

Abstract
Acetylated and methylated lysine residues in histone H3 play important roles in regulating yeast gene expression and other cellular processes. Previous studies have suggested that histone H3 acetylated and methylated lysine residues may functionally interact through interdependent pathways to regulate gene transcription. A common genetic test for functional interdependence is to characterize the phenotype of a double mutant. Using this strategy, we tested the genetic interaction between histone H3 mutant alleles that simultaneously eliminate acetylated or methylated lysine residues. Our results indicate that mutation of histone H3 acetylated lysine residues alleviates growth phenotypes exhibited by the H3 methylated lysine mutant. In contrast, histone H3 acetylated and methylated lysine mutants display largely independent effects on yeast gene expression. Intriguingly, these expression changes are preferentially associated with chromosomal regions in which histone H3 lysine residues are hypoacetylated and hypomethylated. Finally, we show that the acetylated and methylated lysine mutants have strikingly different effects on the binding of Sir4 to yeast telomeres, suggesting that histone H3 acetylated lysine residues regulate yeast silencing through a mechanism independent of SIR binding.
POST-translationally modified residues in histone proteins, such as acetylated or methylated lysine residues, play critical roles in the regulation of gene transcription and silencing (Shilatifard 2006; Shahbazian and Grunstein 2007). Acetylation of the ɛ-amino group of lysine residues in histone proteins is catalyzed by histone acetyltransferases (HATs) (Lee and Workman 2007) and expunged by histone deacetylases (HDACs) (Yang and Seto 2008). In the N-terminal domain of histone H3 the lysine residues K9, K14, K18, K23, and K27 are acetylated. These residues are acetylated predominately by Gcn5 and Sas3 (Howe et al. 2001; Suka et al. 2001), and are deacetylated by Hda1, Hos2, and Rpd3 (Millar and Grunstein 2006). Histone H3 acetylation occurs both in the promoter and coding region of actively transcribed genes (Kurdistani and Grunstein 2003; Millar and Grunstein 2006). The functional consequences of eliminating histone H3 acetylation are varied: mutation or deletion of histone H3 acetylated lysine residues causes a significant increase in the transcription of many genes (Sabet et al. 2003), including GAL1 (Mann and Grunstein 1992; Wan et al. 1995) and genes located in telomeric and subtelomeric heterochromatin (Thompson et al. 1994; Martin et al. 2004). In contrast, deletion of Gcn5 causes a reduction in the transcription of many genes (Holstege et al. 1998; Lee et al. 2000; Durant and Pugh 2006). Histone acetylation also regulates other DNA metabolic processes, including DNA replication (Vogelauer et al. 2002; Aparicio et al. 2004) and repair (Groth et al. 2007; Escargueil et al. 2008).
Histone lysine residues are also methylated by histone methyltransferase enzymes. In Saccharomyces cerevisiae, histone H3 K4, K36, and K79 are methylated by Set1, Set2, and Dot1, respectively. Histone methylation occurs predominately in transcriptionally active chromosomal regions, and the methylation marks are deposited on chromatin during the transcription cycle through the recruitment of the cognate histone methyltransferase (Hampsey and Reinberg 2003; Shilatifard 2006; Li et al. 2007a). Mutations in methylated lysine residues or histone methyltransferase enzymes disrupt epigenetic silencing in yeast (Briggs et al. 2001; Bryk et al. 2002; Krogan et al. 2002; Ng et al. 2002; van Leeuwen and Gottschling 2002; Tompa and Madhani 2007), and can affect yeast viability (Jin et al. 2007).
Accumulating evidence suggests there is considerable interplay between acetylated and methylated lysine residues in histone H3. For example, methylation of H3 K4 is strongly correlated with acetylation of histone H3 N-terminal lysine residues (Zhang et al. 2004; Millar and Grunstein 2006; Taverna et al. 2007). Methylation of H3 K4 recruits the Gcn5 and Sas3 histone acetyltransferase complexes, as these complexes contain subunits (Chd1 and Yng1, respectively) that interact with methylated H3 K4 (Pray-Grant et al. 2005; Martin et al. 2006a,b; Taverna et al. 2006; Lee and Workman 2007). Hence, abrogation of H3 K4 methylation alters the acetylation pattern of adjacent histone H3 lysine residues (Taverna et al. 2006; Jiang et al. 2007). Intriguingly, acetylation of histone H3 N-terminal lysine residues also affects the methylation state of H3 K4 (Jiang et al. 2007). There is evidence that H3 K36 methylation directs the recruitment of the H3-specific Sas3 complex (Martin et al. 2006b). Set2-catalyzed histone H3 K36 methylation also recruits the Rpd3S histone deacetylase complex, which deacetylates histones in the coding regions of actively transcribed genes (Carrozza et al. 2005; Joshi and Struhl 2005; Keogh et al. 2005). Taken together, these studies argue that histone H3 acetylated and methylated lysine residues often function in interdependent molecular pathways to regulate gene transcription (Lee and Workman 2007).
There is considerable genetic evidence, however, suggesting that histone H3 acetylated and methylated lysine residues may regulate gene expression via redundant molecular mechanisms. In a previous study, we found that a significantly greater number of genes were differentially expressed when both a methylated (H3 K4) and acetylated (H3 K9, K14, K18, K23, K27) lysine residues were mutated in combination as opposed to when these residues were mutated individually (Martin et al. 2004). These results suggest that acetylated and methylated lysine residues may act in a redundant manner to regulate gene transcription.
In this study, we have employed a genetic approach to investigate the functional interplay between histone H3 acetylated and methylated lysine residues. We have constructed yeast mutants lacking acetylated lysine residues (H3 K9,14,18,23,27G), methylated lysine residue (H3 K4,36,79G), or both acetylated and methylated lysine residues (H3 K4,9,14,18,23,27,36,79G). Surprisingly, yeast strains harboring each of these mutants were viable. Indeed, mutations in histone H3 acetylated lysine residues rescued the slow growth and other phenotypes (e.g., hydroxyurea sensitivity) exhibited by the H3 methylated lysine mutants. To investigate the interplay between histone H3 acetylation and methylation in gene expression, we used DNA microarrays to profile the changes in transcription in each histone H3 mutant strain. Surprisingly, our results do not support either the interdependent or redundant models, but instead indicate that mutations in acetylated and methylated lysine residues have independent and additive effects on yeast gene expression. As many of the gene expression changes occurred adjacent to yeast telomeres, we also characterized the effects of eliminating histone H3 acetylated and/or methylated lysine residues on the binding of the SIR complex to telomere proximal DNA.
