islet transplantation under the kidney capsule fully corrects the impaired skeletal muscle glucose transport system of streptozocin diabetic rats. J Clin Invest. 1996;97:1389–1397. doi: 10.1172/JCI118559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isshiki K, Haneda M, Koya D, et al. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes. 2000;49:1022–1032. doi: 10.2337/diabetes.49.6.1022. [DOI] [PubMed] [Google Scholar]
- 35.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 36.Way KJ, Isshiki K, Suzuma K, et al. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C beta2 activation and diabetes. Diabetes. 2002;51:2709–2718. doi: 10.2337/diabetes.51.9.2709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.