

Abstract
Hyperphosphorylation and aggregation of the microtubule-binding protein tau characterize a diverse array of neurodegenerative disorders. Most of these lack mutations in the encoding MAPT gene, and the role of tau in disease pathogenesis remains controversial. Among these tauopathies is Niemann–Pick type C disease (NPC), a lysosomal storage disorder characterized by progressive neurodegeneration and premature death, most often caused by an inherited deficiency in the intracellular lipid trafficking protein NPC1. To determine the extent to which tau affects NPC pathogenesis, we generated Npc1−/− mice deficient in tau. Unexpectedly, NPC1/tau double null mutants are generated in markedly smaller litters, exhibit an enhanced systemic phenotype and die significantly earlier than NPC1 single null mutants. As autophagy is up-regulated in NPC and protein degradation through this pathway depends on movement along microtubules, we knocked down MAPT expression in NPC1-deficient human fibroblasts and examined effects on this pathway. We show that an acute reduction of tau expression in a cellular model of NPC decreases induction and flux through the autophagic pathway. Our data establish that MAPT deletion exacerbates the NPC phenotype through a mechanism independent of tau protein aggregation and identifies a critical role for tau in the regulation of autophagy in NPC1-deficient cells.
INTRODUCTION
Niemann–Pick type C disease (NPC) is an autosomal-recessive sphingolipid storage disorder characterized by severe, progressive neurodegeneration, hepatosplenomegaly and early death (1). Most cases are caused by loss-of-function mutations in NPC1 (2), a gene encoding a multipass transmembrane protein that contains a sterol-sensing domain with homology to the regulators of cholesterol metabolism and to the Hedgehog signaling receptor Patched (3). NPC1 is localized primarily to late endosomes and lysosomes, where it is involved in lipid sorting and vesicular trafficking, and is thought to act as an efflux pump for cholesterol from these compartments (4–7). This pathway is essential for the delivery of extracellular, low density lipoprotein-derived cholesterol to the endoplasmic reticulum for esterification and redistribution to other intracellular sites, including the plasma membrane and Golgi apparatus (8–10).
NPC has been studied in a number of models, the best characterized and most widely used of which is a mouse model in which a spontaneous insertion of a retrotransposon into exon 9 of the Npc1 gene results in a frameshift and truncation of the NPC1 protein (11). This model reproduces many aspects of the human disease, including cellular accumulations of cholesterol and glycosphingolipids (12–14). NPC1-deficient mice also exhibit systemic pathology including hepatosplenomegaly and develop progressive neurodegeneration characterized by abnormally swollen axons, demyelination (15), hyperphosphorylation of tau (16) and progressive neuronal loss, most notably of cerebellar Purkinje cells and cortical neurons (17–19). These cellular defects are accompanied by behavioral impairments paralleling the neurological and systemic symptoms of the human disorder, including abnormal gait and rotarod performance, cognitive deficits, weight loss and early death (20).
Children with NPC are among the youngest patients to develop neurofibrillary pathology, similar to that which occurs in Alzheimer's disease and the frontotemporal dementias (21,22). In these disorders, the brain accumulates hyperphosphorylated species of the microtubule-associated protein tau (23). The tau protein is encoded by six alternatively spliced transcripts derived from the MAPT gene and directly binds microtubules through three or four C-terminal tubulin-binding domains (24,25). Experimental evidence suggests that a primary function of tau is to stabilize polymerized microtubules (26). Hyperphosphorylation dissociates tau from microtubules and allows for its self-aggregation into paired helical filaments (PHFs). These PHFs have the ability to form larger aggregates termed neurofibrillary tangles, a pathological hallmark of many tauopathies, and these or other soluble tau species can act through a toxic gain-of-function mechanism to impair neuronal survival (23). The diminished association of hyperphosphorylated tau with microtubules also leads to a loss-of-function that is presumably detrimental to neuronal health due to its impact on axonal transport processes (23); however, the significance of this mechanism in disease has been uncertain. Emerging data additionally suggest novel roles for tau in the regulation of anterograde and retrograde trafficking, which are separate from microtubule stabilization (27,28). Remarkably, mutant mice that lack endogenous tau (Mapt−/− mice) exhibit no overt phenotype (29), likely due to functional redundancy with other microtubule-associated proteins. Clearly, our current understanding of the normal function of tau and its role in neurodegenerative diseases is incomplete.
