

Abstract
The F-box proteins β-TrCP 1 and 2 (β-transducin repeat protein) have 2 and 3 isoforms, respectively, due to alternative splicing of exons encoding the N-terminal region. We identified an extra exon in between the previously known exons 1 and 2 of β-TrCP1 and β-TrCP2. Interestingly, sequence analysis suggested that many more isoforms are produced than previously identified, via the alternative splicing of all possible combination of exons II to V of β-TrCP1 and exons II to IV of β-TrCP2. Different mouse tissues show specific expression patterns of the isoforms, and the level of expression of the isoform that has been used in most published papers was very low. Yeast two-hybrid assays show that β-TrCP1 isoforms containing exon III, which are the most highly expressed isoforms in most tissues, do not interact with Skp1. Indirect immunofluorescence analysis of transiently expressed β-TrCP1 isoforms suggests that the presence of exon III causes β-TrCP1 to localize in nuclei. Consistent with the above findings, isoforms including exon III showed a reduced ability to block ectopic embryonic axes induced via injection of Wnt8 or β-catenin in Xenopus embryos. Overall, our data suggest that isoforms of β-TrCPs generated by alternative splicing may have different biological roles.
Keywords: β-TrCP, isoforms, alternative splicing, β-catenin, Wnt, Ubiquitination
1. Introduction
The ubiquitin proteasome pathway plays pivotal roles in degradation of the majority of eukaryotic proteins [1]. E3 ligases determine substrate specificity and function as adapters that recruit target proteins to a complex containing an E2 enzyme [1]. One means of protein ubiquitination involves the formation of the SCF ligase complex, which contains the proteins Skp1, Cdc53/Cullin, and F-box protein. F-box protein is a type of E3 ligase and contains a highly conserved sequence known as the F-box, which mediates interaction with Skp1 and serves as an adapter to bridge the substrate to Skp1 and Cdc53/Cullin [2-4].
Beta-Transducin repeat-containing proteins (β-TrCPs) are F-box proteins that are highly conserved from Drosophila to human. They contain two essential modular domains, a 42-48 amino acid F-box motif at the N-terminus and seven WD40 repeats at the C-terminus, which are involved in interaction with target substrates [5, 6]. β-TrCP recognizes phosphorylated serine residues of target substrates in DSGXXS motifs, which are processed by the ubiquitin/proteasome pathway [7, 8]. In the case of β-catenin, priming phosphorylation is carried out by CKI and then the protein is sequentially phosphorylated by GSK3β in the Axin-scaffolding complex [9]. Besides β-catenin, β-TrCP 1 and its ortholog β-TrCP 2 serve as E3 ligases for proteins involved in various signal transduction pathways (IκBα for NFκB signaling, Smad3 for TGFβ signaling, Ci for Hedgehog signaling), transcriptional regulation (Rest and Sp1) and cell cycle control (Wee1 and Emi1) [4, 10-13]. Therefore, the regulation of β-TrCP activity is considered to be very important to sustain the balance of a variety of cellular signaling pathways.
Several reports of the regulation of β-TrCP activities have been published. First, an increase of a specific substrate among the β-TrCP substrates may potentially influence the availability of β-TrCP to other substrates. For example, since Vpu functions as a pseudosubstrate for β-TrCP, β-TrCP binding to IκBα can be inhibited [14, 15]. Also, dominant interaction of RASSF1C, an isoform of the tumor suppressor RASSF1A, with β-TrCP promotes the accumulation of β-catenin [16]. Second, the substrate specificity of β-TrCP can be controlled by the localization of β-TrCPs. Endogenous and transiently transfected β-TrCP1 and β-TrCP2 are mainly localized in the nucleus and cytoplasm, respectively [17, 18], although Winston et al. showed that the transiently transfected β-TrCP1 is localized in the cytoplasm [19]. Davis et al. showed that if a substrate, targeted to β-TrCP1, is present in cytoplasm, a pseudosubstrate hnRNP-U exports β-TrCP1 from nucleus to cytoplasm, which allows β-TrCP1 to form a SCF complex for the down-regulation of the substrate [17]. Third, activation of the stress-activated protein kinase pathway increases mRNA levels of β-TrCP1 [20]. Fourth, in some case, alternative splicing of mRNA for certain F-box proteins could be regulated. For example, only an isoform of the F-box protein Skp2 is able to ubiquitinate cyclinD1 [21].
Although the functional differences are not known, β-TrCP isoforms generated by alternative splicing are present in both Xenopus and human, and the genomic organization exons as well as the size and amino acid composition of potential protein domains is evolutionarily conserved [22-24]. Xenopus β-TrCP isoforms show differences in the NH2 region, as well as in the COOH region [22]. The two human genes β-TrCP1 and β-TrCP2 can generate two or three variants by alternative splicing, respectively [23, 24].
In this study, we have identified a new exon for both β-TrCP1 and β-TrCP 2, and we report the presence of many more β-TrCP1/2 isoforms than previously known. We show that sequences encoded by exon III influence the cellular localization of the isoforms and their interaction with Skp1. The isoforms that use exon III are mainly localized in nuclei. Consistent with the fact that β-TrCP mediated degradation of β-catenin occurs mainly in cytoplasm, the isoforms that use exon III are much less able to rescue Wnt8 or β-catenin-induced embryo axis duplication. Overall, our data suggest that isoforms of β-TrCP generated via alternative splicing may have different target specificity and biological roles.
2. Materials and methods
2. 1. Cell cultures and transient transfection
C57mg, HEK 293T and MCF7 cells were grown in Dulbecco’s modified Eagle medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Cambrex Bio Science Walkersville, Inc.) and 1X antibiotic-antimycotic (catalog number 15240; Invitrogen) in humidified 6% CO2. Cells were transiently transfected using either the calcium phosphate precipitation method or WelFect-EX™ PLUS transfection reagent (WelGENE), in accordance with the manufacturer’s instructions. Leptomycin B (LMB) was purchased from Sigma and cells were treated with 50 ng/ml for 8 hours before immunofluorescence analysis.
2.2. Reverse Transcriptase-PCR
Total RNAs isolated from cell lines, different tissues and embryos of CD-1(ICR) mice were reverse-transcribed using random primers (5’-NNNNNN-3’) and ImProme II reverse transcriptase (Promega), and amplification was carried out at 94°C for 2 min, followed by 30 cycles at 94°C for 1 min, at appropriated annealing temperatures for each primer for 1 min, and at 72°C for 50 sec. The PCR products were separated on 2% agarose gel. The primers used for PCR are summarized in supplementary Table I.