MATERIALS AND METHODS
Yeast strain and growth conditions:
The yeast strains used in this study are listed in Table 1. Details of strain construction are available upon request. For genomewide expression analysis of the H3 mutants, duplicate cultures of the H3 mutant strains and wild-type strain were grown in parallel to a final OD600 of 0.3–0.7 in yeast extract/peptone/dextrose (YPD) media, and then harvested as described previously (Holstege et al. 1998). For the spotting assay, yeast cells were grown to mid-log phase, normalized by OD600, fivefold serially diluted, and spotted on appropriate synthetic complete (SC) plates containing various carbon sources and indicated chemicals. All spotted plates were incubated at 30°. Plates were photographed after 2–4 days of incubation. All spotting assays were repeated to ensure reproducibility.
TABLE 1.
List of yeast strains and genotypes used in this study
| Strain | Experiment | Genotype |
|---|---|---|
| WY121 | Wild type | MATaade2-101 his3Δ200 lys2-801 trp1Δ901 ura3-52 hht1,hhf1∷LEU2 hht2,hhf2∷HIS3 plus pJL001 (CEN URA3 HHF2 HHT2) |
| WY139 | Wild type | Isogenic to WY121, plus pJW028 (CEN ADE2 HHF2 HHT2) |
| WY124 | H3 K9,14,18,23,27G | Isogenic to WY121, plus pJL004 (CEN URA3 HHF2 hht2-K9,14,18,23,27G) |
| YJ107 | H3 K9,14,18,23,27G | Isogenic to WY121, plus pYJ028 (CEN ADE2 HHF2 hht2-K9,14,18,23,27G) |
| AMY018 | H3 K4,36,79G | Isogenic to WY121, plus pAM018 (CEN ADE2 HHF2 hht2-K4,36,79G) |
| AMY025 | H3 K4,9,14,18,23,27,36,79G | Isogenic to WY121, plus pAM025 (CEN ADE2 HHF2 hht2-K4,9,14,18,23,27,36,79G) |
| YJ057 | Wild-type Sir4-9xMyc | MATaade2-101 his3Δ200 lys2-801 trp1Δ901 ura3-52 hht1,hhf1∷LEU2 hht2,hhf2∷HIS3 SIR4-9xMyc∷TRP1 plus pJW028 (CEN ADE2 HHF2 HHT2) |
| YJ108 | H3 K9,14,18,23,27G Sir4-9xMyc | Isogenic to YJ057, plus pYJ028 (CEN ADE2 HHF2 hht2-K9,14,18,23,27G) |
| YJ099 | H3 K4,36,76G Sir4-9xMyc | Isogenic to YJ057, plus pAM018 (CEN ADE2 HHF2 hht2-K4,36,79G) |
| YJ098 | H3 K4,9,14,18,23,27,36,79G Sir4-9xMyc | Isogenic to YJ057, plus pAM025 (CEN ADE2 HHF2 hht2-K4,9,14,18,23,27,36,79G) |
| YJ118 | Wild type | MATa, ade2∷hisG his3Δ200, leu2Δ0, lys2Δ0, met15Δ0, trp1Δ63, ura3Δ0, adh4∷URA3-TEL (VIIL), ADE2-TEL (VR), hhf2,hht2∷MET15, hhf1,hht1∷LEU2, plus pYJ029 (CEN TRP1 HHF2 HHT2) |
| YJ119 | H3 K4,36,79G | Isogenic to YJ118, plus pYJ030 (CEN TRP1 HHF2 hht2-K4,36,79G) |
| YJ120 | H3 K9,14,18,23,27G | Isogenic to YJ118, plus pYJ031 (CEN ADE2 HHF2 hht2-K9,14,18,23,27G) |
| YJ121 | H3 K4,9,14,18,23,27,36,79G | Isogenic to YJ118, plus pYJ032 (CEN ADE2 HHF2 hht2-K4,9,14,18,23,27,36,79G) |
Plasmid construction and site-directed mutagenesis:
The QuikChange kit (Stratagene) was used to mutate histone H3 lysine residues to glycine, using plasmid pJL001 or pJL004 (Martin et al. 2004) as a template. Plasmid pJL001 is derived from pRS316 and contains wild-type histone H3 (HHT2) and H4 (HHF2) genes. Plasmid pJL004 contains mutant histone H3 (hht2-K9,14,18,23,27G) and wild-type histone H4 (HHF2) genes. The complete list of mutagenic primer sequences can be found at the following Web site: http://wyrick.sbs.wsu.edu/histoneH3AcetylMethyl/. The resulting mutant histone H3 alleles from site-directed mutagenesis were subsequently cloned into the pRS412 shuttle vector (Brachmann et al. 1998) using the NotI and XhoI restriction enzymes. All mutants were confirmed by DNA sequencing.
Western blot analysis:
Yeast cultures were grown in YPD media to a final OD600 of 0.8–1.0 and harvested by centrifugation. Cell pellets were resuspended in lysis buffer (50 mm HEPES-KOH pH 7.5, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% Na-deoxycholate) containing fresh complete protease inhibitors cocktail (Roche Diagnostics), and then lysed by vigorous agitation with acid-washed glass beads (425–600 μm, Sigma). The crude extract was briefly sonicated and further clarified by centrifugation. The supernatant was heated with Laemmli loading buffer at 95° for 10 min. The protein extracts were resolved on a Tris-HCL 4–20% gradient gel (BioRad) and transferred to Hybond-ECL nitrocellulose membrane (GE Healthcare). Membranes were then probed with appropriate antibodies (anti-histone H3 and anti-tubulin, Abcam). The signals of target proteins were quantified using GelDoc EQ imager with Quantity One software (Bio-Rad).
Genomewide expression profiling:
RNA isolation, cDNA amplification, and biotin-cRNA synthesis were performed as described previously (Martin et al. 2004). The cRNA was hybridized to a single S98 yeast genome oligonucleotide array and scanned following standard protocols (Affymetrix). Fluorescence intensities were determined using GeneChip software (Affymetrix) and a single raw expression level for each gene was calculated. The data from each microarray were normalized by the GeneChip software using a median global intensity scaling method. Differentially expressed genes were identified using previously published criteria: the average change in mRNA levels (up or down) was greater than twofold, the change in mRNA levels in each replicate experiment was greater than 1.5-fold, and the absolute intensity change was above background levels. See the reports by Jin et al. (2007) and Martin et al. (2004) for more details. Microarray data for the H3 K[Ac]G mutant are from a previous study (Martin et al. 2004). Microarray data are available at the following Web site: http://wyrick.sbs.wsu.edu/histoneH3AcetylMethyl/.