In Npc1−/− mice, tau is hyperphosphorylated at multiple sites in a manner similar to human tauopathies, although neurofibrillary tangles do not form (30,31). Cyclin-dependent kinase inhibitors reduce tau phosphorylation and attenuate the phenotype of Npc1−/− mice (32), suggesting a role for tau in disease pathogenesis. However, it remains unknown whether this therapeutic effect is mediated by increased availability of functional tau protein, through decreased formation of toxic tau species or through actions on a different substrate. To define the role of tau in NPC pathogenesis, we generated NPC1 null mice that lack endogenous tau. Here, we show that tau deletion exacerbates the phenotype of NPC mice, resulting in smaller litter sizes, an enhanced systemic phenotype and early death. Furthermore, we demonstrate that tau knockdown in a cellular model of NPC diminishes macroautophagy (hereafter referred to as autophagy), a highly conserved pathway by which cytoplasmic proteins and damaged organelles are sequestered within autophagic vacuoles and targeted to lysosomes for degradation (33). Prior work has implicated this pathway in the pathogenesis of several neurodegenerative disorders (34–37) including NPC (38–40). Functional autophagy is dependent on trafficking along microtubules (41,42), and its impairment is sufficient to cause neurodegeneration in mice (43,44). Our data establish that tau deletion exacerbates the NPC phenotype in mice and suggest that impaired autophagy contributes to this effect.
RESULTS
Deletion of endogenous tau exacerbates the NPC phenotype
To determine the extent to which tau pathology affects the NPC phenotype in vivo, we generated NPC1/tau double null mutant mice (Npc1−/−; Mapt−/−). The first indication of a genetic interaction between Npc1 and Mapt came from breedings, where we noted the occurrence of significantly smaller litters when both parental genotypes were Npc1+/− and Mapt−/−. Although Npc1 heterozygotes deficient in tau generated litters that averaged approximately four pups, the presence of one or two functional tau alleles in both parents resulted in significantly larger litters (Fig. 1A). This effect was not solely attributable to Mapt deficiency, as Npc1+/+; Mapt−/− mice generated litters that averaged approximately eight pups, similar to wild-type (Fig. 1C). We considered the possibility that these smaller litters from Npc1+/−; Mapt−/− mice resulted from the selective loss of NPC1/tau double null mutant pups. However, an analysis of the Npc1 null allele in pups from Npc1+/−; Mapt−/− breedings showed that the percentage of Npc1−/− pups was not significantly different from the expected number when compared with litters from mice with one or two functional tau alleles (Fig. 1B). These results indicate that small litter size was not due to the specific loss of NPC1/tau double null mutants, but rather suggest that it was attributable to other affects on fertility or fetal survival.
Figure 1.
