

Abstract
With the growing epidemic of obesity in an aging population, obstructive sleep apnea (OSA) is increasingly encountered in clinical practice. Given the acute cardiopulmonary stressors consequent to repetitive upper airway collapse, as well as evidence for cardiovascular homeostatic dysregulation in subjects with sleep apnea, there is ample biologic plausibility that OSA imparts increased cardiovascular risk, independent of comorbid disease. Indeed, observational studies have suggested strong associations with multiple disorders, such as systemic hypertension, heart failure, cardiac arrhythmias, and pulmonary hypertension. Further data in the form of longitudinal cohort studies and randomized controlled trials are accruing to add to the body of evidence. This review examines pathophysiologic mechanisms and explores current concepts regarding the impact of OSA and its treatment on selected clinical disease states.
Keywords: sleep-disordered breathing, positive airway pressure, arrhythmia, stroke
OBSTRUCTIVE SLEEP APNEA: ACUTE PATHOPHYSIOLOGIC MECHANISMS
Acute cardiovascular (CV) stressors resulting from repetitive episodes of upper airway narrowing and/or occlusion characteristic of obstructive sleep apnea (OSA) include hypoxemia, reoxygenation, swings in intrathoracic pressure, and central nervous system (CNS) arousals. Plausibly, these effects are cumulative over time, potentially forming the basis for heightened CV risk in individuals with OSA. There is evidence that CV homeostatic mechanisms in subjects with OSA are disrupted, as demonstrated by daytime abnormalities in sympathetic nervous system function and heart rate variability (1).
There is considerable evidence that hypoxemia, in part by stimulation of peripheral arterial chemoreceptors, drives some important aspects of the pathophysiology in OSA. Stimulation of the chemoreflex increases sympathetic efferent traffic during hypoxemic stimulation, as demonstrated by direct peripheral intraneural electrode recordings (2, 3) Those with OSA have been found to have an exaggerated chemoreflex response to hypoxemic stimulation, resulting in acute peripheral vasoconstriction and consequent acute increases in arterial blood pressure (BP). Under conditions of uninterrupted ventilation, lung inflation serves to homeostatically maintain autonomic balance on account of stimulation of lung and chest wall stretch receptors mediated by vagal neural circuits. This sympatholysis is incomplete during the apneas and hypopneas characteristic of OSA (4, 5), thus contributing to heightened sympathetic tone.
Each acute oxyhemoglobin desaturation is coupled to an episode of reoxygenation, a process thought to promote oxidative stress through formation of reactive oxygen species (6, 7), a cascade that may be associated with heightened inflammation (8, 9) and mitochondrial dysfunction (10).
Typical inspiratory efforts against an obstructed upper airway during apneas can result in marked reductions in intrathoracic pressure, as measured by esophageal pressure, and have been associated with acute changes in pulmonary arterial pressures and blood flow (11) and increased cardiac afterload. Enhanced venous return that may occur with reduced intrathoracic pressure can result in acute leftward intraventricular septal shift (12) and alterations in transmural cardiac pressures (13), with impedance of left ventricular (LV) filling (14) and increase in myocardial oxygen demand.
Apneas and hypopneas terminate with CNS arousals, forming the basis for sleep fragmentation and neurocognitive sequelae in OSA (15). CNS arousals are also associated with important effects on CV function, resulting in abrupt increases in sympathetic tone, heart rate, and BP (16, 17).
INTERMEDIARY MECHANISMS OF POTENTIAL IMPORTANCE IN CONFERRING CARDIOVASCULAR RISK
Daytime neural circulatory control is disturbed in subjects with OSA, even in the absence of overt CV disease. In part on the basis of increased tonic chemoreflex drive, heightened sympathetic tone is evident during normal waking hours in some patients with OSA, even under conditions of normoxia (1, 18). Abnormalities in variability of both heart rate and BP, both of which have been found to be markers of future cardiovascular disease in population-based studies (19), are present in OSA (18). The presence of endothelial dysfunction in OSA, as evidenced by a blunted small-vessel dilatory response to vasoactive substances, such as acetylcholine in some (but not all [20]) studies, may also be an important marker of CV risk (21–23). There is evidence to support the role of reduced levels of the vasodilator, nitric oxide, in the mediation of vascular disease and BP regulation in OSA (24), whereas levels of serum endothelin, a potent vasoconstrictor, may be higher in patients with OSA compared with control subjects (25).
Other features of OSA that may indirectly increase the risk for cardiovascular disease include a propensity for glucose intolerance (26), systemic inflammation, as suggested by an increase in serum C-reactive protein levels and up-regulation of leukocyte adhesion factors (27, 28), and abnormalities in coagulation markers (29).
Notwithstanding this and other mechanistic pathways, establishing causality in the relationship between OSA and clinical CV disease has been difficult, in large part because of shared risk factors—in particular, obesity and advancing age, both of which are primary determinants of sleep-disordered breathing, systemic hypertension, heart failure (HF), and pulmonary hypertension (PH), rendering the disentanglement of the independent effects of OSA on clinical disease challenging. Moreover, there is a relative paucity of high-level, evidence-based data, such as interventional treatment trials of OSA in the setting of CV disease. As such, much of the above findings are derived from case–control studies, some of which, it should be noted, have rendered negative associations between OSA and other biomarkers associated with CV risk, including serum levels of brain natriuretic peptide (30) and troponin T (31).
OSA AND SELECTED CARDIOVASCULAR DISEASES
Systemic Hypertension
Disordered breathing events during sleep are associated with well recognized acute peripheral vasoconstriction and attendant rises in BP during sleep (1). Further evidence is mounting to support a probable causative role for OSA in diurnal hypertension as well. Data on the impact of OSA treatment on BP, particularly with continuous positive airway pressure (CPAP) therapy, are accumulating, but are not always consistent.