Microarray data analysis:
An additive model was used to test whether the histone H3 acetylated and methylated lysine mutants had independent effects on yeast gene expression. According to the additive model (Mani et al. 2008), the expected fold change in the H3 K[AcMe]G double mutant is given by the equation
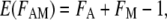 |
(1) |
where FA is the fold change measured in the H3 K[Ac]G mutant, FM is the fold change measured in the H3 K[Me]G mutant, and E(FAM) is the expected fold change in the H3 K[AcMe]G mutant. However, the standard additive model has difficulties with downregulated genes. For example, if FA = 1/3 and FM = 1/3, then according to Equation 1, E(FAM) = −1/3. This is a nonsensical result, as a negative fold change in mRNA levels cannot be interpreted biologically. To use the additive model to analyze microarray data, we made the following modification: for genes downregulated (fold change <1), we first converted the fold change to a fold decrease (e.g., a 0.5-fold change was converted to a twofold decrease) prior to using Equation 1. The calculated fold decrease was then converted to the fold change by taking its inverse.
We also tested for genetic interaction using the log model (Mani et al. 2008), which is given by the following equation:
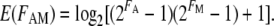 |
(2) |
For genes that are significantly upregulated (FA and FM ≫ 1), the following approximations hold for Equation 2:
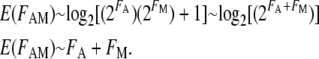 |
Hence, for upregulated genes, the log model approximates the result obtained from the additive model.
Telomere-proximal gene analysis and statistics:
Chromosome plots were conducted as described previously (Jin et al. 2007), except that genes flagged as dubious were included in the analysis, and yeast genes were divided into consecutive 100-gene bins on the basis of their distance from a chromosome end. The fraction of genes upregulated and downregulated in the histone mutant and the average distance of the genes from the telomere were plotted for each bin. Only those genes within ∼200 kb of a telomere were plotted. The significance of enrichment of upregulated genes in telomeric (0–10 kb from a telomere end) or subtelomeric regions (10–20 kb from a telomere end) was determined using a hypergeometric probability distribution.
A Wilcoxon rank sum test was used as an alternative method to test for a bias of upregulated genes adjacent to yeast telomeres. Genes in each histone H3 mutant data set were ranked according to the magnitude of their fold change in mRNA levels. Genes that were called absent, or had inconsistent or background intensity levels were excluded from this analysis. The Wilcoxon rank sum test was employed to test whether genes located adjacent to a telomere end showed a bias in their distribution of ranks in any of the histone H3 mutant strains.
Chromatin immunoprecipitation (ChIP) assay:
ChIP assays were performed as described previously (Jin et al. 2007). Briefly, 50 ml of yeast cells were grown to a final OD600 of 0.8–1.0 in YPD media and cross-linked with 1% formaldehyde. Cells were then lysed by glass beads and sonicated to shear the chromatin to fragment sizes of 150–400 bp. Cross-linked chromatin fragments were immunoprecipitated with anti-Myc antibody (Biosource, AHO0052) bound to magnetic beads (Dynal Biotech). After the cross-links were reversed, the DNA fragments were extracted for PCR analysis. Taq DNA polymerase (New England Biolabs) and appropriate primer pairs were used in the PCR amplification reactions. PCR products were subjected to gel electrophoresis, stained with ethidium bromide, and quantified using GelDoc EQ imager with Quantity One software (Bio-Rad). Primer sequences are available at the following Web site: http://wyrick.sbs.wsu.edu/histoneH3AcetylMethyl/.
Microarray data accession number:
The data discussed in this report have been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession nos. GSE10930 and GSE10933.
RESULTS
Phenotypes of the histone H3 acetylated and methylated lysine mutants:
Previous studies have suggested that acetylation and methylation of histone H3 lysine residues play important and interconnected roles in regulating yeast gene expression and other cellular processes. To further test these hypotheses, we constructed yeast strains containing one of three different histone H3 alleles: H3 K[Ac]G, in which the five histone H3 N-terminal acetylated lysine residues were mutated to glycine (i.e., hht2-K9,14,18,23,27G); H3 K[Me]G, in which the three methylated lysine residues were mutated to glycine (i.e., hht2-K4,36,79G); and H3 K[AcMe]G, in which the acetylated and methylated lysine residues were mutated to glycine (i.e., hht2-K4,9,14,18,23,27,36,79G). Each of these histone H3 mutant strains was viable. While the H3 K[Me]G mutant was not lethal, as was the case for the H3 K[Me]R (i.e., histone H3 K4,36,79R) mutant (Jin et al. 2007), it did exhibit a slower doubling time compared to wild-type in synthetic complete (SC) media (Figure 1A). This result is in accordance with previous studies, which showed that mutating H3 K4 in yeast caused a significant growth defect (Briggs et al. 2001). Intriguingly, this phenotype was suppressed in the H3 K[AcMe]G mutant (Figure 1A), indicating that acetylated lysine mutations rescued the growth defect caused by the methylated lysine mutations.
Figure 1.—

Phenotypic analysis of the histone H3 acetylated and methylated lysine mutants. (A) Wild-type, H3 K[Ac]G, H3 K[Me]G, and H3 K[AcMe]G yeast strains were grown in SC media at 30°. Cell density was monitored by OD600 at consecutive time points. The doubling time was measured on the basis of the growth curve. Each error bar represents the standard deviation of four independent experiments. (B and C) Fivefold serial dilutions of each yeast strain from ∼104 cells were spotted on SC media containing various carbon sources and indicated chemicals.
The histone H3 mutant alleles displayed phenotypes in various growth conditions (Figure 1, B and C). These include growth defects in certain carbon sources (e.g., maltose, glycerol, and ethanol), in high salt (i.e., 0.5 m NaCl), and upon treatment with specific chemicals (e.g., caffeine). In many conditions, the phenotype of the H3 K[AcMe]G mutant reflected the phenotypes of the H3 K[Ac]G and H3 K[Me]G mutants. There were four notable exceptions. The high salt (NaCl), methyl methanesulfonate (MMS), 6-azauracil (6-AU), and hydroxyurea (HU) sensitivities exhibited by the H3 K[Me]G mutant were alleviated in the H3 K[AcMe]G mutant (Figure 1C). Indeed, the H3 K[Ac]G mutant was more resistant to treatment with HU, a DNA replication inhibitor, than the wild-type strain (Figure 1C). Hence, these phenotypes of the histone methylated lysine mutant were rescued upon mutating acetylated lysine residues in histone H3.
It is possible that the mutant phenotypes could be an indirect consequence of changes in the stability of the histone H3 protein. We tested whether the histone H3 mutant alleles affected the cellular levels of histone H3 protein by performing Western blot analysis of yeast extracts using an anti-histone H3 antibody. As shown in Figure 2, the mutant alleles did not affect the levels of histone H3 protein.
Figure 2.—

The level of histone H3 protein in histone H3 mutants. (A) The stability of histone H3 protein from different histone mutants was measured by Western blot analysis. Cell extracts from each strain were resolved by SDS–PAGE and probed with anti-H3 antibody. The level of α-tubulin subunit was used as an internal loading control. (B) Quantification of histone H3 protein level in histone H3 mutants. The signal of histone H3 protein was normalized by the tubulin signal. The calculated signal of wild-type histone H3 protein was set to 100%. Each error bar represents the standard deviation of three independent experiments.