2.3. Construction of plasmids
The RT-PCR products generated with primers TR5 and TR6 were cloned into pCRII vector (Invitrogen) and isoform ‘b’ of full-length β-TrCP1, shown in Table 1, was cloned. More isoforms of the N-terminal region of β-TrCP1 were cloned into pCRII vector after RT-PCR with primers TR5 and TR10. To make full-length β-TrCP1 isoforms, AflII and SacI fragment for the N-terminal region of β-TrCP1 isoforms, obtained via RT-PCR with primers TR5 and TR10, were replaced with the same region of full-length isoform ‘b’ in pCRII vector. To make plasmids for the expression of Myc-tagged β-TrCP1 isoforms in mammalian cells, DNA fragments containing full-length β-TrCP1 isoforms in pCRII vector were excised and cloned into pCS2-MT vector. To generate plasmids for Gal4DB-β-TrCP1 isoforms fusion proteins in yeast, BamHI and BglII fragment (N-terminal to the middle region of first WD domain) of β-TrCP1 isoforms was inserted into the BamHI and BglII sites of pGBT9 vector (Clonetech). pGAD-hSkp1, pGBD-hCullin 1 and hCullin3 were kindly provided from Dr. Xiong [25]. All constructs were sequenced to confirm that the coding sequences for Gal4DB-β-TrCP and Myc-epitope tagged β-TrCP isoforms were in frame. The cDNAs for human skp1, elongin B and elongin C was isolated by RT-PCR using total RNA of HEK 293T cells and cloned into pCMV4-FLAG vector and confirmed by sequencing. The expression plasmid for VHL was obtained from Dr. HS Park (University of Seoul).
Table 1.
N-terminal region of β-TrCP1 isoforms due to alternative splicing of exons II to V
| EST clones in GenBank
|
Interaction with Skp1 & mouse β-TrCP1 isoforms | ||
|---|---|---|---|
| Exons of Isoforms | Mouse | Human | |
|
|
BU612794 | DA792532 | - |
|
|
BY727249 | BX508858 | - |
|
|
BY061122 | BX505115 | -/+ |
|
|
BY279190 | - | - |
|
|
BY294694 | - | - |
|
|
BI103092 | DB453223 | ++ |
|
|
BY109771 | - | - |
|
|
- | - | - |
|
|
- | - | + |
|
|
CJ080331 | - | ND |
|
|
BY713732 | BP289988 | ND |
|
|
BY307082 | CX165131 | ND |
|
|
- | - | ND |
|
|
- | - | - |
|
|
CJ084503 | - | +++ |
|
|
BY290279 | - | ND |
2.4. Identification of BAC clones
A mouse BAC library (RPCI-22M; Invitrogen) was screened with the 5’-end of mouse β-TrCP1 or β-TrCP2 cDNA as a probe. Positive BAC clones (for β-TrCP1: 358E23, 337B6, 437J23 and 298N12; for β-TrCP2: 533I9, 421C11 and 394J6) were identified. BAC DNAs were purified using Large-Construct Kit (QIAGEN) and sequenced with primers specific for individual exons.
2.5. Polyribosomal RNA isolation
Polysomes were isolated using the protocol as described [26] with some modification. Briefly, 200 mg of mouse brain were lysed with a Dounce homogenizer in 2 ml of polysomal lysis buffer (20 mM HEPES-KOH (pH 7.2), 100 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, 50 μg/ml cycloheximide, 0.1 mM phenylmethylsulfonyl fluoride, and 0.5% Nonidet P-40). Cell debris and nuclei were removed by centrifugation for 10 min at 8000 g. The polysomes were pelleted by ultracentrifugation at 4°C for 1.5 h in HITACHI P65ST rotor at 44,000 rpm. Polyribosomal RNAs were isolated using TriZol solution (Invitrogen).
2.6. Yeast two-hybrid analysis
The pGBD-β-TrCP1 isoform plasmids (marker TRP1) were co-transformed with pGAD-hSkp1 (marker LEU2) into the yeast AH109 strain according to the manual from Clontech (Matchmaker™ GAL4 Two-hybrid system). Yeast transformants were grown on synthetic medium lacking leucine, tryptophan, adenine and histidine, and containing p-nitrophenyl-α-D-galactoside (PNP-α-Gal). The expression of HIS, ADE and MEL1 reporter genes in AH109 stain were used to determine plasmids encoding β-TrCP1 isoforms that interact with Skp1.
2.7. Western blot analysis and co-immunoprecipitation
To test interaction between β-TrCP1 isoforms and Skp1, elongin B/C, plasmids encoding these proteins were transfected into HEK 293T cells. Western blot and co-immunoprecipitiation experiments were performed as previously described [27].
2.8. Immunofluorescence analysis
MCF7 cells were plated in six-well plates covered with glass coverslips, then transfected with 2 μg of isoforms of mouse β-TrCP1 cDNAs via WelFect-EX™ PLUS transfection reagent (WelGENE). One day after transfection, the cells were fixed for 20 minutes in 4% formaldehyde in PBS, permeabilized for 20 minutes in 0.4% Triton in PBS, and incubated for 30 minutes at room temperature with blocking solution (5% bovine serum albumin (Sigma) in PBST (0.05% Tween 20 in PBS)). The cells were then incubated for 1 hour at room temperature with anti-FLAG monoclonal antibody (1:200, Calbiochem) in blocking solution. The cells were washed with PBST, incubated in rhodamine-conjugated secondary antibody (1:200, KPL) for 1 hour at room temperature, mounted, and examined via fluorescence microscopy (LEICA). The nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI) after incubation with the secondary antibody.
2.9. Xenopus embryo manipulation, axis duplication assay
Eggs were obtained from Xenopus laevis primed with 800 units of human chorionic gonadotropin (Sigma). They were in vitro fertilized using macerated testis. Developmental stages were determined according to Nieuwkoop and Faber [28]. mRNAs were synthesized using the mMessage mMachine kit (Ambion) following the manufacturer’s protocol. They were injected into the ventral-vegetal one blastomere of 8-cell-stage embryos. Axis duplication was monitored at stage 37, and embryos were classified as complete axis duplication, incomplete axis duplication, DAI 6 or normal. The phenotypes for the classification are followings: complete axis duplication, duplicated trunk and complete secondary head structures including duplicated cement gland and pair of eyes; incomplete axis duplication, duplicated trunk only or duplicated trunk and incomplete head structures; DAI 6, no duplicated trunk but slight dorsalization.
3. Results
3.1. Molecular cloning of alternatively spliced isoforms of β-TrCP1 and analysis of its genomic organization
In the process of cloning mouse β-TrCP1, PCR reactions were performed with primers as shown in Fig. 1A. Unexpectedly, when primers TR5 and TR15 were used for PCR, multiple bands were amplified (Fig. 1B). Two of them were expected, since it is known that the presence or absence of exon III due to alternative splicing of the β-TrCP1 gene could produce two isoforms (Fig. 1A). Sequences analysis of all bands in Fig.1B showed that an extra exon is present in between the previously known exons I and III (the nucleotide and amino acids sequences for this newly identified exon are shown in Fig. 1C) and alternative splicing of the exons II to V may occur randomly and theoretically produce 16 different isoforms (Table 1). Most of the sequences that can be produced via alternative splicing are present in a mouse EST database (Table 1). Genomic DNA analysis using the Blat program at UCSC Genome Bioinformatics Site (http://genome.ucsc.edu/index.html) shows that this new exon sequence is present in between previously known exon I and exon III in chromosome 19 (Fig. 1D). A mouse BAC library was screened using a cDNA fragment encoding the N-terminal end of mouse β-TrCP1 as a probe. Sequences analysis of several positive clones was performed and we found that a clone (clone ID: 437J23) included the 42 bp sequences in between exon I and exon III. These data suggest that this 42 bp sequence is a bona fide exon.