In normal individuals, sleep is associated with a reduced BP when compared with wakefulness, referred to as the “dipping” phenomenon, when systolic and diastolic BP may decline as much as 10–15% (32, 33). Sleep apnea has been found to blunt the dipping of BP during sleep, a finding that may confer heightened cardiovascular risk (34).
Observational studies have shown that hypertension and OSA often coexist and that subjects with OSA tend to have higher BPs than matched controls (35, 36). Longitudinal studies have built on these associations, the most notable from the Wisconsin Sleep Cohort, which provides prospective evidence implicating OSA as a possible causal factor in hypertension (35). Specifically, the presence of hypertension 4 years after initial assessment was found to be dependent upon the severity of OSA at baseline.
CPAP has been shown to acutely attenuate sympathetic drive and nocturnal BP in patients with OSA (1, 37, 38). However, the data regarding effects on daytime BP have been more difficult to interpret. A number of observational studies, often uncontrolled and from highly select populations, have suggested improvements in daytime BP control with the use of CPAP. Because of an apparent true placebo effect realized in measurement of BP, randomized, placebo-controlled studies, a number of which have been published and yielded variable results, may be the best indicator of the antihypertensive effects of CPAP. The largest trial to date comes from Pepperell and colleagues (39), who found a small but significant reduction in BP in a normotensive group of subjects over 4 weeks of CPAP therapy. Data from Becker and colleagues (40), who conducted a controlled trial testing more than 60 days of CPAP treatment, showed the most dramatic reductions in mean BP (9.9 ± 11.4 mm Hg) in a small cohort with severe OSA (mean apnea–hypopnea index [AHI] > 60/h) (Figure 1). Notably, there was a high rate of subject dropout (the data from these subjects were not included in an intention-to-treat analysis), and the majority of subjects were treated with various antihypertensive medications. These two studies were included in a very recent meta-analysis of 12 placebo-controlled, randomized trials (572 patients), which found a statistically significant pooled reduction in mean BP of 1.69 mm Hg associated with CPAP treatment in OSA (41). That most of the trials were limited to normotensive individuals leaves the door open to further research on the BP-lowering properties of OSA treatment in hypertensive populations.
Figure 1.

Changes in mean arterial blood pressure (MAP) over 20 hours after treatment with (A) therapeutic continuous positive airway pressure (CPAP) and (B, lower) subtherapeutic CPAP. Reprinted by permission from Reference 40. nCPAP = nasal CPAP.
Cardiac Arrhythmias and Cardiovascular Mortality
A number of observational studies have shown an association between OSA and various nocturnal arrhythmias. Recent data from the Sleep Heart Health Study, after adjusting for many confounders, showed that, compared with subjects with a respiratory disturbance index less than 5, those with severe OSA (respiratory disturbance index ⩾ 30) had a higher rate of atrial fibrillation, nonsustained ventricular tachycardia, and ectopic ventricular beats (42). Bradyarrhythmias are commonly encountered in OSA, may correlate with the severity of disordered breathing, can occur with a structurally normal heart, and may be attenuated by effective CPAP therapy (43–45). The Sleep Heart Health Study described above, however, found similar rates of bradycardias and conduction delays between those with severe OSA and those without significant OSA.
Mounting data strengthen the association between OSA and atrial fibrillation, two disorders that often coexist (46). Continuous cardiac monitoring with an atrial defibrillator showed that the onset of nearly 75% of episodes of persistent atrial fibrillation in patients with OSA occurred in the overnight hours (8 p.m.–8 a.m.) (47) Retrospective analysis shows that, within 12 months of successful therapeutic electrical cardioversion for atrial fibrillation, untreated subjects with OSA were found to have an arrhythmia recurrence rate double that of patients treated with CPAP (48).
Recent review of 17 years of polysomnographic data from a population-based cohort suggests that nocturnal hypoxemia associated with OSA influences the incidence of atrial fibrillation (49). Because none of these observational data can convincingly implicate OSA as an independent cause of new onset atrial fibrillation, additional longitudinal cohort studies and outcome-based interventional trials are needed to characterize the relationship between OSA and atrial arrhythmias.
Ventricular arrhythmias have been reported in patients with OSA (42, 50), although a causative role for sleep apnea in serious arrhythmias or sudden death has not been definitively proven. Recent data provided by review of polysomnographic measures in 112 patients with sudden death suggest a markedly higher rate of lethal cardiac events between the hours of midnight and 6 a.m. in those with OSA compared with those without, along with a direct correlation between AHI and risk of death during the night (51). Although the study suggests that OSA may influence time of sudden cardiac death, it does not clearly demonstrate that OSA heightens the risk of sudden death from cardiac causes.
In the longest-term prospective cohort study yet published (10 yr), Marin and colleagues (52) demonstrated a higher risk of both fatal and nonfatal cardiovascular events in men with severe OSA who were noncompliant with CPAP therapy compared with snorers, treated patients with OSA, and healthy men (Figure 2). Although biased by potential and difficult-to-measure influences related to treatment noncompliance and imbalances in some confounding variables at baseline (such as prevalence of hypertension and glucose intolerance), this study is among the most persuasive to argue that OSA has detrimental effects on long-term CV outcomes.
Figure 2.