Histone H3 acetylated and methylated lysine mutants have independent effects on gene transcription:
We used Affymetrix oligonucleotide microarrays to profile the changes in gene transcription in each histone mutant strain. The results indicate that the H3 K[AcMe]G mutant caused an increase in the mRNA levels of 250 genes and a decrease in the mRNA levels of 81 genes (Figure 3, A and B). The differentially expressed genes function in a variety of cellular processes, including vitamin metabolism (P = 6.3 × 10−5). The microarray data sets for the H3 K[Ac]G (Martin et al. 2004) and H3 K[Me]G affected the expression of similar but smaller sets of genes (Figure 3, A and B).
Figure 3.—

Effects of histone H3 acetylated and methylated lysine mutants on gene transcription. Venn diagrams were used to compare the sets of (A) upregulated genes and (B) downregulated genes among the histone H3 mutants. The circle size is proportionate to the number of genes in a data set. The degree of overlap between two circles is proportionate to the number of genes shared between data sets. Data for histone H3 K[Ac]G mutant is from Martin et al. (2004). (C) The microarray data were displayed as a log2 ratio of mRNA fold change (mutant/wild type) using the Treeview software. The data sets in each column correspond to the indicated histone H3 mutant. Each row represents a single gene; the log2 ratio of the fold change is represented by a color (see color scale). Genes that are differentially expressed among the histone H3 mutants are displayed. Each vertical color bar indicates the corresponding subset of genes in the Venn diagrams at the top. (D) The histone H3 acetylated and methylated lysine residues make independent contributions to gene transcription. The observed gene expression changes in the H3 K[AcMe]G mutant were compared with the calculated gene expression changes on the basis of the H3 K[Ac]G and H3 K[Me]G data sets using a modified additive model. The comparison is viewed as a scatter plot of the log2 ratios for all yeast genes.
Comparison of the data using Venn diagrams indicated that there were significant overlaps in the sets of genes regulated in each mutant (Figure 3, A and B). Many of the genes upregulated in the H3 K[AcMe]G mutant were also affected in the H3 K[Ac]G or H3 K[Me]G mutants (or both). Significant overlaps were also observed between the sets of downregulated genes, though to a lesser extent (Figure 3B). It is important to note, however, that a few genes showed altered expression only in the H3 K[Ac]G or H3 K[Me]G. For example, 34 genes were upregulated and 20 genes were downregulated only in the H3 K[Ac]G mutant (Figure 3, A–C). Presumably, the expression of these genes was not altered in the H3 K[AcMe]G double mutant due to epistatic effects exerted by the H3 K[Me]G mutant. We also observed that a significant number of genes were up- or downregulated only in the H3 K[AcMe]G mutant (123 and 51 genes, respectively). However, inspection of the microarray data revealed that most of these genes were also affected in the H3 K[Ac]G and H3 K[Me]G mutants, though not to the level of the twofold threshold (Figure 3C). Hence, the greater number of genes differentially expressed in the H3 K[AcMe]G mutant may simply be due to the cumulative effects of the H3 K[Ac]G and H3 K[Me]G mutants.
The microarray data allow us to test which model of genetic interaction best explains the gene expression changes observed in the H3 K[AcMe]G double mutant. The null hypothesis is that the H3 K[Ac]G and H3 K[Me]G mutants make independent, and thus additive, contributions to the gene expression changes in the H3 K[AcMe]G double mutant. This additive model has been commonly used to test for genetic interactions between transcription factors and other genes (Carey et al. 1990; Lin et al. 1990; Hertel et al. 1997; Mani et al. 2008). Significant deviations from the additive model would support either the interdependence model or the redundancy model, depending upon the direction of the deviation (see discussion section for more details).
We compared the observed gene expression changes in the H3 K[AcMe]G mutant with the expected changes on the basis of an additive (i.e., independent) model. It is important to note that the standard additive model cannot be readily fit to microarray expression data that includes downregulated genes (see materials and methods for more details). For this reason, we used a modified version of the additive model to fit the data, as described in materials and methods. The results shown in Figure 3D indicate that calculated and observed gene expression changes were highly correlated (r = 0.78; P < 10−100). This correlation was even higher if just the differentially expressed genes were examined (r = 0.90; P < 10−100). Indeed, the calculated and observed gene expression changes among the differentially expressed genes displayed a linear relationship (slope and R2 are 0.96 and 0.81, respectively). This analysis indicates that the gene expression changes in the H3 K[AcMe]G mutant can largely be explained by a model assuming that the histone H3 acetylated and methylated lysine residues make independent and additive contributions to gene transcription.
As an alternative method to test the independence model, we used a log model to test for genetic interaction. For significantly upregulated genes, the log model closely approximates the predictions of the additive model (see materials and methods for more details), but unlike the standard additive model, the log model does not encounter difficulties handling data for downregulated genes. Thus, the log model can be readily applied to microarray data, and has been previously validated for genetic interaction testing (Mani et al. 2008). We find that calculated expression changes on the basis of the log model were highly correlated with the observed expression changes in the H3 K[AcMe]G mutant, both for differentially expressed genes (r = 0.90; P < 10−100), and for the entire genome (supplemental Figure 1). Taken together, these results do not support the alternative hypotheses of interdependence or redundancy, but instead support the model that histone H3 acetylated and methylated lysine residues make independent contributions to transcription regulation.
Histone H3 lysine mutants preferentially affect telomere-proximal gene expression:
Both methylated and acetylated lysine residues in histone H3 have been implicated in telomeric silencing in yeast. Hence, we examined whether the histone H3 mutants had a particular effect on telomeric gene expression, which we defined as genes located from 0 to 10 kb from a chromosomal end (Wyrick et al. 1999; Martin et al. 2004). Chromosome plots of the genomewide expression data for the histone H3 acetylated and methylated lysine mutants are shown in Figure 4. We observed a significant cluster of upregulated genes in telomeric regions (0–10 kb) in the H3 K[Me]G mutant (P = 9.5 × 10−9), but not in the H3 K[Ac]G mutant (P = 0.1). In contrast, both the H3 K[Ac]G (P = 1.8 × 10−4) and H3 K[Me]G (P = 1.2 × 10−4) mutants significantly upregulate the expression of subtelomeric genes (defined as genes located 10–20 kb from a telomere end) (Wyrick et al. 1999; Martin et al. 2004). The H3 K[AcMe]G mutant shows a striking increase in telomeric gene expression (see Figure 4), likely due to the cumulative effects of mutating the acetylated and methylated lysine residues in combination.
Figure 4.—

Histone H3 mutants preferentially affect telomere-proximal gene expression. Chromosome plots of the genomewide expression data for the H3 K[Ac]G, H3 K[Me]G, and H3 K[AcMe]G mutants. For each histone H3 mutant, a histogram of the fraction of genes whose mRNA levels are up/downregulated is plotted as a function of their distance from a chromosome end.