Fig. 1.
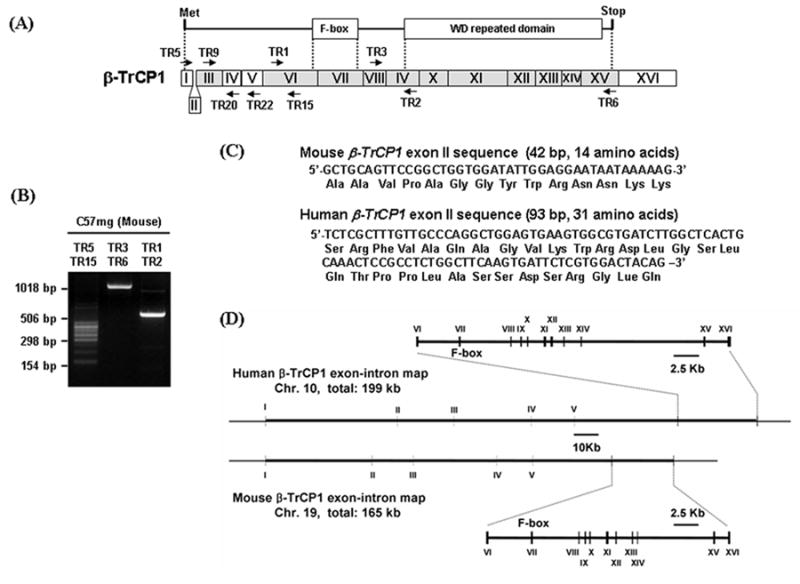
Identification of an extra exon of β-TrCP1 and its genomic organization. A. Two major domains of β-TrCP1, the F-box that interacts with skp1 and the WD repeat domain that binds to substrate, are shown in the top diagram. Organization of exons for β-TrCP1 is shown in the bottom diagram. The regions of exons encoding F-box and WD repeated domain are indicated by dotted lines. The exon identified in this work is designated as exon II and shown in between exon I and III. Shaded boxes show exons with high amino acid sequence similarity between β-TrCP1 and β-TrCP2. Arrows show the primers used for RT-PCR. B. RT-PCR products with RNA isolated from C57mg cells with primers indicated on top of the gel. C. Sequences of the newly identified ‘Exon II’ and encoded peptide for mouse and human β-TrCP1. D. Genomic organization of β-TrCP1 as determined using the BLAT program.
When we searched human EST database to examine the presence of exon II that was found in mouse β-TrCP1, we could not find similar sequences. However, instead of having the 42 bp exon II sequence as in mouse, human β-TrCP1 has a 93 bp exon II sequence which may encode 31 amino acids and the sequence of human exon II does not show any homology to that of mouse (Fig. 1C). The human exon II sequence is present in many EST clones and it exists in between human β-TrCP1 exon I and III.
3.2. Molecular cloning of alternatively spliced isoforms of β-TrCP2 and analysis of its genomic organization
It has been shown that the β-TrCP1 related gene, β-TrCP2 has three isoforms due to alternative splicing of exons III and IV (Fig. 2A, [24]). One isoform includes exon III, another includes exon IV and the other includes neither exon III nor IV. Since we found extensive alternative splicing in β-TrCP1, we examined whether β-TrCP2 also has more isoforms than previously known. A similar analysis of β-TrCP2 revealed that β-TrCP2 also contains an extra exon (162 or 156 bp in human and mouse, respectively) in between the previously known exons I and III and encodes eight isoforms, instead of the previously known three isoforms (Fig. 2A and Table 2). The genomic organization of human and mouse β-TrCP2 is shown in Fig. 2C.
Fig. 2.
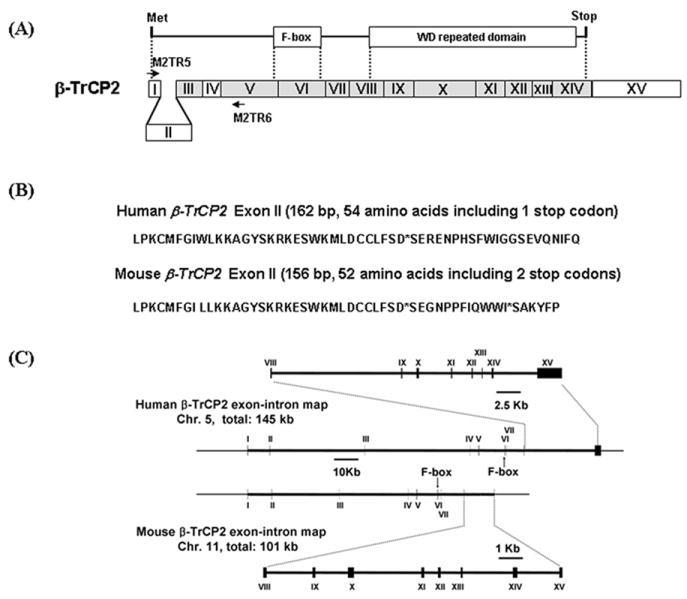
Identification of an extra exon of β-TrCP1 and its genomic organization. A. F-box and WD repeated domain of β-TrCP2 are shown in the top diagram. Organization of exons for β-TrCP2 is shown in the bottom diagram. The regions of exons encoding F-box and WD repeated domain are indicated by dotted lines. The exon identified in this work is designated exon II and shown in between exon I and III. Shaded boxes show exons with high amino acid sequence similarity between β-TrCP2 and β-TrCP1. Arrows show the primers used for RT-PCR. B. Sequences of the newly identified ‘Exon II’ and encoded peptide for mouse and human β-TrCP1. C. Genomic organization of β-TrCP2 as determined using the BLAT program.
Table 2.
N-terminal region of β-TrCP2 isoforms due to alternative splicing of exons II to IV
Interestingly, the new exon II contains one and two stop codons, in human and mouse respectively, in the proper open reading frame from the first exon (Fig. 2B). When the exon II sequences were queried in EST database, many ESTs, which have the same exon II sequences along with other exons, were found. Similar to the case of β-TrCP1, ESTs for most of the possible alternatively spliced exons in II to IV (Table 2) were found. The presence of stop codons in exon II was confirmed via sequencing of mouse BAC clones. A clone (clone ID: 394J6) has sequences for exon II to IV and the exon II has the same stop codons that were identified in ESTs.
3.3. Tissue specific distribution of alternatively spliced isoforms of β-TrCP1 and β-TrCP2
Total RNAs from different mouse tissues were examined by RT-PCR to determine the tissue-specific distribution of alternatively spliced isoforms of β-TrCP1 and β-TrCP2. Surprisingly, while the ‘a’ and ‘b’ isoforms (Table 1) were present as major bands in all tissues, the isoform ‘f’, which has been used in most of published papers by other researchers [14, 18, 19], was very low in all tissues except for colon and testis. The relative expression pattern of isoforms is somewhat different from tissue to tissue (Fig. 3A).