Cumulative percentage of men who had (A) fatal and (B) nonfatal cardiovascular events over more than 10 years of follow-up. The upper tracing in each graph represents men who had severe obstructive sleep apnea–hypopnea and were noncompliant with continuous positive airway pressure (CPAP) therapy. CVS = cardiovascular system; OSAH = obstructive sleep apnea–hypopnea. Reprinted by permission from Reference 52.
Cerebrovascular Disease and Stroke
Several studies have investigated the association between stroke and sleep-disordered breathing (53–55). A large prospective study showed self-reported snoring to be an independent risk factor for stroke in women (56). Until recently, associations with OSA have been reported primarily in cross-sectional and case-control studies, so it is unclear if OSA is a direct contributor to stroke incidence, as comorbidities and risk factors are commonly seen in both diseases. However, reports from two observational cohorts have helped strengthen this association. Using data from the Wisconsin Sleep Cohort, Arzt and colleagues (57) showed moderate to severe sleep-disordered breathing to be a risk factor for prevalent stroke and, with serial polysomnographic data, demonstrated that the preexisting sleep disorder may be a risk factor for incident stroke. Yaggi and colleagues (58) reported longitudinal data (mean follow-up, 3.4 yr) on mortality from stroke and other causes in more than 1,000 patients with preexisting OSA, showing an increasing risk of events with OSA severity. Although not powered to detect potential differences related to treatment of OSA, and in contrast to findings in the Marin cohort (52), there did not appear to be treatment effects in more than half of patients who were either treated with CPAP, lost weight, or underwent upper airway surgery.
It is feasible that stroke, particularly as represented in case–control studies, may itself predispose to sleep-disordered breathing. This may relate to disruption of central respiratory control mechanisms, leading to central sleep apnea or brainstem-mediated upper airway reflexes that may cause obstructive apneas or hypopneas. Indeed, in a report of 161 inpatients with acute stroke or transient ischemic attack who underwent studies at baseline and after 3 months, over 70% had an AHI greater than 10 (59). Nearly one-third of apneas were central in origin during the acute phase. At 3 months, however, the central apneas were significantly reduced, whereas the obstructive events remained stable. This could suggest that OSA preceded, and perhaps contributed to, stroke, whereas central apneas resulted from the acute neurologic event.
In addition to effects on atherogenesis and blood vessel function noted previously here, a number of other mechanisms may predispose to stroke in OSA. The strong association with atrial fibrillation may confer a heightened risk of embolic events. Furthermore, OSA has been shown to promote thrombosis, as evidenced by enhanced platelet aggregation (60) and activation (61), elevated fibrinogen levels (62), and diminished fibrinolytic activity (63). Finally, Doppler measurements have suggested that apneic events are associated with reduced cerebral blood flow (64, 65), which can result in cerebral hypoxia (66). Although CPAP treatment has been shown to reverse some of these findings (67, 68), the impact of treatment on the occurrence of stroke and death, as suggested by the study be Yaggi and colleagues (58), may be limited and needs further evaluation.
HEART FAILURE
By their strong associations with aging and obesity, HF and OSA are closely linked, with the prevalence of OSA, approaching 40% in patients with HF referred to a clinical sleep laboratory (69). Further evidence linking OSA to HF comes from the Framingham study, which showed that increasing body mass index (BMI) is directly correlated with incident HF (70), an effect that may be mediated, at least in part, by OSA. Incident atrial fibrillation, an important risk factor for HF, is associated with the degree of oxyhemoglobin desaturation in OSA (49). The cascade of physiological responses to repetitive upper airway closure in OSA may exert deleterious effects on cardiac function, particularly in the already compromised heart. Despite advances in treatment with drugs, lifestyle modifications, and therapeutic devices, mortality from HF continues to rise. Thus, there is increasing interest in the role of OSA treatment on outcomes in HF.
Two controlled, interventional trials of CPAP for OSA in the setting of HF have been cited frequently for their positive impact on various CV variables (71, 72). Using a randomized, parallel comparative design, the control groups in both studies were comprised of subjects optimally medically managed, though not subjected to placebo. Kaneko and colleagues (71) reported an approximately 9% increase in LV ejection fraction (LVEF) and significant reductions in BP after just 1 month of CPAP therapy. Mansfield and colleagues (72), studying a group of subjects with somewhat less severe degrees of both HF and OSA than those reported in the Kaneko paper, applied CPAP therapy for 3 months and showed significant improvements in LVEF and reductions in urinary catecholamines, but no changes in BP.
In an unexpected turn of events, Smith and colleagues (73) very recently published the results of a rigorous, placebo-controlled cross-over study of CPAP in a population with a similar degree of HF as the previous trials, and found no improvement in any parameter of CV function, including LVEF, BP, and exercise tolerance. Although these findings may relate in part to methodologic limitations, such as the lack of a follow-up polysomnogram to confirm treatment efficacy with autotitrating CPAP (74), the currently limited data regarding the impact of OSA treatment on important HF endpoints calls for further interventional trials.
PULMONARY HYPERTENSION
In 1947, Motley and colleagues (75) demonstrated that breathing a gas mixture containing 10% oxygen induced a rise in pulmonary arterial pressure (PAP). This hypoxic pulmonary vasoconstriction is a critical autoregulatory mechanism important in maintaining an appropriate 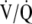 relationship (76). Over time, hypoxic vasoconstriction may result in pulmonary vascular remodeling, which may or may not be reversible (77), potentially contributing to the development of PH, as demonstrated in populations with advanced lung disorders, such as chronic obstructive pulmonary disease. It may follow then, that repetitive upper airway collapse and oxyhemoglobin desaturations characteristic of OSA could also provide a pathophysiologic basis for chronic elevations in PAP.
relationship (76). Over time, hypoxic vasoconstriction may result in pulmonary vascular remodeling, which may or may not be reversible (77), potentially contributing to the development of PH, as demonstrated in populations with advanced lung disorders, such as chronic obstructive pulmonary disease. It may follow then, that repetitive upper airway collapse and oxyhemoglobin desaturations characteristic of OSA could also provide a pathophysiologic basis for chronic elevations in PAP.