It is possible that the H3 K[Ac]G mutant might affect the expression of telomeric genes, but not to the level of the twofold threshold used to analyze the microarray data. To test this possibility, we used a rank sum statistical test to measure whether telomeric genes showed a quantitative increase in expression in the H3 K[Ac]G mutant. The results shown in Table 2 and supplemental Table 1 indicate that there is a significant increase in the expression of telomeric genes in the H3 K[Ac]G mutant, although to a lesser extent than the H3 K[Me]G mutant. This conclusion is supported by data from telomeric silencing assays. The histone H3 K[Ac]G mutant significantly disrupts the silencing of a telomere-located URA3 reporter gene (Figure 5A), in accordance with the results from a previous study (Thompson et al. 1994).
TABLE 2.
Effects of histone H3 mutant strains on telomere-proximal gene expression
| Telomeric genesa
|
Random gene set
|
|||
|---|---|---|---|---|
| Mutants | Rank percentile (%) | P-value | Rank percentile (%) | P-value |
| H3 K[Ac]G | 65 | <10−5 | 51 | 0.74 |
| H3 K[Me]G | 78 | <10−16 | 50 | 0.92 |
| H3 K[AcMe]G | 82 | <10−21 | 50 | 0.90 |
Genes located <10 kb from a telomere.
Figure 5.—

(A) Histone H3 lysine mutants disrupt telomere silencing. Telomeric silencing of a URA3 reporter gene was measured in histone H3 mutant strains. Fivefold serial dilutions of each yeast strain were spotted on SC −TRP media or SC −TRP with 5-FOA as indicated. (B and C) Effects of the histone H3 acetylated and methylated lysine mutants on the binding of the SIR silencing complex near the chromosome telomere regions. The abundance of Sir4 at chromosome V-L (Chr V-L) and chromosome VI-R (Chr VI-R) telomere regions in wild-type strains and H3 mutant strains were determined by ChIP analysis. An untagged strain was used as control. Sir4 binding ratio at Chr V-L is normalized to an internal control (ACT1) and the input DNA. Sir4 binding ratio at Chr VI-R is normalized to an internal control (SPS2) and the input DNA. Each error bar represents the standard deviation of three independent ChIP experiments. A t-test was applied to generate the P-values. Three stars indicate P < 0.001 compared to wild type.
We next examined how these mutants affected the binding of the SIR silencing complex to DNA regions adjacent to telomeres. We used ChIP assays to measure the binding of the Sir4 protein to genomic regions adjacent to telomere V-L (372 bp away) and telomere VI-R [525 bp away; see Xu et al. (2007)] in the histone H3 mutant strains (Figure 5, B and C). The H3 K[Me]G mutant caused a striking decrease in Sir4 binding at each telomere proximal region relative to wild type (P < 0.001). In contrast, the H3 K[Ac]G mutant caused only a slight, insignificant decrease in Sir4 binding (P > 0.05). The decrease in Sir4 binding in the H3 K[AcMe]G mutant was similar to the decrease observed in the H3 K[Me]G mutant.
For comparison, we used RT–PCR to examine the changes in the mRNA levels of the YEL077C gene in the histone H3 mutants. YEL077C is located directly adjacent to telomere V-L and overlaps with the PCR probes used for the Sir4 ChIP experiments. The results shown in supplemental Figure 2 indicate that the mRNA levels of YEL077C showed a small but significant increase in the H3 K[Me]G and H3 K[Ac]G mutants, although the expression change in the H3 K[Ac]G mutant was somewhat marginal (P < 0.05). Importantly, although relatively little if any Sir4 binding was observed in either the H3 K[Me]G or H3 K[AcMe]G mutants (see Figure 5B), the expression of YEL077C was significantly greater in the H3 K[AcMe]G mutant than in the H3 K[Me]G mutant (supplemental Figure 2). Hence, the transcriptional induction observed in telomeric genes upon mutating the histone H3 acetylated lysine residues appears largely independent of any effect on Sir4 binding. In contrast, the histone H3 methylated lysine residues regulate telomere silencing principally through their effects on the binding of the SIR complex.
Histone modification status of genes regulated by histone H3 lysine residues:
The histone H3 K[Ac]G, H3 K[Me]G, and H3 K[AcMe]G mutants eliminate both modified and unmodified lysine residues in genomic chromatin. Hence, the transcriptional changes in these mutants could theoretically be due to the loss of the modified version of the histone H3 lysine residues or the unmodified version. To distinguish between these two possibilities, we used ChromatinDB (O'Connor and Wyrick 2007) to compare the gene expression changes in the histone H3 mutants with published ChIP-chip data (Bernstein et al. 2002; Kurdistani et al. 2004; Rao et al. 2005) for histone acetylation and methylation.
We first compared ChIP-chip data for acetylation of H3 K9, K14, K18, K23, and K27 (Kurdistani et al. 2004) with the expression data for the H3 K[Ac]G and H3 K[AcMe]G mutant strains. For simplicity, we analyzed the average acetylation level for the five acetylated H3 lysine residues. Figure 6A indicates that the genes upregulated in the H3 K[Ac]G mutant were predominately those that originally had low levels of promoter histone acetylation in wild-type cells. This trend was even more pronounced in the H3 K[AcMe]G mutant (Figure 6A). Each of these trends was statistically significant, as judged using a Wilcoxon rank sum test (P < 10−3 and P < 10−10 for the H3 K[Ac]G and H3 K[AcMe]G data sets, respectively). A similar result was obtained when we tested the acetylation levels of each histone H3 lysine residue individually (data not shown). However, this trend was not observed in the coding regions of the upregulated genes. In summary, disruption of hypoacetylated chromatin was correlated with derepression in the H3 K[Ac]G mutant.
Figure 6.—

Correlation between histone modification levels and gene expression changes in the histone H3 mutants. (A) For the H3 K[Ac]G and H3 K[AcMe]G mutants, the fraction of genes whose mRNA levels are up/downregulated is plotted against their average promoter H3 acetylation levels in wild-type cells. The ChIP-chip data has been normalized for nucleosome levels, as described (O'Connor and Wyrick 2007). (B) For the H3 K[Me]G and H3 K[AcMe]G mutants, the fraction of genes whose mRNA levels were up/downregulated is plotted against their average level of nucleosome-normalized histone H3 K4 dimethylation in wild-type cells.
A similar pattern was observed when we compared ChIP-chip data for methylation of H3 K4 and H3 K36 (Bernstein et al. 2002; Rao et al. 2005) with the expression data for the H3 K[Me]G and H3 K[AcMe]G mutant strains. Figure 6B shows that genes upregulated in the H3 K[Me]G mutant were biased toward those genes that had low levels of H3 K4 dimethylation in their promoter regions (P < 10−8). Again, this effect was exaggerated in the H3 K[AcMe]G mutant (P < 10−22). Low levels of H3 K36 dimethylation in the coding regions of genes was also correlated with gene expression changes in the H3 K[Me]G and H3 K[AcMe]G mutant strains (P < 10−4 and P < 10−23, respectively), as was H3 K4 dimethylation (data not shown).