Fig. 3.
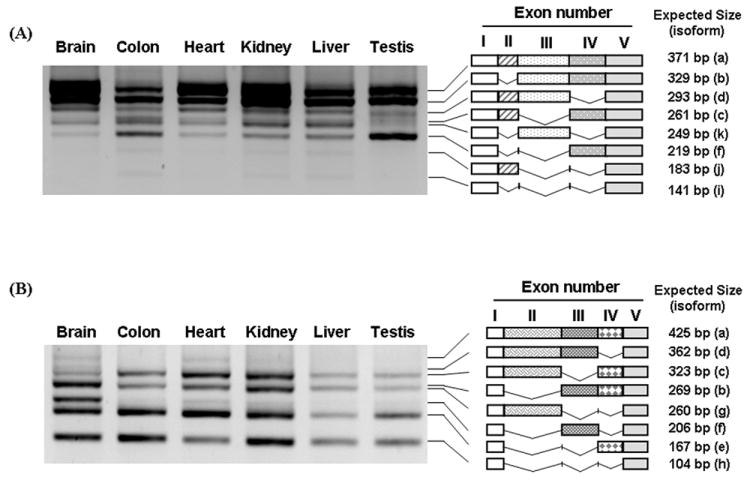
Expression pattern of β-TrCP isoforms in different tissues. Total RNAs from different mouse tissues were isolated and RT-PCR was performed with the primers specific to exon I and Exon V of β-TrCP1 and 2. The PCR products are marked with schematic diagrams of isoforms that are similar to the expected size of β-TrCP1 (A) and β-TrCP2 isoforms (B). The expected sizes of DNA are shown in the right panel and the name of each isoforms is shown in parenthesis.
Similar expression patterns of β-TrCP2 isoforms were found in tissues examined in our experiments except that more isoform ‘f’ and less isoform ‘c’ is expressed in brain than other tissues (Fig. 3B and Table 2). The isoforms that contain exon II were also found in all tissues. Since we identified one or two stop codons, for human and mouse β-TrCP2, respectively, in the same open reading frame from the initiation codon in exon I, the cDNAs that contain exon II may not encode functional β-TrCP2 isoforms (Fig. 2A and B). This raises the interesting possibility that alternative splicing (i.e., inclusion of exon II) could serve as a regulatory mechanism to control the level of β-TrCP2.
We tested whether the expression pattern of β-TrCPs can be changed in several different conditions. As shown in supplementary Fig. 1, the expression pattern of isoforms did not vary at different developmental stage of mouse embryos.
3.4. The detection of β-TrCP1/2 isoforms in polyribosomal fraction
To rule out the possibility that some isoforms detected by RT-PCR are splicing intermediates rather than real, mature mRNAs, we tried to generate antibodies that could specifically detect endogenous β-TrCP1/2 isoforms, but were unsuccessful. In the absence of isoform-specific antibodies, we sought additional evidence supporting idea that these isoforms are mature forms. One source of evidence was the presence of these RNAs in the cytosolic polyribosomal fraction. The pattern of PCR products generated by RT-PCR with polyribosomal RNA was almost identical with that generated using total RNA (Fig. 4). These data suggest that alternatively spliced isoforms of β-TrCPs detected by RT-PCR represent bona fide mRNAs which are translated in vivo. However, as we cannot be sure that all the isoforms that we found via RT-PCR are normally translated in vivo, we performed functional studies mainly using the isoforms that are published by others and present in GenBank as full length cDNAs (isoforms ‘a’, ‘b’ and ‘f’).
Fig. 4.
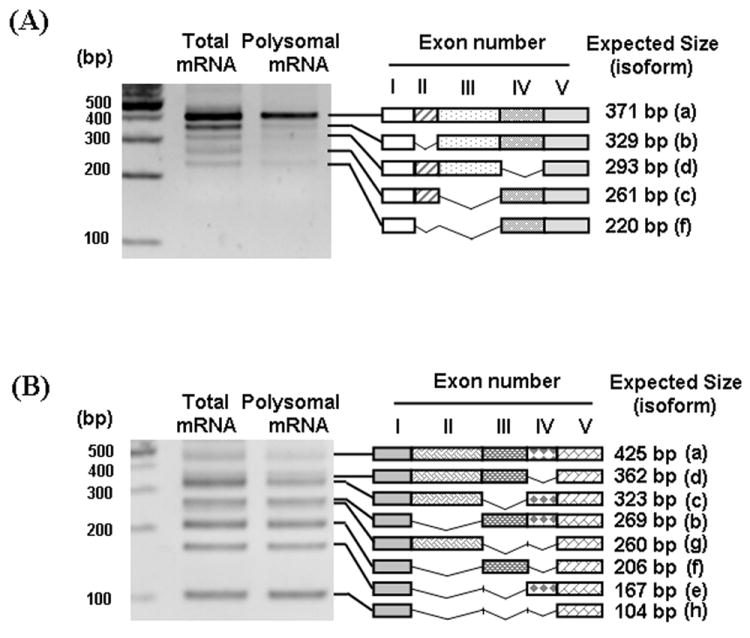
Presence of β-TrCP1/2 isoforms in polyribosomal fraction. To determine if the alternatively spliced isoforms are translated in vivo, polyribosomal RNAs were isolated from the mouse brain and RT-PCR was performed with the primers specific to exon I (TR5 and M2TR5) and exon V (TR22 and M2TR6) of β-TrCP1 (A) and β-TrCP2 (B), respectively. Schematic diagrams of isoforms that are similar to the expected size of β-TrCP1 and β-TrCP1 isoforms were shown on the right side of gels.
3.5. Different cellular localization of β-TrCP1 isoforms
It has been reported that β-TrCP1 is mainly localized in nuclei while β-TrCP2 is present in cytoplasm [17, 18]. This result was unexpected, since cytoplasmic β-catenin is generally considered as a target of β-TrCP1. We tested whether the subcellular localizations of isoforms of β-TrCP1 are different. We tested 2 isoforms for each group; ‘a’ and ‘b’, which contain exon III, and ‘f’ and ‘o’, which do not have exon III. Indirect immunofluorescence after transfection of these constructs into MCF7 cells showed that isoform ‘a’ or ‘b’ tends to be localized mainly in nuclei, while in contrast, isoform ‘f’ or ‘o’ was mainly localized either in cytoplasm only, or in both cytoplasm and nuclei (Fig. 5). Because the antibody used by others to examine the localization of endogenous β-TrCP1 detects all isoforms, it detected mainly the most abundant isoforms that contain exon III (Fig. 3A and Fig. 5). Transiently transfected Skp1 is localized in both cytoplasm and nuclei and β-TrCP2 is mainly localized in cytoplasm (Fig. 5). Leptomycin B (LMB) is known to block shuttling of proteins from the nucleus to the cytoplasm [29]. Interestingly, in cells transfected with isoforms ‘a’ or ‘b’, LMB increased the nuclear localization of these isoforms, while it had no effect on the pattern of localization in cells transfected with isoforms ‘f’ or ‘o’ (Fig. 5). The nuclear localization of isoforms ‘a’ and ‘b’, which do not interact with Skp1 (see Fig.6), suggests that these isoforms may not be involved in the down-regulation of β-catenin in cytoplasm. These results support the idea that β-TrCP1 isoforms may have different biological roles in the regulation of Wnt/β-catenin signaling in vivo.