Precisely defining the role of OSA in the genesis of PH has been difficult for a number of reasons. First, one limitation has been the various methods by which the diagnosis of PH is made in studies of subjects with OSA, many by way of Doppler echocardiography, with varying right heart/PAP thresholds. Currently, PH is defined as a mean PAP greater than 25 mm Hg at rest or 30 mm Hg with exercise, as measured by right heart catheterization (78). Previous definitions were based upon systolic PAP greater than 40 mm Hg and echocardiographic Doppler measurements, which may be particularly challenging to obtain in obese patients with OSA. Second, as in other disease states mentioned previously here, PH and OSA share common risk factors—namely, obesity and aging, which confound risk factor associations. In fact, a pulmonary artery systolic pressure (PASP) greater than 40 mm Hg is found in 6% of otherwise normal individuals age 50 years or older, and in 5% of individuals with a BMI greater than 30 kg/m2 (79). Third, finding appropriate control groups with which to compare endpoints (i.e., matched subjects with PH but no OSA) is challenging. Nevertheless, based upon some literature examining the relationship between OSA and PH, the latest revision of the Clinical Classification of Pulmonary Hypertension identifies sleep-disordered breathing as part of the category of respiratory disorders associated with PH (80). Limited epidemiologic data, coming from numerous case series, comprised primarily of male patients, suggest a prevalence of PH in OSA ranging from 17 to 52% (81–83). The largest published sample to date numbers 220 subjects with OSA, of whom 17% met diagnostic criteria for PH (82). Population-based data are currently lacking.
Papers dating back more than three decades have documented increases in PAP associated with sleep-related hypoxemia. Coccagna and colleagues (84) continuously measured PAP during sleep in 10 patients with sleep-disordered breathing and found sleep stage–dependent increases in PAP, with more marked changes occurring during rapid eye movement sleep. Most early clinical studies suggested that abnormalities in underlying lung function sufficient to induce daytime hypoxemia were required for the development of PH and right heart failure (85, 86). Further supporting this notion was the finding that the severity of sleep-disordered breathing, as measured by the AHI, and PAP elevations often failed to correlate. It should also be noted that not all studies adequately excluded increases in left atrial pressure, suggested by elevated pulmonary capillary wedge pressures, as a contributor to the development of daytime increases in PAP (87–89).
In an attempt to control for some of these confounding variables, Sajkov and colleagues (90) studied 27 patients with OSA in whom clinically significant cardiac and pulmonary disease had been excluded. A total of 11 (41%) were found to have PH, with a mean PAP of 26 mm Hg. No difference was noted between patients with PH and those without PH in AHI, BMI, smoking history, or lung function. However, those with PH were found to be more hypoxemic during daytime wakefulness than patients without PH, a finding that could either contribute to or result from PH.
Treatment in the form of tracheostomy or supplemental oxygen has been shown to reduce PAP in patients with chronic obstructive pulmonary disease and nocturnal hypoxemia (91). In 1978, the Stanford group reported an approximate 50% reduction in PAP in six selected patients with OSA who underwent tracheostomy, some of whom may have had comorbid disease (92).
There are very limited data on the effects of CPAP treatment of OSA on PAP. Alchanatis and colleagues (93) studied a group of 29 patients with OSA (with no evidence of pulmonary or cardiac disease) with Doppler echocardiography before and after 6-month CPAP treatment. Comparisons were made with 12 healthy subjects. A total of 20% of the patients with OSA had PH that was clinically mild (mean PAP, 25.6 mm Hg). Greater age and increased BMI distinguished these from the patients with OSA without PH; 6 months of CPAP treatment was associated with reductions in mean PAP in both patients with OSA with PH (25.6 ± 4.0 to 19.5 ± 1.5 mm Hg) and those without PH (14.9 ± 2.2 to 11.5 ± 2.0 mm Hg).
Sajkov and colleagues (94) treated 20 patients with OSA (without coexistent pulmonary or cardiac disease) with 4 months of CPAP therapy. Only 5 patients met criteria for PH, with a mean PAP for the whole group of 16.8 mm Hg. To assess the reversibility of PH in these patients, PAP was measured by echocardiography at three levels of inspired oxygen (50, 21, and 11%). After 4 months of CPAP therapy, PAP (for all patients) decreased to a mean of 13.9 mm Hg. CPAP may also affect vasoreactivity, as the pulmonary artery pressor response to hypoxia was attenuated.
Finally, in the first placebo-controlled trial of treatment of OSA in PH, Arias and colleagues (95) recently reported the results of a randomized cross-over trial of CPAP and sham CPAP over 12 weeks in 23 patients with OSA. A total of 10 patients with PH (defined as PASP > 30 mm Hg by Doppler echocardiography) were more obese, had more ventilatory limitation (reduced FVC), and more severe sleep apnea (by AHI and mean oxygen saturation) than the 13 patients without PH. CPAP therapy reduced PASP in all patients with OSA, though more so in those with PH at baseline (mean reductions, 8.5 vs. 2.6 mm Hg) (Figure 3). Although the baseline differences in obesity and lung function between groups preclude the attribution of PH to OSA alone, these data are the first to show, in a placebo-controlled fashion, the positive impact of CPAP therapy on PH in a small group of patients with OSA. Further research is needed to assess the durability of CPAP therapy on PAP and right heart function and how CPAP therapy fits into the treatment paradigm amid an ever-increasing arsenal of pharmacologic treatments for PH.
