

Abstract
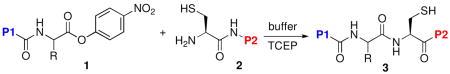
A direct oxo-ester peptide ligation method has been developed. Through the use of an activated C-terminal para-nitrophenyl ester (cf. 1), it is possible to achieve direct cysteine ligations (1 +2 → 4). Peptide substrates incorporating bulky C-terminal amino acids (cf. 1, R) can be accommodated with high reaction efficiency.
The landmark disclosure, in 1901, of the synthesis of the dipeptide glycylglycine through hydrolysis of glycine is considered to mark the beginning of the field of peptide synthetic chemistry.i The past 100 years have seen major advances in the development of enabling peptide assembly techniques. Complex proteins are now viewed to be viable targets for total synthesis. Perhaps the most innovative and central of the advances, from a technical standpoint, was the introduction of solid phase peptide synthesis (SPPS) by Merrifield, in 1963.ii Currently, automated SPPS is commonly employed for the assembly of large, homogeneous peptide fragments. Despite the significant gains made in SPPS methodology, practical issues remain, as the technique is generally limited to peptide chains incorporating fewer than ca. 50 amino acids. In 1994, another major breakthrough in the field was made with the disclosure by Kent and coworkers of a general technique, termed native chemical ligation (NCL), which enables the coupling of large peptidic fragments.iii This method, outlined in Scheme 1a, is based on the principles employed in a 1953 disclosure of Wieland and coworkers.iv As shown, the C-terminus of one fragment (Peptide 1) is equipped with a thioester. The coupling partner (Peptide 2) possesses an N-terminal cysteine residue. The rate limiting phase of the sequence is widely accepted to be the intermolecular transthioesterification (step 1), which generates a unimolecular intermediate that is well-positioned to undergo intramolecular S→N acyl transfer, generating the coupled peptide fragment.v
Scheme 1.