Fig. 5.
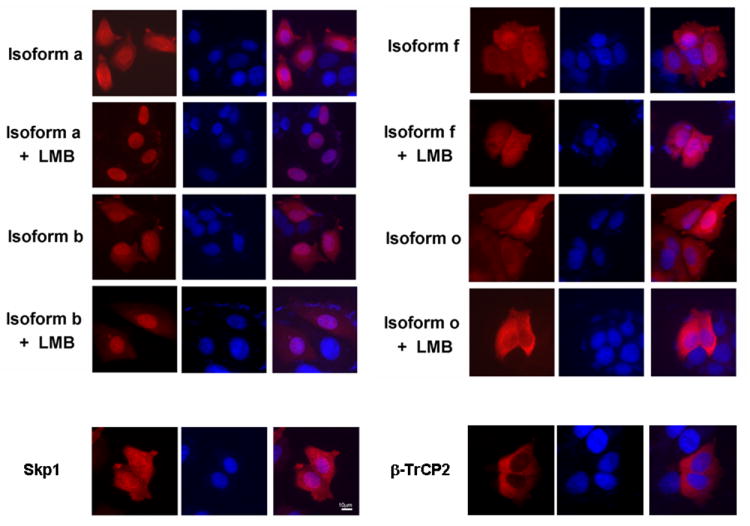
Differential cellular localization of β-TrCP1 isoforms. Isoforms of β-TrCP1, Skp1 or β-TrCP2 were transiently transfected into MCF7 cells and the cellular localization was analyzed. Rhodamine-conjugated secondary antibody was used to detect the localization of transfected proteins (left panel) and nuclei were stained with DAPI (middle panel). The two merged pictures are shown in the right panel. Isoforms ‘a’ and ‘b’, which have exon III, are mainly localized in nuclei and treatment of cells with LMB enhances nuclear localization. In contrast, isoforms ‘f’ and ‘o’ are present in both cytoplasm and nuclei and treatment of LMB does not alter the localization of these isoforms. Skp1 is present in both cytoplasm and nuclei and β-TrCP2 isoform ‘h’ is mainly localized in cytoplasm.
Fig. 6.
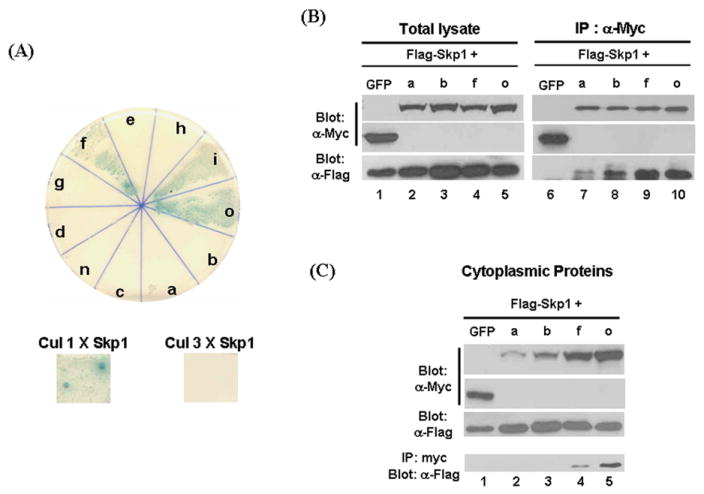
Tests for interaction of β-TrCP1 isoforms with Skp1. A. Yeast two-hybrid analysis was used to examine differential interaction of Skp1 with isoforms of β-TrCP1. Yeast strain AH109 was co-transfected with pGBD-β-TrCP1 isoforms and pGAD-Skp1 and plated on Leu, Trp, His and Ade drop-off plate with PNP-α-Gal. Introduction of pGBD-β-TrCP1 and pGAD-Skp1 vectors allows AH109 cells to grow on the plate lacking Trp and Leu, respectively. If β-TrCP1 interacts with Skp1, AH109 cells are able to grow on the plate lacking on Leu, Trp, His and Ade. In addition, the colonies will show blue color due to breakdown of PNP-α-Gal by the induced expression of α-galactosidase. Only isoforms that lack exon III show interaction with Skp1 (top panel and Table 1). Since it has been shown that Cullin 1 interacts with Skp1 while Cullin 3 doesn’t, pGBD-Cullin 1 with pGAD-Skp1 and pGBD-Cullin 3 with pGAD-Skp1 were used positive and negative controls for interaction, respectively (bottom panel.). B. HEK 293T cells were co-transfected with myc-epitope tagged β-TrCP1 isoforms and Flag-epitope tagged Skp1 and co-immunoprecipitation experiments were performed. In contrast to the results of yeast two-hybrid assays, each isoform did not show obvious differential interaction with Skp1. C. Isoforms of β-TrCP1 and Skp1 were transfected into HEK 293T cells and cytoplasmic fractions were isolated. The western blot data is consistent with result shown in Fig. 6. Co-immunoprecipitation experiments suggest that β-TrCP1 isoforms ‘a’ and ‘b’ cannot form a complex with Skp1 for the down-regulation of β-catenin in cytoplasm.
3.6. Differential interaction of β-TrCP1 isoforms with Skp1
We hypothesized that amino acid changes in the N-terminal region of β-TrCP1, generated via alternative splicing, may cause conformational changes and thus influence the interaction between β-TrCP1 isoforms and Skp1. To test this hypothesis, we used the yeast-two hybrid system. As none of full-length β-TrCP1 isoforms interacted with Skp1 in this assay, we suspected that β-TrCP1 might be too big to use in the yeast-two hybrid assay. Therefore, we instead cloned the sequences encoding the region N-terminal to the middle of the first WD domain of β-TrCP1 isoforms into yeast two-hybrid vector (pGBD vector) and examined their ability to interact with Skp1 (in pGAD vector). Isoforms a, b, c, d, e, f, g, h, i, n and o were tested for interaction with Skp1. Interestingly, only isoforms f, i and o showed a strong positive signal, whose intensity was similar to that due to the interaction between Skp1 and Cullin 1, which was used as a positive control ([25]. Fig. 6A and Table 1). In our repeated experiments isoform ‘c’ showed variable results (Table 1). Thus, the isoforms containing exon III (isoforms a, b, d, e, g, h and n) did not interact with Skp1 at all, while the presence of exon II (in isoform c) had a subtle negative effect on the interaction (Fig. 6 and Table 1). Overall, the results suggest that the presence of peptide sequences encoded by exon III via alternative splicing in the N-terminal region of β-TrCP1 inhibits the interaction of β-TrCP1 isoforms with Skp1.