Figure 3.

Pulmonary artery systolic pressure (PASP) after sham and therapeutic continuous positive airway pressure (CPAP). Reprinted by permission from Reference 95.
Supported by National Institutes of Health grants HL-65176, HL-70302, HL-73211, and M01-RR00585 (V.K.S), and the Mayo Clinic, a grant from the ResMed Foundation, and research support from Restore Medical.
Conflict of Interest Statement: J.M.G. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. V.K.S. has served as a consultant for Sepracor, Respironics, ResMed, and Cardiac Concepts. He has also received research grants from the Respironics Sleep and Breathing Foundation ($120,000), and is a coinvestigator on a grant from the ResMed Foundation. S.M.C. has received research grant support from the ResMed Foundation for investigator initiated research protocols.
References
- 1.Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 1995;96:1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Somers V, Mark A, Abboud F. Interaction of baroreceptor and chemoreceptor control of sympathetic nerve activity in normal humans. J Clin Invest 1991;87:1953–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valbo AB, Hagbarth KE, Torebjörk HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 1979;59:919–957. [DOI] [PubMed] [Google Scholar]
- 4.Smith ML, Niedermaier ONW, Hardy SM, Decker MJ, Strohl KP. Role of hypoxemia in sleep apnea-induced sympathoexcitation. J Auton Nerv Syst 1996;56:184–190. [DOI] [PubMed] [Google Scholar]
- 5.Somers VK, Dyken ME, Skinner JL. Autonomic and hemodynamic responses and interactions during the Mueller maneuver in humans. J Auton Nerv Syst 1993;44:253–259. [DOI] [PubMed] [Google Scholar]
- 6.Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea: impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 2000;162:566–570. [DOI] [PubMed] [Google Scholar]
- 7.Lavie L. Obstructive sleep apnoea syndrome–an oxidative stress disorder. Sleep Med Rev 2003;7:35–51. [DOI] [PubMed] [Google Scholar]
- 8.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005;112:2660–2667. [DOI] [PubMed] [Google Scholar]
- 9.Shamsuzzaman AS, Winnicki M, Lanfranchi P, Wolk R, Kara T, Accurso V, Somers VK. Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 2002;105:2462–2464. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol 2002;282:C227–C241. [DOI] [PubMed] [Google Scholar]
- 11.Bonsignore MR, Marrone O, Insalaco G, Bonsignore G. The cardiovascular effects of obstructive sleep apnoeas: analysis of pathogenic mechanisms. Eur Respir J 1994;7:786–805. [DOI] [PubMed] [Google Scholar]
- 12.Shiomi T, Guilleminault C, Stoohs R, Schnittger I. Leftward shift of the interventricular septum and pulsus paradoxus in obstructive sleep apnea syndrome. Chest 1991;100:894–902. [DOI] [PubMed] [Google Scholar]
- 13.Virolainen J, Ventila M, Turto H, Kupari M. Effect of negative intrathoracic pressure on left ventricular pressure dynamics and relaxation. J Appl Physiol 1995;79:455–460. [DOI] [PubMed] [Google Scholar]
- 14.Brinker JA, Weiss JL, Lappe DL, Rabson JL, Summer WR, Permutt S, Weisfeldt ML. Leftward septal displacement during right ventricular loading in man. Circulation 1980;61:626–633. [DOI] [PubMed] [Google Scholar]
- 15.Gleeson K, Zwillich CW, White DP. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis 1990;142:295–300. [DOI] [PubMed] [Google Scholar]
- 16.Morgan BJ, Crabtree DC, Puleo DS, Badr MS, Toiber F, Skatrud JB. Neurocirculatory consequences of abrupt change in sleep state in humans. J Appl Physiol 1996;80:1627–1636. [DOI] [PubMed] [Google Scholar]
- 17.Blasi A, Jo J, Valladares E, Morgan BJ, Skatrud JB, Khoo MC. Cardiovascular variability after arousal from sleep: time-varying spectral analysis. J Appl Physiol 2003;95:1394–1404. [DOI] [PubMed] [Google Scholar]
- 18.Narkiewicz K, van de Borne PJ, Montano N, Dyken ME, Phillips BG, Somers VK. Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation 1998;97:943–945. [DOI] [PubMed] [Google Scholar]
- 19.Tsuji H, Larson MG, Venditti FJ Jr, Manders ES, Evans JC, Feldman CL, Levy D. Impact of reduced heart rate variability on risk for cardiac events: the Framingham Heart Study. Circulation 1996;94:2850–2855. [DOI] [PubMed] [Google Scholar]
- 20.Kraiczi H, Hedner J, Peker Y, Carlson J. Increased vasoconstrictor sensitivity in obstructive sleep apnea. J Appl Physiol 2000;89:493–498. [DOI] [PubMed] [Google Scholar]
- 21.Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation 2005;111:363–368. [DOI] [PubMed] [Google Scholar]
- 22.Carlson J, Rangemark C, Hedner J. Attenuated endothelium-dependent vascular relaxation in patients with sleep apnoea. J Hypertens 1996;14:577–584. [DOI] [PubMed] [Google Scholar]
- 23.Kato M, Roberts-Thomson P, Phillips B. Impairment of endothelium-sependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 2000;102:2607–2610. [DOI] [PubMed] [Google Scholar]