Previous studies have shown that histone H3 acetylated and methylated lysine residues are depleted of these modifications at telomeric and subtelomeric genomic regions (Bernstein et al. 2002; Robyr et al. 2002; Millar and Grunstein 2006). Hence, we classified the genes upregulated in the histone H3 mutants into telomere-proximal (i.e., genes located in telomeric or subtelomeric genomic regions) and nontelomere-proximal (i.e., genes located further than 20 kb from a telomere end) groups and repeated the analysis described above. This analysis indicated that the nontelomere-proximal groups of upregulated genes were also significantly associated with low levels of histone H3 acetylation and H3 K4 methylation (P < 10−3 and P < 10−5, respectively). It is important to note that these unmodified, upregulated genes were distributed throughout the genome (on the basis of visual inspection, data not shown) and did not appear to be an indirect effect of the loss of telomeric silencing in these mutants, on the basis of our analysis of a sir2Δ microarray data set (Wyrick et al. 1999; Martin et al. 2004). Hence, the correlation with unmodified histone H3 lysine residues was not limited to silenced genes, but was a feature of genes located throughout the genome.
DISCUSSION
Previous studies have suggested that acetylated and methylated lysine residues in histone H3 functionally interact to regulate gene expression and other cellular processes. In this study, we have used a genetic approach to characterize the functional interactions between acetylated and methylated lysine residues in histone H3. We constructed histone H3 mutant alleles that simultaneously eliminate acetylated lysine residues (H3 K[Ac]G), methylated lysine residues (H3 K[Me]G), or both acetylated and methylated lysine residues (H3 K[AcMe]G). Analysis of the growth phenotypes and gene expression changes exhibited in these mutant strains argues against the models of either functional interdependence or redundancy; instead, our results are consistent with the model that acetylated and methylated histone lysine residues function through distinct mechanisms to make independent contributions to yeast gene expression and silencing.
For the purposes of this article, we have classified the histone H3 lysine residues into a group that is predominately acetylated (H3 K9, K14, K18, K23, K27) and a group that is predominately methylated (H3 K4, K36, K79). It should be noted, however, that a recent report suggests that histone H3 K36 is also acetylated (Morris et al. 2007).
Mutations in histone H3 methylated lysine residues (e.g., H3 K4) or histone methyltransferase enzymes (e.g., Set1) have been previously shown to exhibit various growth phenotypes, including slow growth defects and sensitivity to the DNA replication inhibitor hydroxyurea (HU) (Briggs et al. 2001; Miller et al. 2001; Fingerman et al. 2005). We observed similar growth defects in the H3 K4,36,79G mutant strain (H3 K[Me]G, see Figure 1), including a slowed doubling time in SC media, and striking sensitivities to the transcription elongation inhibitor 6-AU, and the DNA replication inhibitor and damaging agent HU and MMS. Strikingly, the simultaneous mutation of H3 acetylated lysine residues rescues these growth phenotypes in the H3 methylated lysine mutant strain. Indeed, the H3 K[Ac]G mutant suppresses the HU sensitivity of a SET1 deletion mutant (data not shown) and is more resistant to HU than wild type. Previous studies have shown that histone acetylation can regulate the firing of replication origins (Vogelauer et al. 2002; Aparicio et al. 2004); hence, it is possible that the H3 K[Ac]G mutant directly modulates the process of DNA replication.
Alternatively, the different phenotypes in the H3 K[Ac]G or H3 K[Me]G mutants could be due to differing effects on yeast gene expression. While the H3 K[Ac]G and H3 K[Me]G mutants have fairly similar effects on gene expression, there are a number of genes whose expression change is different in the mutant strains. These genes are listed in supplemental Table 3. Inspection of the gene lists suggests a number of potential candidate genes involved in DNA repair (e.g., MRE11, FYV6, and REV1), whose expression change could be responsible for the differing HU and MMS phenotypes in the histone mutant strains.
Comparison of the H3 K[Ac]G, H3 K[Me]G, and H3 K[AcMe]G microarray data sets enabled us to test whether these histone residues genetically interact to regulate transcription. As our null hypothesis (i.e., neutrality function), we used a modified version of the additive model. If the measured expression data for the H3 K[AcMe]G significantly deviated from this model, this would indicate a genetic interaction. For example, if histone H3 acetylated and methylated lysine residues were functionally redundant, then the change in transcription in the H3 K[AcMe]G mutant should be significantly greater than the sum of the effects observed in the H3 K[Ac]G and H3 K[Me]G mutants. On the other hand, if the interdependence model were correct, then the expression changes in the H3 K[AcMe]G should be significantly smaller than predicted by the additive model, as mutating either the acetylated or methylated lysine residues would disrupt their interdependent functions.
Our analysis revealed that the gene expression changes in the H3 K[AcMe]G “double” mutant could be accurately modeled as the sum of the effects of the H3 K[Ac]G and H3 K[Me]G mutants, and thus fit the additive model. There were some exceptions: for a few genes, the increase in mRNA levels observed in the H3 K[Ac]G mutant was suppressed upon mutating the histone H3 methylated lysine residues, suggesting an epistatic interaction. Despite these exceptions, the data as a whole supported the additive model. In summary, our data suggest that the acetylated and methylated H3 lysine mutants make independent contributions to yeast gene expression. This finding is in accordance with the conclusion of a previous study, which showed that mutations in histone H4 acetylated lysine residues (H4 K5, K8, K12) had a cumulative effect on yeast gene expression (Dion et al. 2005).
Previous studies have shown that mutations that affect H3 K36 methylation can stimulate aberrant internal transcription to generate cryptic transcripts (Carrozza et al. 2005; Li et al. 2007b). Comparison of our microarray data with a profile of cryptic transcripts generated in a set2Δ mutant (Li et al. 2007b) revealed a significant overlap with each of the histone mutants, particularly the H3 K[Me]G mutant data set (supplemental Figure 3). This suggests that a subset of the upregulated genes observed in the histone mutants may be due to an increase in internally initiated cryptic sense transcripts, as expected from previous studies.