To test differential interaction of β-TrCP1 isoforms with Skp1 in mammalian cells, β-TrCP1 isoforms were cloned in a mammalian expression vector. Myc-epitope tagged mouse β-Trcp1 isoforms ‘a or b’, which have exon III, or isoforms ‘f or o’, which do not, were co-transfected with Flag epitope-tagged Skp1 into HEK 293T cells and co-immunoprecipitation experiments were performed. However, contrary to the results of yeast two hybrid assays, all isoforms interacted with Skp1, although isoforms ‘a or b’ seemed to have much weaker interaction (Fig. 6B). While we cannot explain this discrepancy, we think that yeast two-hybrid assay is more stringent to test interaction of proteins than co-immunoprecipitation experiments of overexpressed two proteins. The co-immunoprecipitation assay may not show subtle differences in the ability of β-TrCP isoforms to interact with Skp1.
Since down-regulation of β-catenin mainly occurs in the cytoplasm, the interaction in cytoplasm between β-TrCP and Skp1 may be critical for the function of β-TrCP1. HEK 293T cells were transfected with Flag epitope-tagged Skp1 and myc epitope-tagged β-TrCP1 isoforms and co-immunoprecipitation experiments were performed with cytoplasmic proteins (Fig. 7C). Consistent with the data shown in Fig.6, the level of isoforms ‘a or b’ is much less than that of ‘f or o’ in the cytoplasmic fraction (Fig. 7C, top row)). As expected, there is no Skp1 co-immunopreciptated with isoforms ‘a or b’. Thus, although isoforms ‘a and b’ are capable of forming a weak complex with Skp1 (figure 7B), because of the low level in the cytoplasm, isoforms ‘a or b’ do not appear to be involved in the down-regulation of β-catenin in cytoplasm.
Fig. 7.
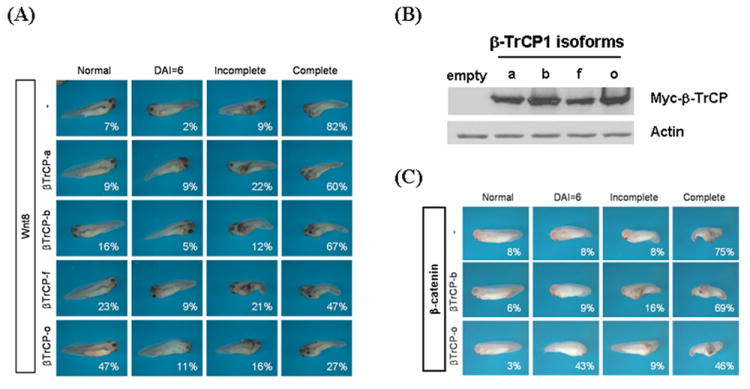
β-TrCP1 isoforms have different effect on the Wnt-mediated induction of embryonic axis. A. Injection of 5 pg of Wnt8 into ventral side of Xenopus egg leads to embryonic axis duplication (82% of complete axis duplication and 7 % normal embryos, respectively, first row). Co-injection of Wnt8 and β-TrCP1 isoform ‘a’ or ‘b’ neither significantly inhibit the formation of complete axis duplication nor increase the formation of normal embryos (second and third rows). In contrast, co-injection of Wnt8 and β-TrCP1 isoform ‘f’ or ‘o’ not only severely reduces the formation of complete axis duplication (47% and 27%, respectively) but increase the normal embryos (23% and 47 %, respectively, third and fourth rows). B. The level of β-TrCP1 isoforms injected into Xenopus embryos was almost identical. Total lysates were isolated from Xenopus embryos 18 h after injection of RNAs and western blot analysis was performed. C. Axis duplication induced by the injection of β-catenin is rescued by isoform ‘o’ but not by ‘b’.
We realized that the amino acid sequences encoded by exon III of β-TrCP1 and β-TrCP2 include a conserved sequence known as a BC box ([30] and Supplementary Fig. 2). It is known that elongin C, which is a Skp1 family protein, forms a complex with elongin B and binds to BC box-containing E3 ubiquitin ligase proteins such as VHL [30]. We tested whether isoform ‘b’, which contains exon III, could interact with elongin B/C complex. While elongin B/C could be co-immunopecipitated with VHL, β-TrCP1 isoform ‘b’ did not interact with elongin B/C complex (Supplementary Fig. 2). Therefore, the peptide sequences encoded by exon III are not sufficient for the interaction with elongin B/C.
In summary, the results from yeast two-hybrid analysis and co-immunoprecipitation experiments using cytoplasmic proteins suggest that the isoforms that contains exon III do not normally form a complex with Skp1 in vivo and may not be involved in the down-regulation of β-catenin in cytoplasm. However, it is still possible that these isoforms interact with Skp1 in the nuclei and control the level of β-catenin.
3.7. Isoforms have different abilities to inhibit Wnt signaling
Since β-TrCP is known to down-regulate the level of cytoplasmic β-catenin and Tcf reporter activity, we tested whether different isoforms have differential effects on the level of cytoplasmic β-catenin. Contrary to previous reports [8, 31], our results were highly variable even with the isoform ‘f’ that others have used to show down-regulation of β-catenin. Sometimes ectopic expression of β-TrCP1 could block Wnt-induced luciferase activity as published, but it enhanced Wnt-mediated luciferase activity in most cases in HEK 293T cells (data not shown.). However, these data are consistent with the data published by Belaidouni et al. [32], which showed that overexpressed β-TrCP1 acts as a dominant negative and enhances the level of β-catenin. Other isoforms also showed similar patterns in reporter assays, and there were no apparent differences among isoforms (data not shown.). Our luciferase assay was reliable, since we could easily detect the blocking of Wnt or β-catenin induced luciferase activity by the ectopic expression of Axin, which is a well-known inhibitor of Wnt signaling (data not shown).
To examine the differential roles of β-TrCP1 isoforms in vivo, experiments were performed using Xenopus embryos (Fig. 7). Injection of 5 pg of Wnt8 into the ventrovegetal side of Xenopus egg leads to embryonic axis duplication. Interestingly, co-injection of Wnt8 and 500 pg of β-TrCP1 isoforms that do not contain exon III and are mainly localized in cytoplasm (isoforms ‘f’ and ‘o’) significantly reduce Wnt8-induced embryonic axis duplication, while the isoforms ‘a’ and ‘b’ that contain exon III and are mainly localized in nuclei do not (Fig. 7A). For example, the percentage of embryos displaying complete rescue to “normal” was increased by only 2 and 9 % by the co-injection of isoforms ‘a’ and ‘b’, while it was increased by 16 and 40% by the isoforms ‘f’ and ‘o’, respectively. The protein level of injected β-TrCP1 isoforms was almost identical in all cases (Fig. 7B). Interestingly, injection of 4-fold more of the β-TrCP1 isoforms (2 ng instead of 500 pg) failed to rescue significantly (data not shown.), which confirms that overexpression of β-TrCP1 does not down-regulate the cytoplasmic level of β-catenin, as described above. Axis duplication induced by the injection of β-catenin could be rescued much more efficiently by the co-injection of isoform ‘o’ than isoform ‘b’ (Fig. 7C). The percentage of the completely rescued phenotype is not increased, but the percentage of embryos similar to normal (DAI, dorsal–anterior index = 6) is increased from 8 to 43 by the co-injection of isoform ‘o’ (Fig. 7C). To determine whether the rescue of axis duplication is specifically caused by the down-regulation of Wnt/β-catenin signaling, we tested if TcfVP16 (Tcf conjugated with VP16 activation domain) mediated axis duplication could be rescued by the co-injection of β-TrCP1 isoforms. None of β-TrCP1 isoforms could rescue TcfVP16-mediated axis duplication (Supplementary Fig. 3). Overall, these data strongly suggest that specific isoforms of β-TrCP1 have the ability to inhibit Wnt/β-catenin signaling in vivo.