- 24.Ip MS, Lam B, Chan LY, Zheng L, Tsang KW, Fung PC, Lam WK. Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am J Respir Crit Care Med 2000;162:2166–2171. [DOI] [PubMed] [Google Scholar]
- 25.Phillips BG, Narkiewicz K, Pesek CA, Haynes WG, Dyken ME, Somers VK. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J Hypertens 1999;17:61–66. [DOI] [PubMed] [Google Scholar]
- 26.Harsch IA, Schahin SP, Radespiel-Troger M, Weintz O, Jahreiss H, Fuchs FS, Wiest GH, Hahn EG, Lohmann T, Konturek PC, et al. Continuous positive airway pressure treatment rapidly improves insulin sensitivity in patients with obstructive sleep apnea syndrome. Am J Respir Crit Care Med 2004;169:156–162. [DOI] [PubMed] [Google Scholar]
- 27.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 2002;165:934–939. [DOI] [PubMed] [Google Scholar]
- 28.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 2004;27:123–128. [PubMed] [Google Scholar]
- 29.von Kanel R, Dimsdale JE. Hemostatic alterations in patients with obstructive sleep apnea and the implications for cardiovascular disease. Chest 2003;124:1956–1967. [DOI] [PubMed] [Google Scholar]
- 30.Svatikova A, Shamsuzzaman AS, Wolk R, Phillips BG, Olson LJ, Somers VK. Plasma brain natriuretic peptide in obstructive sleep apnea. Am J Cardiol 2004;94:529–532. [DOI] [PubMed] [Google Scholar]
- 31.Gami AS, Svatikova A, Wolk R, Olson EJ, Duenwald CJ, Jaffe AS, Somers VK. Cardiac troponin T in obstructive sleep apnea. Chest 2004;125:2097–2100. [DOI] [PubMed] [Google Scholar]
- 32.Staessen J, Bulpitt CJ, O'Brien E, Cox J, Fagard R, Stanton A, Thijs L, Van Hulle S, Vyncke G, Amery A. The diurnal blood pressure profile: a population study. Am J Hypertens 1992;5(6 Pt 1):386–392. [DOI] [PubMed] [Google Scholar]
- 33.Staessen JA, Fagard RH, Lijnen PJ, Thijs L, Van Hoof R, Amery AK. Mean and range of the ambulatory pressure in normotensive subjects from a meta-analysis of 23 studies. Am J Cardiol 1991;67:723–727. [DOI] [PubMed] [Google Scholar]
- 34.Davies CW, Crosby JH, Mullins RL, Barbour C, Davies RJ, Stradling JR. Case-control study of 24 hour ambulatory blood pressure in patients with obstructive sleep apnoea and normal matched control subjects. Thorax 2000;55:736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000;342:1378–1384. [DOI] [PubMed] [Google Scholar]
- 36.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002;165:1217–1239. [DOI] [PubMed] [Google Scholar]
- 37.Ali N, Davies R, Fleetham J, Stradling J. The acute effects of continuous positive airway pressure and oxygen administration on blood pressure during obstructive sleep apnea. Chest 1992;101:1526–1532. [DOI] [PubMed] [Google Scholar]
- 38.Dimsdale JE, Loredo JS, Profant J. Effect of continuous positive airway pressure on blood pressure: a placebo trial. Hypertension 2000;35:144–147. [DOI] [PubMed] [Google Scholar]
- 39.Pepperell JC, Ramdassingh-Dow S, Crosthwaite N, Mullins R, Jenkinson C, Stradling JR, Davies RJ. Ambulatory blood pressure after therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised parallel trial. Lancet 2002;359:204–210. [DOI] [PubMed] [Google Scholar]
- 40.Becker HF, Jerrentrup A, Ploch T, Grote L, Penzel T, Sullivan CE, Peter JH. Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation 2003;107:68–73. [DOI] [PubMed] [Google Scholar]
- 41.Haentjens P, Van Meerhaeghe A, Moscariello A, De Weerdt S, Poppe K, Dupont A, Velkeniers B. The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med 2007;167:757–764. [DOI] [PubMed] [Google Scholar]
- 42.Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, Sahadevan J, Redline S; Sleep Heart Health Study. Association of nocturnal arrhythmias with sleep-disordered breathing: the Sleep Heart Health Study. Am J Respir Crit Care Med 2006;173:910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guilleminault C, Connolly SJ, Winkle RA. Cardiac arrhythmia and conduction disturbances during sleep in 400 patients with sleep apnea syndrome. Am J Cardiol 1983;52:490–494. [DOI] [PubMed] [Google Scholar]
- 44.Grimm W, Koehler U, Fus E, Hoffmann J, Menz V, Funck R, Peter JH, Maisch B. Outcome of patients with sleep apnea-associated severe bradyarrhythmias after continuous positive airway pressure therapy. Am J Cardiol 2000;86:688–692. [DOI] [PubMed] [Google Scholar]
- 45.Becker H, Brandenburg U, Peter J, Von Wichert P. Reversal of sinus arrest and atrioventricular conduction block in patients with sleep apnea during nasal continuous positive airway pressure. Am J Respir Crit Care Med 1995;151:215–218. [DOI] [PubMed] [Google Scholar]
- 46.Gami AS, Pressman G, Caples SM, Kanagala R, Gard JJ, Davison DE, Malouf JF, Ammash NM, Friedman PA, Somers VK. Association of atrial fibrillation and obstructive sleep apnea. Circulation 2004;110:364–367. [DOI] [PubMed] [Google Scholar]