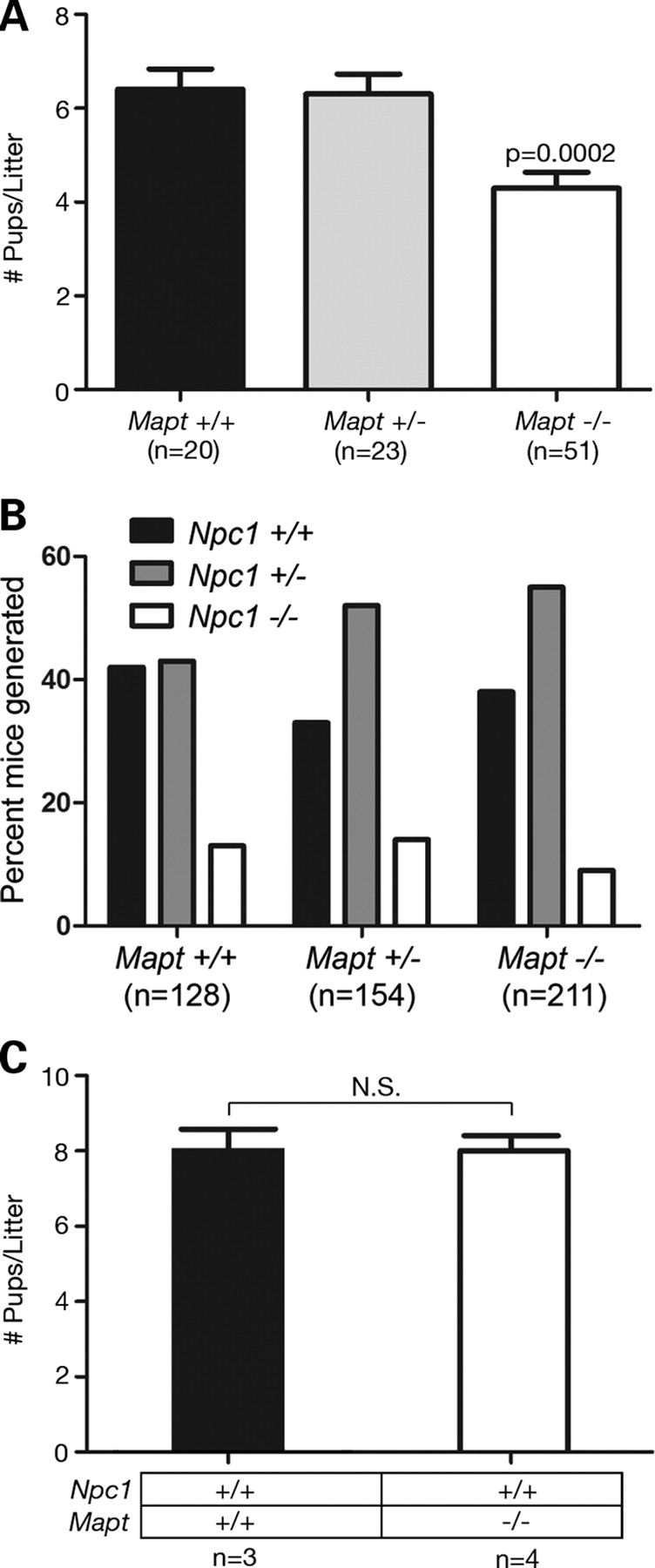
Mapt deletion decreases litter size of NPC1-deficient mice. (A) Litter sizes (mean±SEM) generated by Npc1+/− mice, stratified by parental Mapt genotype. Npc1+/− and Mapt−/− breedings generated fewer pups on average (P = 0.0002 by ANOVA with Newman–Keul's multiple comparison test) than mice with at least one functional Mapt allele (n, number of litters). (B) Npc1 null allele distribution in pups generated by NPC1 haploinsufficient mice, stratified by parental Mapt genotype. The percentage of NPC1/tau double null pups generated by Npc1+/−; Mapt−/− breeding was not significantly different from expected (P = 0.2 by χ2 analysis) (n, number of mice). (C) Litter size (mean±SEM) of wild-type (black bar) and Mapt−/− (white bar) mice. N, number of litters; n.s., not significant (P > 0.05) by unpaired Student's t-test.
NPC1/tau double null mutants displayed a more severe phenotype than NPC1 single null mutants. Female NPC1 null mutants at 5 weeks of age that were haploinsufficient or deficient in tau weighed significantly less than NPC1 single null mutants (Fig. 2A, left panel); similar findings were observed in animals at 4 and 6 weeks of age (data not shown). Additionally, both male and female double null mutants died significantly earlier. Although NPC1 single null mutants succumbed to disease at 8–12 weeks in our colony, approximately half of the NPC1/tau double null mutants died by 5 weeks (Fig. 2B and C). Although age of death exhibited a bimodal distribution, earlier death did not correlate with lower weight or the appearance of systemic features described below. Mice haploinsufficient for Mapt (Npc1−/−; Mapt +/−) also demonstrated this robust shift in the survival curve, establishing that even partial loss of tau expression was detrimental to the survival of NPC mice.