a) Native Chemical Ligation
b) NCL Phenolic Ester Variation
c) Proposed Direct Oxo-Ester Ligation
Our laboratory has been interested in developing novel and reliable methods for the assembly of complex glycopeptides and glycoproteins bearing multiple carbohydrate domains. Part of this focus stems from our intensive program directed toward the de novo total synthesis of the glycoprotein, erythropoietin alpha (EPO) as a homogeneous entity (Figure 1). EPO, a 166-residue glycoprotein bearing four different carbohydrate domains (three of which are N-linked, and one of which is O-linked), is a medicinally important agent that is commonly used in the treatment of anemia.vi It is well understood that the carbohydrate domains play a crucial role in conferring efficacy and stability to the glycoprotein. However, rigorous comparisons of the efficacy of various EPO glycoforms have been largely hindered by difficulties associated with isolating homogeneous EPO from natural sources. A de novo synthesis program could well provide access to valuable homogeneous material for comparative investigations. Our EPO total synthesis program has served to stimulate ongoing efforts to develop enabling advances in peptide elongation and ligation techniques.
Figure 1.
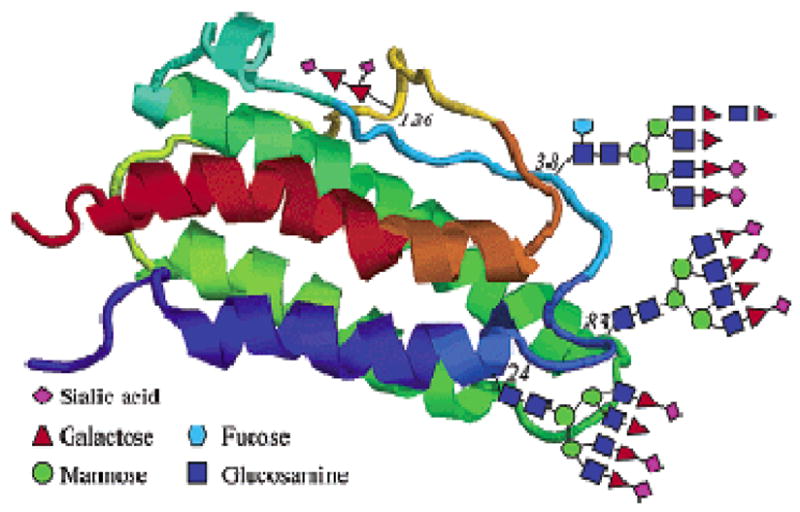
Structure of Erythropoietin Alpha (EPO).
It was in this context that our laboratory developed, in 2004, a novel and broadly useful NCL variant, which allows for the merger of glycopeptide fragments through the use of a relatively inert C-terminal ortho-thiophenolic ester (Scheme 1b).vii Upon cleavage of the S–S bond, the thiophenolic ester apparently undergoes intramolecular O→S transfer, as shown. The intermediate thioester is then trapped intermolecularly by an N-terminal cysteine residue of the peptide or glycopeptide coupling partner. Finally, S→N acyl transfer provides the final coupled peptide.
Although cysteine-based NCL is a powerful method, it too suffers from a number of limitations. First, cysteine residues are found relatively infrequently in naturally occurring peptides and proteins. There have been attempts to circumvent this issue through various approaches, including the development of homocysteineviii and seleno-cysteineix ligation protocols, through post-ligation desulfurization techniques,x and through the use of cysteine-free auxiliaries which may be cleaved following ligation.xi
A second type of issue arises from the limitations imposed by the C-terminal amino acid residue of the thioester coupling fragment. Dawson and coworkers have demonstrated that the rate of ligation is strongly dependent on the identity of the C-terminal amino acid.xii Ligations at sterically hindered, β-branched C-terminal sites, such as Pro, Val, Ile, and Thr, are prohibitively slow. Even in the wake of NCL, the development of efficient and reliable methods for achieving hindered peptide ligations remains an immense practical challenge.
Since the rate-limiting S→S acyl transfer (Scheme 1a, step 1) appears to be highly dependent on the steric bulk of the C-terminal amino acid, we pursued the possibility of developing a modified ligation method that would proceed directly from an oxo-ester C-terminal peptide, without the intermediacy of the problematic thioester (see Scheme 1c).xiii In this regard, we took note of a 1955 disclosure by Bodanszky and coworkers on the synthesis of small peptides through aminolysis of para-nitrophenyl esters.xiv Such esters are sufficiently activated to facilitate direct peptide coupling.xv,xvi However, a number of issues had limited the general applicability of the method, including: (1) the relative instability of the C-terminal nitrophenyl ester, (2) the potential for racemization of the C-terminal amino acid residue, and (3) the lack of strong selectivity for the α-amino group vs. the ε-amino group of Lys. These issues would clearly need to be overcome in the development of a broadly useful direct oxo-ester ligation method.
Activated esters, such as para-nitrophenyl ester, are known to be strong acyl donors. We first evaluated the selectivity of the activated ester for the cysteine functionality through a simple competition experiment. Thus, as outlined in Scheme 2, Boc-Phe-p-NO2Ph was added to a solution containing equal parts Cys, Ala, and β-Ala. Dipeptide Boc-Phe-Cys-OH was the only observed product, thus strongly suggesting that prior thiol capture of Cys predominates under these conditions.
Scheme 2.

In our initial studies, we investigated NCL at less hindered C-termini. However, though ligated products could be isolated, undesired hydrolysis of the acyl donor was found to be a major competing factor, even at lower pH ranges (data not shown). We speculated that ester hydrolysis would be less problematic in peptides with more hindered C-terminal amino acids. In order to maximally suppress hydrolysis, para-NO2-phenol was added to guanidine buffer. Ligation studies were conducted in a buffered solution at pH 6.3–6.5, at 25–30 °C. As outlined in Table 1, under these conditions, oxo-ester mediated ligation proceeded smoothly with a variety of hindered C-terminal amino acid residues, including Thr, Val, Ile, D-allo-Ile, and Pro (entries 1–5). In each case, the reaction times were relatively short and the yields were generally quite good.
Table 1.
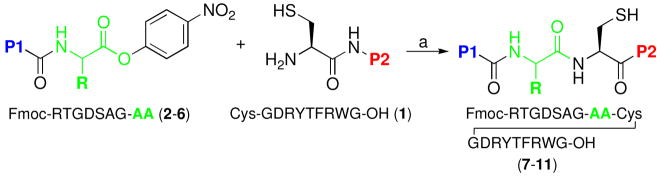 | |||
|---|---|---|---|
| Entry | C-terminal AA | Time (h) | Yield |
| 1 | Thr (2) | 2 | 79% (7) |
| 2 | Val (3) | 6 | 69% (8) |
| 3 | Ile (4) | 7 | 70% (9) |
| 4 | D-allo-Ile (5) | 6 | 60% (10) |
| 5 | Pro (6) | 15 | 50% (11) |
Reagents and conditions: (a) Buffer at pH= 6.3–6.5 (6.0 M Gn·HCl, 188.8 mM Na2HPO4, 7.2 mM p-NO2PhOH), TCEP, RT
For comparative purposes, the efficiency of a traditional thioester-mediated NCL was also examined (Scheme 3). While the analogous nitrophenyl ester ligation proceeded in 7h to afford 9 in 70% yield (entry 3), the thioester ligation took 72h to generate 9 in only 58% yield. It is known that such prolonged reaction times can lead to the formation of undesired side products.
Scheme 3.