4. Discussion
Alternative splicing is a major regulatory mechanism to control gene usage and increase coding capacity, and misregulation of this process is known to underlie the pathogenesis of diverse human diseases [33]. Alternative splicing is known to be a regulatory mechanism for F-box proteins, which are responsible for the regulation of proteins that control diverse cellular signaling events. A Skp2 isoform generated by alternative splicing of the C-terminal region alters Skp2 function by blocking interaction with Skp1 and its localization into the nucleus [21]. In addition, alternative splicing isoforms of Fbw7 (α, β, γ) localize to discrete subcellular compartments and differ in their ability to specifically degrade cyclin E [34, 35]. Therefore, alternative splicing of F-box proteins may add another level of complexity to the regulation of target proteins.
It has been reported that β-TrCP1 and β-TrCP2 have 2 or 3 isoforms, respectively, while the significance of the presence of multiple isoforms was not examined [22-24]. Since most researchers did not pay attention to the isoforms of β-TrCP, it is not clear which isoforms were used in their studies, although we think that β-TrCP1 isoform ‘f’ and β-TrCP2 isoform ‘h’, shown in Table 1 and 2, are used in most published papers [22, 36, 37]. In this report, we show that an extra exon is present in between the previously known exons 1 and 2 and extensive alternative splicing occurs in the N-terminal region of β-TrCPs. Cloning of RT-PCR products and sequence analysis of ESTs in the GenBank database suggest that many more isoforms are generated via almost all the possible combinations of alternative splicing in exons II to V for β-TrCP1 and exons II to IV for β-TrCP2. The presence of most isoforms in the polyribosomal fraction suggests that these mRNAs are not intermediates of splicing, but are translated mRNAs (Fig. 4). To determine if these isoforms are indeed expressed in vivo, we tried to generate isoform specific antibodies, but were not successful. However, a previous report [38] showed that a β-TrCP1 specific antibody recognizes three isoform bands, and we obtained similar data when we used the same antibody (data not shown.). In addition, since many full length cDNAs have been reported for isoforms ‘a’, ‘b’ and ‘f’, we focused mainly on these three isoforms in our functional studies. While we do not know whether individual β-TrCP1 isoforms have different biological roles, their differential ability to interact with Skp1 (measured by the yeast two hybrid assay), differences in cellular localization, and differing ability to rescue Wnt/β-catenin induced axis duplication of Xenopus embryos (which depends on the presence of peptide sequences encoded by exon III), suggest that at least these two classes of isoforms (‘a’ and ‘b’ vs. ‘f’) may have different biological roles in vivo (Fig. 5-7).
The differential activities for rescuing Wnt-mediated Xenopus embryonic axis duplication clearly suggest that β-TrCP1 isoforms have different biological roles in vivo. Our data suggest that the β-TrCP1 isoforms such as ‘f’ and ‘o’ that are localized in the cytoplasm are involved in the down-regulation of β-catenin. What, then, are the roles of isoforms that are mainly localized in nuclei? Several target proteins for β-TrCP, such as ATF, Emi1, REST and Sp1 [11-13, 18], are known to be localized in nuclei. It is possible that the isoforms localized in nuclei specifically control these target proteins, although we did not test that possibility here. If the isoforms that have peptide sequences encoded by exon III do not interact with Skp1, as suggested by the yeast two-hybrid assay, how can these isoforms behave as E3 ligases? Is there an unknown protein that works as Skp1? Further studies are needed to understand the regulation of alternative splicing and other roles of β-TrCPs in vivo.
Supplementary Material
Expression pattern of β-TrCP1 isoforms in different developmental stages of mouse embryos. Total RNAs from three different developmental stages of mouse embryo were isolated and RT-PCR was performed with the primers specific to exon I and V of β-TrCP1. No variation was seen in the overall pattern of alternatively spliced isoforms.
Testing interaction of isoforms of β-TrCP1 with Skp1 or elongin B/C by co-immunoprecipitation. A. Conservation of elongin B/C box sequences encoded by exon III of β-TrCP1. The conserved sequences known as B/C box are marked with bold letters. B. Lack of interaction between isoforms of β-TrCP1 and elongin B/C. HEK 293T cells were transiently transfected with Flag epitope-tagged Elongin B and C and with Myc epitope-tagged isoforms of β-TrCP1 or HA-epitope tagged VHL, which is known to interact with Elongin B and C. Lanes 1-4, western blot analysis shows the proper expression of each transfected protein. Lanes 5-8, after immunoprecipitation with anti-Myc antibody, the blot was probed with anti-Myc and anti-HA (top panel) anti-Flag antibody (bottom panel). Elongin B/C does not interact with β-TrCP1 isoforms, while they interact with VHL. Each expressed protein is marked on the right side of the blot.
Injection of 5pg of Tcf3-VP16 induces ectopic axis formation similarly to Wnt8 does (first row). However, co-injection of any of the β-TrCP1 isoforms neither significantly inhibits the formation of axis duplication nor increases the percentage of normal embryos (second to fifth rows).