Figure 2.

Decreased weight and early death of NPC1/tau double null mutant mice. (A) Weight (mean±SEM) of 5-week-old female (left panel) and male (right panel) mice. A significant difference between NPC1 single nulls (Npc1−/− and Mapt+/+), and NPC1 nulls haploinsufficient or deficient in tau was observed in females, whereas all Npc1−/− males were observed to have decreased weights when compared with wild-type and Mapt−/− controls. *, ** and *** signify P-values of <0.05, <0.01 and <0.001, respectively, by ANOVA with Newman–Keul's multiple comparison test. (B) Survival curve. Npc1−/−; Mapt +/− (green line) and Npc1−/−; Mapt−/− (blue line) were significantly different (P < 0.0001 by log-rank test) from wild-type, Mapt null (black line) and Npc1−/−; Mapt +/+ (red line) mice. (C) Age at death for Npc1−/−; Mapt +/+ (red triangles), Npc1−/−; Mapt +/− (blue squares) and Npc1−/−; Mapt−/− (black triangles) mice.
Although the severity of the phenotype displayed by NPC1/tau double null mutants varied, many of the animals (approximately half) exhibited mild facial dysmorphia (shortened snout), kyphosis (Fig. 3A) and an abnormal gait in which mice failed to press their paw pads down while walking (Fig. 3B and C). This gait abnormality was present in mice as young as 3–4 weeks and preceded the occurrence of ataxia in NPC1 null mutants. Additionally, almost all of the double null mutant males that survived to at least 5 weeks developed penile prolapse, a phenotype rarely exhibited by NPC1 single null mutants (Fig. 3D). This constellation of features, together with effects on weight and survival of double null mutants, demonstrates that tau deletion worsens disease severity in NPC1-deficient mice. Histopathological examination of the brain and liver of NPC1/tau double null mutants compared with NPC1 single null mutants did not reveal exacerbated Purkinje cell loss, axonal pathology, microgliosis or astrogliosis or increased accumulation of lipid-laden macrophages in the liver (Supplementary Material, Figs S1 and S2), suggesting that cellular dysfunction contributed to the more severe phenotypic abnormalities. Notably, the exacerbation of phenotypes in NPC1/tau double null mutants occurred in the complete absence of tau, excluding tau hyperphosphorylation or PHF formation as mediators of these effects.
Figure 3.

Systemic phenotypes associated with NPC1/tau double null mice. (A) Double null mice (Npc1−/−; Mapt−/−) demonstrate shortened snout and kyphosis when compared with control mice (Npc1+/+; Mapt−/−). (B) Still images of video reveal abnormal gait of double null mice (Npc1−/−; Mapt−/−, bottom panel). (C) Footprint analysis demonstrates toe walking gait of 4-week-old double null mice (Npc1−/−; Mapt−/−) when compared with NPC1 single null (Npc1−/−; Mapt +/+) and control mice (Npc1+/+; Mapt−/−) of the same age and sex. (D) Nearly, all NPC1/tau double null males that survive 5 weeks or longer develop penile prolapse, whereas NPC1 single null males and WT males rarely exhibit this phenotype (P = 0.038 by ANOVA).
Tau knockdown impairs autophagy in NPC1-deficient cells
We next sought for to establish the mechanism by which tau deletion exacerbates the NPC phenotype. We hypothesized that impairment of critical microtubule-dependent processes might underlie the worsened phenotype of double null mutants. Prior work established that NPC1 deficiency increases basal autophagy (40) and that transport along microtubules is required for autophagic protein degradation (41,42). We therefore examined whether tau deletion affects autophagy in NPC1-deficient mice and cells.