- 47.Mitchell ARJ, Spurrell PAR, Sulke N. Circadian variation of arrhythmia onset patterns in patients with persistent atrial fibrillation. Am Heart J 2003;146:902–907. [DOI] [PubMed] [Google Scholar]
- 48.Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, Shamsuzzaman AS, Somers VK. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 2003;107:2589–2594. [DOI] [PubMed] [Google Scholar]
- 49.Gami AS, Hodge DO, Herges RM, Olson EJ, Nykodym J, Kara T, Somers VK. Obstructive sleep apnea, obesity, and the risk of incident atrial fibrillation. J Am Coll Cardiol 2007;49:565–571. [DOI] [PubMed] [Google Scholar]
- 50.Shepard JW Jr, Garrison MW, Grither DA, Evans R, Schweitzer PK. Relationship of ventricular ectopy to nocturnal oxygen desaturation in patients with chronic obstructive pulmonary disease. Am J Med 1985;78:28–34. [DOI] [PubMed] [Google Scholar]
- 51.Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 2005;352:1206–1214. [DOI] [PubMed] [Google Scholar]
- 52.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea–hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005;365:1046–1053. [DOI] [PubMed] [Google Scholar]
- 53.Mohsenin V, Valor R. Sleep apnea in patients with hemispheric stroke. Arch Phys Med Rehabil 1995;76:71–76. [DOI] [PubMed] [Google Scholar]
- 54.Dyken ME, Somers VK, Yamada T, Ren Z-Y, Zimmerman MB. Investigating the relationship between stroke and obstructive sleep apnea. Stroke 1996;27:401–407. [DOI] [PubMed] [Google Scholar]
- 55.Bassetti C, Aldrich MS. Sleep apnea in acute cerebrovascular diseases: final report on 128 patients. Sleep 1999;22:217–223. [DOI] [PubMed] [Google Scholar]
- 56.Hu FB, Willett WC, Manson JE, Colditz GA, Rimm EB, Speizer FE, Hennekens CH, Stampfer MJ. Snoring and risk of cardiovascular disease in women. J Am Coll Cardiol 2000;35:308–313. [DOI] [PubMed] [Google Scholar]
- 57.Arzt M, Young T, Finn L, Skatrud JB, Bradley TD. Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med 2005;172:1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034–2041. [DOI] [PubMed] [Google Scholar]
- 59.Parra O, Arboix A, Bechich S, García-Eroles L, Montserrat JM, López JA, Ballester E, Guerra JM, Sopeña JJ. Time course of sleep-related breathing disorders in first-ever stroke or transient ischemic attack. Am J Respir Crit Care Med 2000;161:375–380. [DOI] [PubMed] [Google Scholar]
- 60.Bokinsky G, Miller M, Ault K, Husband P, Mitchell J. Spontaneous platelet activation and aggregation during obstructive sleep apnea and its response to therapy with nasal continuous positive airway pressure: a preliminary investigation. Chest 1995;108:625–630. [DOI] [PubMed] [Google Scholar]
- 61.Eisensehr I, Noachtar EB, Korbett S, Byrne K, Mc A, Auley A, Palabrica T. Platelet activation, epinephrine, and blood pressure in obstructive sleep apnea syndrome. Neurology 1998;51:188–195. [DOI] [PubMed] [Google Scholar]
- 62.Wessendorf TE, Thilmann AF, Wang YM, Schreiber A, Konietzko N, Teschler H. Fibrinogen levels and obstructive sleep apnea in ischemic stroke. Am J Respir Crit Care Med 2000;162:2039–2042. [DOI] [PubMed] [Google Scholar]
- 63.Rångemark C, Hedner JA, Carlson JT, Gleerup G, Winther K. Platelet function and fibrinolytic activity in hypertensive and normotensive sleep apnea patients. Sleep 1995;18:188–194. [DOI] [PubMed] [Google Scholar]
- 64.Netzer N, Werner P, Jochums I, Lehmann M, Strohl KP. Blood flow of the middle cerebral artery with sleep-disordered breathing: correlation with obstructive hypopneas. Stroke 1998;29:87–93. [DOI] [PubMed] [Google Scholar]
- 65.Balfors EM, Franklin KA. Impairment of cerebral perfusion during obstructive sleep apneas. Am J Respir Crit Care Med 1994;150:1587–1591. [DOI] [PubMed] [Google Scholar]
- 66.Hayakawa T, Terashima M, Kayukawa Y, Ohta T, Okada T. Changes in cerebral oxygenation and hemodynamics during obstructive sleep apneas. Chest 1996;109:916–921. [DOI] [PubMed] [Google Scholar]
- 67.Diomedi M, Placidi F, Cupini LM, Bernardi G, Silvestrini M. Cerebral hemodynamic changes in sleep apnea syndrome and effect of continuous positive airway pressure treatment. Neurology 1998;51:1051–1056. [DOI] [PubMed] [Google Scholar]
- 68.Chin K, Ohi M, Kita H, Noguchi T, Otsuka N, Tsuboi T, Mishima M, Kuno K. Effects of NCPAP therapy on fibrinogen levels in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 1996;153:1972–1976. [DOI] [PubMed] [Google Scholar]
- 69.Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 1999;160:1101–1106. [DOI] [PubMed] [Google Scholar]
- 70.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med 2002;347:305–313. [DOI] [PubMed] [Google Scholar]
- 71.Kaneko Y, Floras JS, Usui K, Plante J, Tkacova R, Kubo T, Ando S-i, Bradley TD. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med 2003;348:1233–1241. [DOI] [PubMed] [Google Scholar]