Acknowledgments
This work was supported by the National R&D Program for Cancer Control and the Biomedical Brain Research Center of the Korea Health 21 R&D Project funded by the Ministry of Health and Welfare, Republic of Korea (Grant 0520230 and A040042 to E. Jho), Advanced Basic Research Laboratory Program of KRF(R14-2002-012-01001-0 to JK Han) and the National Institutes of Health (grant 5R01HD044265 to F.C.). E. Seo, H. Kim and M. Kim were supported by the Brain Korea 21 program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deshaies RJ. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 2.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. Cell. 1996;86(2):263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 3.Patton EE, Willems AR, Tyers M. Trends Genet. 1998;14(6):236–243. doi: 10.1016/s0168-9525(98)01473-5. [DOI] [PubMed] [Google Scholar]
- 4.Maniatis T. Genes Dev. 1999;13(5):505–510. doi: 10.1101/gad.13.5.505. [DOI] [PubMed] [Google Scholar]
- 5.Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. Nature. 1994;371(6495):297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs SY, Spiegelman VS, Kumar KG. Oncogene. 2004;23(11):2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 7.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Embo J. 1997;16(13):3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hart M, Concordet JP, Lassot I, Albert I, del los Santos R, Durand H, Perret C, Rubinfeld B, Margottin F, Benarous R, Polakis P. Curr Biol. 1999;9(4):207–210. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 9.Kimelman D, Xu W. Oncogene. 2006;25(57):7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. Proc Natl Acad Sci U S A. 2004;101(13):4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann JD, Jackson PK. Dev Cell. 2003;4(6):813–826. doi: 10.1016/s1534-5807(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 12.Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, Leng Y, Maehr R, Shi Y, Harper JW, Elledge SJ. Nature. 2008;452(7185):370–374. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spengler ML, Guo LW, Brattain MG. Cell Cycle. 2008;7(5):623–630. doi: 10.4161/cc.7.5.5402. [DOI] [PubMed] [Google Scholar]
- 14.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. Mol Cell. 1998;1(4):565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 15.Bour S, Perrin C, Akari H, Strebel K. J Biol Chem. 2001;276(19):15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 16.Estrabaud E, Lassot I, Blot G, Le Rouzic E, Tanchou V, Quemeneur E, Daviet L, Margottin-Goguet F, Benarous R. Cancer Res. 2007;67(3):1054–1061. doi: 10.1158/0008-5472.CAN-06-2530. [DOI] [PubMed] [Google Scholar]
- 17.Davis M, Hatzubai A, Andersen JS, Ben-Shushan E, Fisher GZ, Yaron A, Bauskin A, Mercurio F, Mann M, Ben-Neriah Y. Genes Dev. 2002;16(4):439–451. doi: 10.1101/gad.218702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lassot I, Segeral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, Benarous R, Margottin-Goguet F. Mol Cell Biol. 2001;21(6):2192–2202. doi: 10.1128/MCB.21.6.2192-2202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. Genes Dev. 1999;13(3):270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spiegelman VS, Stavropoulos P, Latres E, Pagano M, Ronai Z, Slaga TJ, Fuchs SY. J Biol Chem. 2001;276(29):27152–27158. doi: 10.1074/jbc.M100031200. [DOI] [PubMed] [Google Scholar]
- 21.Ganiatsas S, Dow R, Thompson A, Schulman B, Germain D. Oncogene. 2001;20(28):3641–3650. doi: 10.1038/sj.onc.1204501. [DOI] [PubMed] [Google Scholar]
- 22.Ballarino M, Marchioni M, Carnevali F. Biochim Biophys Acta. 2002;1577(1):81–92. doi: 10.1016/s0167-4781(02)00416-5. [DOI] [PubMed] [Google Scholar]
- 23.Chiaur DS, Murthy S, Cenciarelli C, Parks W, Loda M, Inghirami G, Demetrick D, Pagano M. Cytogenet Cell Genet. 2000;88(34):255–258. doi: 10.1159/000015532. [DOI] [PubMed] [Google Scholar]
- 24.Koike J, Sagara N, Kirikoshi H, Takagi A, Miwa T, Hirai M, Katoh M. Biochem Biophys Res Commun. 2000;269(1):103–109. doi: 10.1006/bbrc.2000.2241. [DOI] [PubMed] [Google Scholar]
- 25.Michel JJ, Xiong Y. Cell Growth Differ. 1998;9(6):435–449. [PubMed] [Google Scholar]
- 26.Yang C, Li YC, Kuter DJ. Br J Haematol. 1999;105(2):478–485. [PubMed] [Google Scholar]
- 27.Fagotto F, Jho E, Zeng L, Kurth T, Joos T, Kaufmann C, Costantini F. J Cell Biol. 1999;145(4):741–756. doi: 10.1083/jcb.145.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis (Daudin) New York: Garland Publishing; 1994. [Google Scholar]
- 29.Henderson BR, Eleftheriou A. Exp Cell Res. 2000;256(1):213–224. doi: 10.1006/excr.2000.4825. [DOI] [PubMed] [Google Scholar]
- 30.Botuyan MV, Koth CM, Mer G, Chakrabartty A, Conaway JW, Conaway RC, Edwards AM, Arrowsmith CH, Chazin WJ. Proc Natl Acad Sci U S A. 1999;96(16):9033–9038. doi: 10.1073/pnas.96.16.9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spiegelman VS, Slaga TJ, Pagano M, Minamoto T, Ronai Z, Fuchs SY. Mol Cell. 2000;5(5):877–882. doi: 10.1016/s1097-2765(00)80327-5. [DOI] [PubMed] [Google Scholar]
- 32.Belaidouni N, Peuchmaur M, Perret C, Florentin A, Benarous R, Besnard-Guerin C. Oncogene. 2005;24(13):2271–2276. doi: 10.1038/sj.onc.1208418. [DOI] [PubMed] [Google Scholar]
- 33.Caceres JF, Kornblihtt AR. Trends Genet. 2002;18(4):186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 34.Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. Curr Biol. 2004;14(20):1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Koepp DM. Mol Cancer Res. 2006;4(12):935–943. doi: 10.1158/1541-7786.MCR-06-0253. [DOI] [PubMed] [Google Scholar]
- 36.Cenciarelli C, Chiaur DS, Guardavaccaro D, Parks W, Vidal M, Pagano M. Curr Biol. 1999;9(20):1177–1179. doi: 10.1016/S0960-9822(00)80020-2. [DOI] [PubMed] [Google Scholar]
- 37.Kroll M, Margottin F, Kohl A, Renard P, Durand H, Concordet JP, Bachelerie F, Arenzana-Seisdedos F, Benarous R. J Biol Chem. 1999;274(12):7941–7945. doi: 10.1074/jbc.274.12.7941. [DOI] [PubMed] [Google Scholar]
- 38.Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. J Biol Chem. 2005;280(12):11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression pattern of β-TrCP1 isoforms in different developmental stages of mouse embryos. Total RNAs from three different developmental stages of mouse embryo were isolated and RT-PCR was performed with the primers specific to exon I and V of β-TrCP1. No variation was seen in the overall pattern of alternatively spliced isoforms.
Testing interaction of isoforms of β-TrCP1 with Skp1 or elongin B/C by co-immunoprecipitation. A. Conservation of elongin B/C box sequences encoded by exon III of β-TrCP1. The conserved sequences known as B/C box are marked with bold letters. B. Lack of interaction between isoforms of β-TrCP1 and elongin B/C. HEK 293T cells were transiently transfected with Flag epitope-tagged Elongin B and C and with Myc epitope-tagged isoforms of β-TrCP1 or HA-epitope tagged VHL, which is known to interact with Elongin B and C. Lanes 1-4, western blot analysis shows the proper expression of each transfected protein. Lanes 5-8, after immunoprecipitation with anti-Myc antibody, the blot was probed with anti-Myc and anti-HA (top panel) anti-Flag antibody (bottom panel). Elongin B/C does not interact with β-TrCP1 isoforms, while they interact with VHL. Each expressed protein is marked on the right side of the blot.
Injection of 5pg of Tcf3-VP16 induces ectopic axis formation similarly to Wnt8 does (first row). However, co-injection of any of the β-TrCP1 isoforms neither significantly inhibits the formation of axis duplication nor increases the percentage of normal embryos (second to fifth rows).