To monitor the autophagic pathway, we used the microtubule-associated protein 1 light chain 3 (LC3) (45,46). This protein is modified from its LC3-I cytosolic form to LC3-II, a lipidated form that migrates more rapidly on SDS–PAGE and is associated with autophagosome formation and the induction of autophagy. Immunohistochemical analysis of livers from 10-week-old mice demonstrated frequent LC3-positive vacuoles, particularly in the peri-portal regions in NPC1/tau double null mutants compared with NPC1 single null mutants and wild-type controls (Fig. 4A). Both groups of mutant mice showed LC3-positive vesicles in the cerebellum (Supplementary Material, Fig. S3), where they accumulated in occasional swollen axons and cell bodies. A similar accumulation of LC3-positive vacuoles has been associated with impaired autophagic flux in several model systems (47), including those used in the study of lysosomal storage diseases (48–50), where decreased autophagosome—lysosome fusion results in an increased number of autophagosomes.
Figure 4.

LC3 and p62 expressions in NPC1 and NPC1/tau null mice. (A) Immunohistochemical staining demonstrates frequent LC3-positive vacuoles in liver of 10-week-old double null mutant (Npc1−/−; Mapt−/−) when compared with single null mutant (Npc1−/−; Mapt +/+) or wild-type mice (original magnification, 1000×). (B) Liver lysates from 10-week-old mice were probed by western blot for expression of LC3. Both double null (Npc1−/−; Mapt−/−) and NPC1 single null mice (Npc1−/−; Mapt +/+) demonstrate increased levels of LC3-II compared WITH controls. GAPDH serves as a loading control. (C–E) Lysates from the telencephalon (C) and cerebellum (D and E) of NPC1 single null, and NPC1/tau double null mice demonstrate increased levels of LC3-II and SDS-insoluble p62 compared with controls.
These results led us to examine the autophagic pathway in tissue lysates from NPC1 and tau null mutants, using LC3-II and p62 to monitor its activity. Although LC3-II is a robust marker of autophagy induction, p62 can be used to assess autophagic flux. p62 links ubiquitinated proteins to LC3 and is itself incorporated into autophagosomes and degraded upon fusion with lysosomes (51). Therefore, impaired flux through the autophagic pathway can lead to the accumulation of insoluble p62, as shown in mice in which autophagy is disrupted by genetic deletion of Atg7 (51). The formation of ubiquitin and p62-containing protein aggregates in these autophagy-deficient mice occur through a process that is dependent on p62 (51). In NPC1-deficient mice, western blot analyses demonstrated increased soluble LC3-II in liver (Fig. 4B) and brain (Fig. 4C–E) and increased SDS-insoluble p62 in brain (Fig. 4C–E). These data are consistent with the previously published findings that NPC1 deficiency in vivo leads to the robust activation of autophagy (38–40). Nonetheless, ubiquitinated proteins (18,22,39) and insoluble p62 (Fig. 4C–E) accumulate in the brains of NPC1-deficient mice, consistent with the notion that autophagic flux is not sufficient to handle the quantity of proteins targeted for degradation. Situations in which induction and flux are disproportionate to each other may lead to autophagic stress, a possible mediator of cell dysfunction and a precursor to cell death (52). Similar alterations in LC3-II and p62 expressions were observed in tissue lysates from NPC1 single null mutants as young as 4 weeks (data not shown), prior to phenotype onset, and in NPC1/tau double null mutants.
As the in vivo deletion of tau in NPC mice led to limited changes in autophagic markers (Fig. 4A), perhaps due to compensatory mechanisms that blunted these qualitative responses in mice, we were prompted to examine the impact of tau on autophagy in an NPC model system that allows for more quantitative analyses following acute changes in tau expression. The acute down-regulation of tau in vitro markedly reduced both autophagic induction and flux in NPC1-deficient cells (Fig. 5). To reduce MAPT expression, we used pooled, targeted siRNAs to specifically knockdown MAPT in control and NPC1-deficient primary human fibroblasts. This cell type was selected for the analysis as prior studies demonstrated that it expresses all human tau isoforms (53). Treatment with MAPT siRNAs significantly decreased endogenous MAPT mRNA levels (Fig. 5A). The reduction of tau in NPC1-deficient fibroblasts, but not in control cells, decreased the amount of LC3-II to levels equivalent to control cells treated with non-targeted siRNA (Fig. 5B). Our results demonstrate that tau is required for the induction of autophagy in response to NPC1 deficiency, supporting a functional role for tau in regulating the activity of the autophagic pathway in this cellular NPC model.