- 72.Mansfield DR, Gollogly NC, Kaye DM, Richardson M, Bergin P, Naughton MT. Controlled trial of continuous positive airway pressure in obstructive sleep apnea and heart failure. Am J Respir Crit Care Med 2004;169:361–366. [DOI] [PubMed] [Google Scholar]
- 73.Smith LA, Vennelle M, Gardner RS, McDonagh TA, Denvir MA, Douglas NJ, Newby DE. Auto-titrating continuous positive airway pressure therapy in patients with chronic heart failure and obstructive sleep apnoea: a randomized placebo-controlled trial. Eur Heart J 2007;28:1221–1227. [DOI] [PubMed] [Google Scholar]
- 74.Caples SM, Somers VK. CPAP treatment for obstructive sleep apnoea in heart failure: expectations unmet. Eur Heart J 2007;28:1184–1186. [DOI] [PubMed] [Google Scholar]
- 75.Motley HL, Cournand A, Werko L, Himmelstein A, Dresdale D. The influence of short periods of induced acute anoxia upon pulmonary artery pressures in man. Am J Physiol 1947;150:315–320. [DOI] [PubMed] [Google Scholar]
- 76.Voelkel NF. Mechanisms of hypoxic pulmonary vasoconstriction. Am Rev Respir Dis 1986;133:1186–1195. [DOI] [PubMed] [Google Scholar]
- 77.Presberg KW, Dincer HE. Pathophysiology of pulmonary hypertension due to lung disease. Curr Opin Pulm Med 2003;9:131–138. [DOI] [PubMed] [Google Scholar]
- 78.Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43(12 Suppl S):40S–47S. [DOI] [PubMed] [Google Scholar]
- 79.McQuillan BM, Picard MH, Leavitt M, Weyman AE. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation 2001;104:2797–2802. [DOI] [PubMed] [Google Scholar]
- 80.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43(12 Suppl S):5S–12S. [DOI] [PubMed] [Google Scholar]
- 81.Bady E, Achkar A, Pascal S, Orvoen-Frija E, Laaban JP. Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax 2000;55:934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chaouat A, Weitzenblum E, Krieger J, Oswald M, Kessler R. Pulmonary hemodynamics in the obstructive sleep apnea syndrome: results in 220 consecutive patients. Chest 1996;109:380–386. [DOI] [PubMed] [Google Scholar]
- 83.Krieger J, Sforza E, Apprill M, Lampert E, Weitzenblum E, Ratomaharo J. Pulmonary hypertension, hypoxemia, and hypercapnia in obstructive sleep apnea patients. Chest 1989;96:729–737. [DOI] [PubMed] [Google Scholar]
- 84.Coccagna G, Mantovani M, Brignani F, Parchi C, Lugaresi E. Continuous recording of the pulmonary and systemic arterial pressure during sleep in syndromes of hypersomnia with periodic breathing. Bull Physiopathol Respir (Nancy) 1972;8:1159–1172. [PubMed] [Google Scholar]
- 85.Bradley TD, Phillipson EA. Pathogenesis and pathophysiology of the obstructive sleep apnea syndrome. Med Clin North Am 1985;69:1169–1185. [DOI] [PubMed] [Google Scholar]
- 86.Fletcher EC, Luckett RA, Miller T, Costarangos C, Kutka N, Fletcher JG. Pulmonary vascular hemodynamics in chronic lung disease patients with and without oxyhemoglobin desaturation during sleep. Chest 1989;95:757–764. [DOI] [PubMed] [Google Scholar]
- 87.Buda AJ, Schroeder JS, Guilleminault C. Abnormalities of pulmonary artery wedge pressures in sleep-induced apnea. Int J Cardiol 1981;1:67–74. [DOI] [PubMed] [Google Scholar]
- 88.Hetzel M, Kochs M, Marx N, Woehrle H, Mobarak I, Hombach V, Hetzel J. Pulmonary hemodynamics in obstructive sleep apnea: frequency and causes of pulmonary hypertension. Lung 2003;181:157–166. [DOI] [PubMed] [Google Scholar]
- 89.Sanner BM, Doberauer C, Konermann M, Sturm A, Zidek W. Pulmonary hypertension in patients with obstructive sleep apnea syndrome. Arch Intern Med 1997;157:2483–2487. [PubMed] [Google Scholar]
- 90.Sajkov D, Cowie RJ, Thornton AT, Espinoza HA, McEvoy RD. Pulmonary hypertension and hypoxemia in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 1994;149:416–422. [DOI] [PubMed] [Google Scholar]
- 91.Fletcher EC, Levin DC. Cardiopulmonary hemodynamics during sleep in subjects with chronic obstructive pulmonary disease: the effect of short- and long-term oxygen. Chest 1984;85:6–14. [DOI] [PubMed] [Google Scholar]
- 92.Motta J, Guilleminault C, Schroeder JS, Dement WC. Tracheostomy and hemodynamic changes in sleep-inducing apnea. Ann Intern Med 1978;89:454–458. [DOI] [PubMed] [Google Scholar]
- 93.Alchanatis M, Tourkohoriti G, Kakouros S, Kosmas E, Podaras S, Jordanoglou JB. Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration 2001;68:566–572. [DOI] [PubMed] [Google Scholar]
- 94.Sajkov D, Wang T, Saunders NA, Bune AJ, McEvoy RD. Continuous positive airway pressure treatment improves pulmonary hemodynamics in patients with obstructive sleep apnea. Am J Respir Crit Care Med 2002;165:152–158. [DOI] [PubMed] [Google Scholar]
- 95.Arias MA, Garcia-Rio F, Alonso-Fernandez A, Martinez I, Villamor J. Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure: a randomized, controlled cross-over study. Eur Heart J 2006;27:1106–1113. [DOI] [PubMed] [Google Scholar]