The functional independence of the histone H3 acetylated and methylated lysine residues is highlighted in their effects on telomeric silencing. Previous studies have indicated that both methylated and acetylated lysine residues in histone H3 regulate telomeric silencing. While we find that both the methylated and acetylated lysine mutants affect telomeric gene expression, they appear to do so through independent mechanisms. The H3 K[Me]G mutant causes a significant increase in telomeric gene transcription, and a significant decrease in Sir4 binding to telomere regions. In contrast, the H3 K[Ac]G mutant causes a small but significant increase in telomeric gene expression and disrupts the silencing of a telomere-located reporter gene, but has only a slight, insignificant effect on Sir4 binding. While it is formally possible that the H3 K[Ac]G mutant could affect the binding of other members of the SIR complex (e.g., Sir2 or Sir3), we do not favor this model. Our results show that binding of Sir4 (and presumably the SIR complex) is similarly disrupted in the H3 K[Me]G and H3 K[AcMe]G mutants, yet the H3 K[AcMe]G mutant has a significantly greater effect on telomeric gene expression than the H3 K[Me]G mutant. Taken together, these results suggest that acetylated lysine residues in histone H3 regulate telomeric silencing through a mechanism independent of SIR binding.
The histone H3 acetylated and methylated lysine mutants also affect the expression of genes located elsewhere in the yeast genome. Intriguingly, we observed that each of the histone H3 mutants upregulates a significant fraction of genes located in HAST chromosomal domains (P < 0.001). These domains were previously identified as subtelomeric regions that were depleted for histone acetylation by the Hda1 histone deacetylase (Robyr et al. 2002). This analysis suggests that acetylated and methylated lysine residues in histone H3 may play a direct role in regulating the expression of these genes.
Finally, our analysis indicates that the transcriptional repression exerted by histone H3 acetylated and methylated lysine residues is associated with the unmodified versions of these lysine residues. Hence, the genetic interactions we describe cannot necessarily be attributed to the loss of histone acetylation or methylation. Previous studies have shown that the SIR silencing proteins bind preferentially to the unmethylated forms of histone lysine residues (van Leeuwen and Gottschling 2002; Ng et al. 2003; Santos-Rosa et al. 2004). This observation could explain why telomeric genes are upregulated in the histone H3 lysine-to-glycine mutants, as this mutant eliminates the unmethylated lysine side chain, and thus should disrupt SIR binding. It is intriguing to speculate that the mutation of unacetylated lysine residues in histone H3 could disrupt the binding of other repressor proteins. Likely possibilities include the Tup1 repressor (Malave and Dent 2006) or SANT domain-containing proteins (Boyer et al. 2004), as these proteins bind preferentially to unacetylated chromatin (Edmondson et al. 1996; Davie et al. 2002; Boyer et al. 2004; Malave and Dent 2006).
Acknowledgments
We thank Julie Stanton and McKenna Kyriss for helpful comments on the manuscript. We thank Derek Pouchnik for technical assistance. We are grateful to Michael Grunstein for the gift of yeast strains and plasmids. This work was supported by American Cancer Society grant RSG-03-181-01-GMC. A.R. was supported by National Institutes of Health postdoctoral fellowship GM074541-01.
References
- Aparicio, J. G., C. J. Viggiani, D. G. Gibson and O. M. Aparicio, 2004. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol. Cell. Biol. 24 4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, B. E., E. L. Humphrey, R. L. Erlich, R. Schneider, P. Bouman et al., 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 99 8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, L. A., R. R. Latek and C. L. Peterson, 2004. The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 5 158–163. [DOI] [PubMed] [Google Scholar]
- Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li et al., 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14 115–132. [DOI] [PubMed] [Google Scholar]
- Briggs, S. D., M. Bryk, B. D. Strahl, W. L. Cheung, J. K. Davie et al., 2001. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk, M., S. D. Briggs, B. D. Strahl, M. J. Curcio, C. D. Allis et al., 2002. Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2-independent mechanism. Curr. Biol. 12 165–170. [DOI] [PubMed] [Google Scholar]
- Carey, M., Y. S. Lin, M. R. Green and M. Ptashne, 1990. A mechanism for synergistic activation of a mammalian gene by GAL4 derivatives. Nature 345 361–364. [DOI] [PubMed] [Google Scholar]
- Carrozza, M. J., B. Li, L. Florens, T. Suganuma, S. K. Swanson et al., 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123 581–592. [DOI] [PubMed] [Google Scholar]
- Davie, J. K., R. J. Trumbly and S. Y. Dent, 2002. Histone-dependent association of Tup1-Ssn6 with repressed genes in vivo. Mol. Cell. Biol. 22 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion, M. F., S. J. Altschuler, L. F. Wu and O. J. Rando, 2005. Genomic characterization reveals a simple histone H4 acetylation code. Proc. Natl. Acad. Sci. USA 102 5501–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant, M., and B. F. Pugh, 2006. Genome-wide relationships between TAF1 and histone acetyltransferases in Saccharomyces cerevisiae. Mol. Cell. Biol. 26 2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson, D. G., M. M. Smith and S. Y. Roth, 1996. Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev. 10 1247–1259. [DOI] [PubMed] [Google Scholar]
- Escargueil, A. E., D. G. Soares, M. Salvador, A. K. Larsen and J. A. Henriques, 2008. What histone code for DNA repair? Mutat. Res. 658 259–270. [DOI] [PubMed] [Google Scholar]
- Fingerman, I. M., C. L. Wu, B. D. Wilson and S. D. Briggs, 2005. Global loss of Set1-mediated H3 Lys4 trimethylation is associated with silencing defects in Saccharomyces cerevisiae. J. Biol. Chem. 280 28761–28765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth, A., W. Rocha, A. Verreault and G. Almouzni, 2007. Chromatin challenges during DNA replication and repair. Cell 128 721–733. [DOI] [PubMed] [Google Scholar]
- Hampsey, M., and D. Reinberg, 2003. Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell 113 429–432. [DOI] [PubMed] [Google Scholar]
- Hertel, K. J., K. W. Lynch and T. Maniatis, 1997. Common themes in the function of transcription and splicing enhancers. Curr. Opin. Cell. Biol. 9 350–357. [DOI] [PubMed] [Google Scholar]
- Holstege, F. C., E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner et al., 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95 717–728. [DOI] [PubMed] [Google Scholar]
- Howe, L., D. Auston, P. Grant, S. John, R. G. Cook et al., 2001. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 15 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L., J. N. Smith, S. L. Anderson, P. Ma, C. A. Mizzen et al., 2007. Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail. J. Biol. Chem. 282 27923–27934. [DOI] [PubMed] [Google Scholar]
- Jin, Y., A. M. Rodriguez, J. D. Stanton, A. A. Kitazono and J. J. Wyrick, 2007. Simultaneous mutation of methylated lysine residues in histone H3 causes enhanced gene silencing, cell cycle defects, and cell lethality in Saccharomyces cerevisiae. Mol. Cell. Biol. 27 6832–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, A. A., and K. Struhl, 2005. Eaf3 chromodomain interaction with methylated H3–K36 links histone deacetylation to Pol II elongation. Mol. Cell 20 971–978. [DOI] [PubMed] [Google Scholar]