Figure 5.
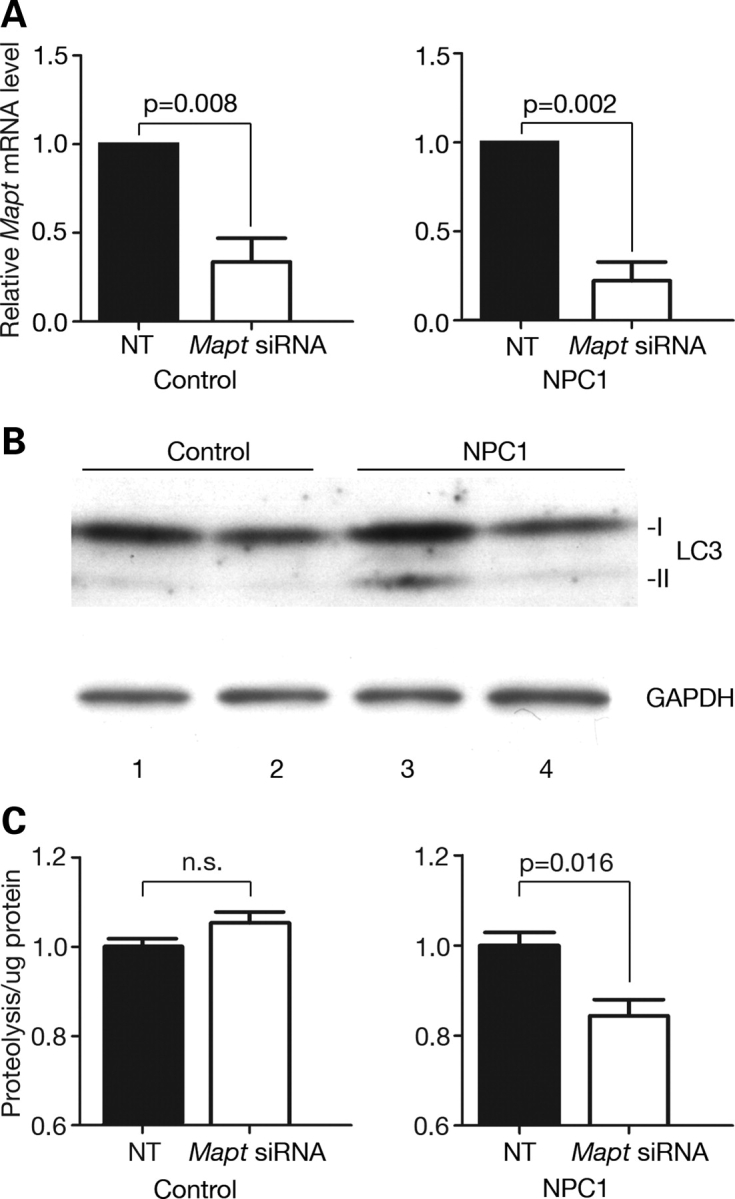
Tau knockdown decreases autophagic induction and flux in NPC1-deficient cells. (A) Relative MAPT mRNA levels (mean±SEM) 72 h post-treatment with non-targeted (black bars) or MAPT siRNAs (white bars), as determined by qPCR. Targeted siRNAs significantly reduced endogenous MAPT mRNA expression in control and NPC1-deficient primary human fibroblasts by unpaired Student's t-test. (B) Human fibroblast lysates from control (lanes 1 and 2) and NPC1-deficient (lanes 3 and 4) cells were analyzed by western blot for the expression of LC3 (top) and GAPDH (bottom) 72 h post-treatment with non-targeted (lanes 1 and 3) or MAPT siRNAs (lanes 2 and 4). (C) Degradation of long-lived proteins in control (left panel) and NPC1-deficient human fibroblasts (right panel), following transfection with non-targeted (black bars) or MAPT siRNAs (white bars). Data (mean±SEM) are reported relative to non-targeted siRNA-transfected cells at 72 h. Relative proteolysis is significantly decreased in NPC1-deficient cells (P = 0.016 by unpaired Student's t-test), but not in controls (P > 0.05).