Reagents and conditions: (a) buffer at pH = 6.3–6.5 (6.0 M Gn•HCl, 188.8 m M Na2HPO4, 18.8 m M TCEP•HCl), PhSH, TCEP, 30 °C.
“Racemization” of the C-terminal amino acid residue is known to be an issue in peptide coupling.xvii Presumably, racemization proceeds through the pathway outlined in Scheme 4. An analysis of the extent of racemization in the peptides presented in Table 1 revealed less than 5–6% racemization at the C-terminal Leu, Ile, and Val sites.xviii
Scheme 4.

Due to the high reactivity of the p-nitrophenolic ester, it is conceivable that resident free lysine residues could be prone to undergo competitive direct aminolysis with the ester to provide undesired cyclic peptide adducts. To evaluate the selectivity of the ligation protocol described above for the α-amino group over the lysine ε-amine, we synthesized peptide 13, which incorporates an unprotected Lys residue on the fragment bearing the C-terminal nitrophenyl ester (Scheme 5). In the event, we were pleased to find that ligation with 1 readily afforded coupled product, 14, in 61% yield. Only a small amount of the undesired cyclic peptide was observed under our conditions.
Scheme 5.

Reagents and conditions: (a) buffer at pH= 6.3–6.5 (6.0 M Gn•HCl, 188.8 mM Na2HPO4, 7.2 mM p-NO2PhOH), TCEP, RT, 1 h, 61% isolated yield.
Finally, in an effort to probe the limits of our system in a discerning way, we sought to investigate couplings of C-terminal nitrophenyl ester peptides with the very hindered unnatural amino acid penicillamine (Pen, 15).xix This commercially available amino acid, which may serve as a surrogate for Val, can provide a viable alternative to Cys in cases where ligation at a Cys site is not practical. In the event, we were pleased to observe that both peptides 16 and 2 readily underwent ligation with Pen (15) to afford the corresponding peptides in acceptable yields (Scheme 6). Exposure to thiol reduction conditions then served to efficiently transform Pen to Val. A comparative coupling reaction was performed on the corresponding thio-ester (Fmoc-RTGDSAG-Thr-SPh). After 24h, only 4% of compound 19 was generated, based on LCMS integration.
Scheme 6.