- Keogh, M. C., S. K. Kurdistani, S. A. Morris, S. H. Ahn, V. Podolny et al., 2005. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123 593–605. [DOI] [PubMed] [Google Scholar]
- Krogan, N. J., J. Dover, S. Khorrami, J. F. Greenblatt, J. Schneider et al., 2002. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 277 10753–10755. [DOI] [PubMed] [Google Scholar]
- Kurdistani, S. K., and M. Grunstein, 2003. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 4 276–284. [DOI] [PubMed] [Google Scholar]
- Kurdistani, S. K., S. Tavazoie and M. Grunstein, 2004. Mapping global histone acetylation patterns to gene expression. Cell 117 721–733. [DOI] [PubMed] [Google Scholar]
- Lee, K. K., and J. L. Workman, 2007. Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8 284–295. [DOI] [PubMed] [Google Scholar]
- Lee, T. I., H. C. Causton, F. C. Holstege, W. C. Shen, N. Hannett et al., 2000. Redundant roles for the TFIID and SAGA complexes in global transcription. Nature 405 701–704. [DOI] [PubMed] [Google Scholar]
- Li, B., M. Carey and J. L. Workman, 2007. a The role of chromatin during transcription. Cell 128 707–719. [DOI] [PubMed] [Google Scholar]
- Li, B., M. Gogol, M. Carey, S. G. Pattenden, C. Seidel et al., 2007. b Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 21 1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. S., M. Carey, M. Ptashne and M. R. Green, 1990. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature 345 359–361. [DOI] [PubMed] [Google Scholar]
- Malave, T. M., and S. Y. Dent, 2006. Transcriptional repression by Tup1-Ssn6. Biochem. Cell Biol. 84 437–443. [DOI] [PubMed] [Google Scholar]
- Mani, R., R. P. St Onge, J. L. Hartman, IV, G. Giaever and F. P. Roth, 2008. Defining genetic interaction. Proc. Natl. Acad. Sci. USA 105 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, R. K., and M. Grunstein, 1992. Histone H3 N-terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO J. 11 3297–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, A. M., D. J. Pouchnik, J. L. Walker and J. J. Wyrick, 2004. Redundant roles for histone H3 N-terminal lysine residues in subtelomeric gene repression in Saccharomyces cerevisiae. Genetics 167 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. G., K. Baetz, X. Shi, K. L. Walter, V. E. MacDonald et al., 2006. a The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell. Biol. 26 7871–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. G., D. E. Grimes, K. Baetz and L. Howe, 2006. b Methylation of histone H3 mediates the association of the NuA3 histone acetyltransferase with chromatin. Mol. Cell. Biol. 26 3018–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar, C. B., and M. Grunstein, 2006. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 7 657–666. [DOI] [PubMed] [Google Scholar]
- Miller, T., N. J. Krogan, J. Dover, H. Erdjument-Bromage, P. Tempst et al., 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 98 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, S. A., B. Rao, B. A. Garcia, S. B. Hake, R. L. Diaz et al., 2007. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J. Biol. Chem. 282 7632–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, H. H., Q. Feng, H. Wang, H. Erdjument-Bromage, P. Tempst et al., 2002. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 16 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, H. H., D. N. Ciccone, K. B. Morshead, M. A. Oettinger and K. Struhl, 2003. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc. Natl. Acad. Sci. USA 100 1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, T. R., and J. J. Wyrick, 2007. ChromatinDB: a database of genome-wide histone modification patterns for Saccharomyces cerevisiae. Bioinformatics 23 1828–1830. [DOI] [PubMed] [Google Scholar]
- Pray-Grant, M. G., J. A. Daniel, D. Schieltz, J. R. Yates, III and P. A. Grant, 2005. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433 434–438. [DOI] [PubMed] [Google Scholar]
- Rao, B., Y. Shibata, B. D. Strahl and J. D. Lieb, 2005. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell. Biol. 25 9447–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robyr, D., Y. Suka, I. Xenarios, S. K. Kurdistani, A. Wang et al., 2002. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109 437–446. [DOI] [PubMed] [Google Scholar]
- Sabet, N., F. Tong, J. P. Madigan, S. Volo, M. M. Smith et al., 2003. Global and specific transcriptional repression by the histone H3 amino terminus in yeast. Proc. Natl. Acad. Sci. USA 100 4084–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa, H., A. J. Bannister, P. M. Dehe, V. Geli and T. Kouzarides, 2004. Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J. Biol. Chem. 279 47506–47512. [DOI] [PubMed] [Google Scholar]
- Shahbazian, M. D., and M. Grunstein, 2007. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76 75–100. [DOI] [PubMed] [Google Scholar]
- Shilatifard, A., 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75 243–269. [DOI] [PubMed] [Google Scholar]
- Suka, N., Y. Suka, A. A. Carmen, J. Wu and M. Grunstein, 2001. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 8 473–479. [DOI] [PubMed] [Google Scholar]
- Taverna, S. D., S. Ilin, R. S. Rogers, J. C. Tanny, H. Lavender et al., 2006. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell 24 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna, S. D., B. M. Ueberheide, Y. Liu, A. J. Tackett, R. L. Diaz et al., 2007. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. USA 104 2086–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. S., X. Ling and M. Grunstein, 1994. Histone H3 amino terminus is required for telomeric and silent mating locus repression in yeast. Nature 369 245–247. [DOI] [PubMed] [Google Scholar]
- Tompa, R., and H. D. Madhani, 2007. Histone H3 lysine 36 methylation antagonizes silencing in Saccharomyces cerevisiae independently of the Rpd3S histone deacetylase complex. Genetics 175 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen, F., and D. E. Gottschling, 2002. Genome-wide histone modifications: gaining specificity by preventing promiscuity. Curr. Opin. Cell Biol. 14 756–762. [DOI] [PubMed] [Google Scholar]
- Vogelauer, M., L. Rubbi, I. Lucas, B. J. Brewer and M. Grunstein, 2002. Histone acetylation regulates the time of replication origin firing. Mol. Cell 10 1223–1233. [DOI] [PubMed] [Google Scholar]
- Wan, J. S., R. K. Mann and M. Grunstein, 1995. Yeast histone H3 and H4 N termini function through different GAL1 regulatory elements to repress and activate transcription. Proc. Natl. Acad. Sci. USA 92 5664–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick, J. J., F. C. Holstege, E. G. Jennings, H. C. Causton, D. Shore et al., 1999. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 402 418–421. [DOI] [PubMed] [Google Scholar]
- Xu, F., Q. Zhang, K. Zhang, W. Xie and M. Grunstein, 2007. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol. Cell 27 890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. J., and E. Seto, 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K., J. S. Siino, P. R. Jones, P. M. Yau and E. M. Bradbury, 2004. A mass spectrometric “Western blot” to evaluate the correlations between histone methylation and histone acetylation. Proteomics 4 3765–3775. [DOI] [PubMed] [Google Scholar]