To independently confirm these results, we sought to determine whether MAPT knockdown decreased flux through the autophagic pathway by measuring the degradation of long-lived proteins (54). This assay provides a quantitative, functional readout of autophagic flux (55,56). Control and NPC1-deficient cells were treated with MAPT or non-targeted siRNAs and then labeled for 48 h with 3H-leucine. Cells were washed and re-fed, and trichloroacetic acid soluble radioactive counts released into the media were measured 24 h after labeling. NPC1-deficient fibroblasts treated with MAPT siRNAs demonstrated significantly lower levels of long-lived protein degradation than cells treated with non-targeted siRNAs (Fig. 5C, right panel). In contrast, long-lived protein degradation was unchanged in similarly treated control cells (Fig. 5C, left panel). We conclude that tau is required for the induction of autophagy in response to NPC1 deficiency in primary fibroblasts. In contrast, diminished tau expression in mice or cells possessing functional NPC1 is not sufficient to alter basal autophagy.
Trafficking along microtubules is required for efficient protein quality control through processes that include, in addition to autophagy, protein degradation by the ubiquitin–proteasome pathway (57). To determine whether tau deletion also affected this pathway, we evaluated proteasome function in NPC1 single null and NPC1/tau double null mutants. Liver lysates were isolated and used as a source of active 20S proteasomes. Cleavage of Suc-LLVY-AMC, a substrate for the chymotrypsin-like activity of the proteasome, was unchanged by Npc1 and Mapt genotypes (Fig. 6A). Similarly, the expression of 20S proteasome subunits was unaltered in single and double null mutants compared with wild-type controls (Fig. 6B). Additionally, no induction of heat shock proteins, as observed during a stress response, was detected in single or double null mutants (Fig. 6B). Our analyses support the conclusion that the function of the ubiquitin–proteasome pathway is intact in NPC1/tau double null mutant mice and suggest that tau deletion primarily impairs protein degradation through autophagy in NPC1 deficiency.
Figure 6.
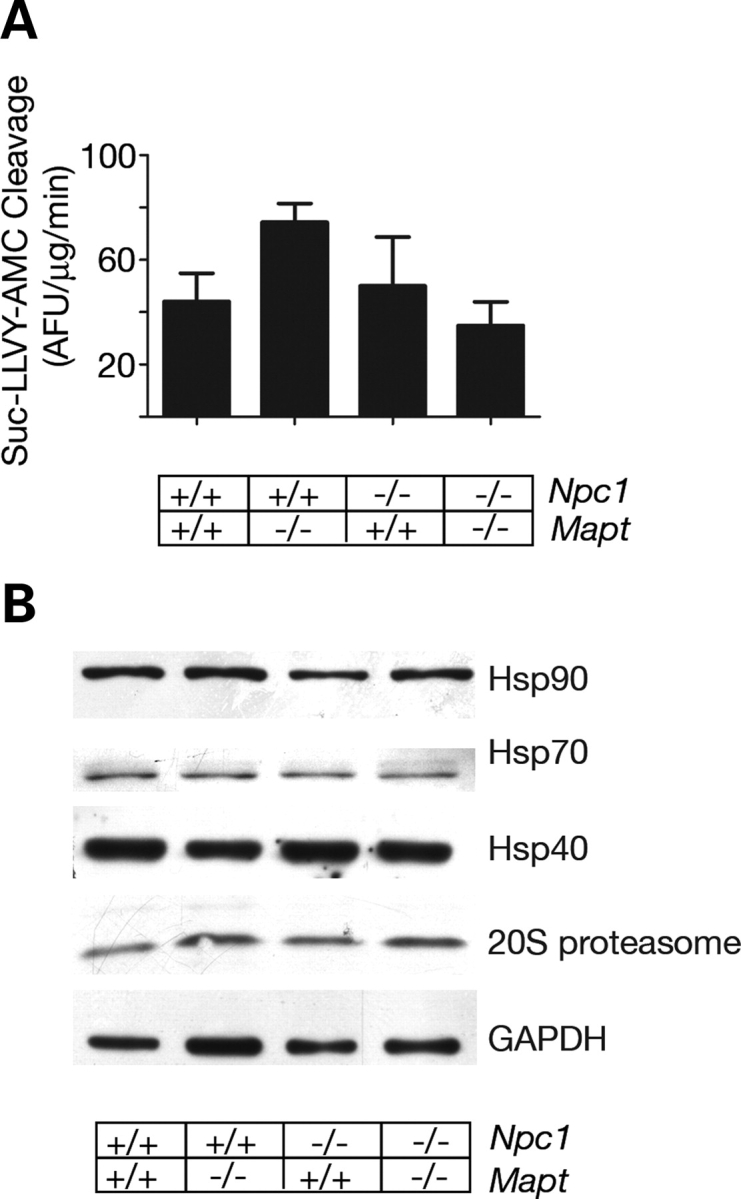
The ubiquitin–proteasome pathway is intact in NPC1-deficient animals. (A) Activity of the 20S proteasome, assayed in liver lysates of 10-week-old mice (mean±SEM). P > 0.05 by ANOVA. (B) Western blot analyses of liver lysates from 10-week-old mice demonstrate equivalent expression of hsp40, stress-inducible hsp70, hsp90 and 20S proteasome subunits. GAPDH serves as a loading control.
DISCUSSION