aKey: a) buffer at pH= 6.3–6.5 (6.0 M Gn•HCl, 188.8 mM Na2HPO4, 7.2 mM p-NO2PhOH), TCEP, 30 °C, 2–3h; b) buffer at pH= 6.3–6.5 (6.0 M Gn•HCl, 188.8 mM Na2HPO4, 18.8 mM TCEP•HCl), TCEP, VA-044, 37 °C, 2–3h.
In conclusion, we have described above a major simplification in the field of native chemical ligation. Hitherto, the success of this method had been assumed to be dependent on the formation of a thioester acyl donor. Through proper balancing of acyl donor potential with substrate stability, we have demonstrated the feasibility of NCL with oxo- rather than thio-esters. Further applications of this method, as well as this extended logic, to targets of major biological interest will be described in due course.
Supplementary Material
Supporting Information Available: Experimental procedures and compound characterization data (PDF) This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
Support for this research was provided by the National Institutes of Health (CA28824). A postdoctoral fellowship is gratefully acknowledged by Q. W. (Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center, SKI). We thank Dr. George Sukenick, Ms. Sylvi Rusli and Ms. Hui Fang of the Sloan-Kettering Institute’s NMR core facility for mass spectral and NMR spectroscopic analysis (SKI core grant no.: CA02848). We thank Ms. Rebecca Wilson for editorial counsel.
References
- (i).Fischer E, Fourneau E. Ber Dtsch Chem Ges. 1901;34:2868–2879. [Google Scholar]
- ii.Merrifield RB. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- iii.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- iv.Wieland T, Bokelmann E, Bauer L, Lang HU, Lau H. Justus Liebigs Ann Chem. 1953;583:129–149. [Google Scholar]
- v.Johnson ECB, Kent SBH. J Am Chem Soc. 2006;128:6640–6646. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- vi.Lai P, Everett R, Wang F, Arakawa T, Goldwasser E. J Biol Chem. 1986;261:3116–3121. [PubMed] [Google Scholar]
- vii.(a) Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J Am Chem Soc. 2004;126:6576–6578. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]; (b) Chen G, Warren JD, Chen J, Wu B, Wan Q, Danishefsky SJ. J Am Chem Soc. 2006;128:7560–7462. doi: 10.1021/ja061588y. [DOI] [PubMed] [Google Scholar]; (c) Chen J, Warren JD, Wu B, Chen G, Wan Q, Danishefsky SJ. Tetrahedron Lett. 2006;47:1969–1972. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.Tam JP, Yu Q. Biopolimer. 1998;46:319–327. [Google Scholar]
- ix.Quaderer R, Sewing A, Hilvert D. Helvetica Chimica Acta. 2001;84:1197–1025. [Google Scholar]
- x.(a) Yan LZ, Dawson PE. J Am Chem Soc. 2001;128:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]; (b) Pentelute BL, Kent SBH. Org Lett. 2007;9:687–690. doi: 10.1021/ol0630144. [DOI] [PubMed] [Google Scholar]; (c) Crich D, Banerjee A. J Am Chem Soc. 2007;129:10064–10065. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]; (d) Botti P, Tchertchian S. WO/2006/133962 ; (e) Wan Q, Danishefsky SJ. Angew Chem Int Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- xi.(a) Low DW, Hill MG, Carrasco MR, Kent SBH. Proc Natl Acad Sci USA. 2001;98:6554–6559. doi: 10.1073/pnas.121178598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew Chem Int Ed. 2006;45:4416–4425. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]; (c) Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew Chem Int Ed. 2007;46:7383–7387. doi: 10.1002/anie.200702865. [DOI] [PubMed] [Google Scholar]; (d) Payne RJ, Fichet S, Greenberg WA, Wong C-H. Angew Chem Int Ed. 2008;47:4411–4415. doi: 10.1002/anie.200705298. [DOI] [PubMed] [Google Scholar]; (e) Okamato R, Kajihara Y. Angew Chem Int Ed. 2008;47:5402–5406. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- xii.Hackeng TM, Griffin JH, Dawson PE. Proc Natl Acad Sci USA. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. Kemp also observed similar results. Kemp DS, Galakatos NG, Dranginis S, Asthon C, Fotouhi N, Curran TP. J Org Chem. 1986;51:3320–3324.
- xiii.Oxo-esters were prepared according to the general procedures described in references 7a, 7b, and 10e. See Supporting Information for full synthetic details.
- xiv.Bodanszky M. Nature. 1955;175:685. doi: 10.1038/175685a0. [DOI] [PubMed] [Google Scholar]
- xv.Gagnon P, Huang X, Therrien E, Keillor JW. Tetrahedron Lett. 2002;43:7717–7719. [Google Scholar]
- xvi.Hojo H, Aimoto S. Bull Chem Soc Jpn. 1991;64:111–17. [Google Scholar]
- xvii.For discussion of C-terminal amino acid racemization in thioester-mediated NCL, please see references 12a and 20a.
- xviii.Goodman M, McGahren WJ. Tetrahedron. 1967;23:2031–2050. doi: 10.1016/0040-4020(67)80037-1. [DOI] [PubMed] [Google Scholar]
- xix.Following submission of this communication, Seitz and co-workers reported a ligation at valine, using penicillamine as the valine precursor. Haase C, Rohde H, Seitz O. Angew Chem Int Ed. 2008;47:6807–6810. doi: 10.1002/anie.200801590. Based on the results of using penicillamine (15) as a valine surrogate, described in this communication, our group performed further research directed to a more reactive thiol based acyl acceptor culminating in a valine equivalence, which is described in the following reference Chen J, Wan Q, Yuan Y, Zhu JL, Danishefsky SJ. Angewandte Chemie Int Ed. 2008 doi: 10.1002/anie.200803523. In Press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental procedures and compound characterization data (PDF) This material is available free of charge via the Internet at http://pubs.acs.org