The tauopathies are a large and diverse collection of neurodegenerative disorders that have in common the accumulation of hyperphosphorylated and aggregated tau species (23,58). This group of diseases includes forms of frontotemporal lobar dementia caused by MAPT mutations, establishing a role for tau pathology in human disease (59–61). However, most tauopathies, including NPC, lack these mutations, and the contribution of tau to the pathogenesis of these disorders has remained poorly defined. Using NPC as an example of this latter group of disorders, here we show that tau deletion exacerbates the disease phenotype in vivo. Through the generation of NPC1/tau double null mutants, we establish that tau deficiency decreases litter size and survival and worsens the systemic phenotype of NPC1 null mutants. Our findings show that diminishing functional tau has dire consequences in the setting of certain neurodegenerative disorders. Although Mapt−/− mice exhibit no overt phenotype, our analyses indicate that tau deletion sensitizes mice to the stresses of NPC1 deficiency. We suggest that tau-dependent pathways normally exert a compensatory effect in NPC1 deficiency and that defining these pathways may identify targets where interventions could result in effective treatments. The effects of Mapt deletion on the phenotype of NPC mice are distinct from toxicity mediated by tau protein aggregation, a mechanism that has attracted widespread attention. In contrast to the results reported here, a recent study demonstrated that reduction of endogenous tau partially ameliorates the phenotype of Alzheimer's disease mice (62), demonstrating that the stresses imposed by the elimination of functional tau do not invariably lead to an exacerbation of a neurodegenerative phenotype. This suggests that both tau loss-of-function and toxic gain-of-function mechanisms modulate disease progression in tauopathies, implying the existence of two distinct subclasses within this large and diverse group of neurodegenerative disorders.
We observed that knockdown of MAPT expression decreases autophagic induction and flux in NPC1-deficient fibroblasts. We conclude that tau contributes to the regulation of autophagy in response to NPC1 deficiency and that loss of functional tau impairs this pathway in fibroblasts. Future studies in neurons are required to confirm the extent to which tau similarly contributes to the regulation of autophagy in the NPC1-deficient nervous system. Our findings build on published data, showing that NPC1 deficiency robustly increases basal autophagy (38–40) and stimulates flux through the autophagic pathway in a Beclin-1 dependent manner (40). As diagrammed in Figure 7, we propose that this induction of basal autophagy acts as a pro-survival response to facilitate recycling of critically required cellular components in NPC1-deficient cells. Tau deletion diminishes this response, perhaps through affects on microtubule-based trafficking. The resulting exacerbation of systemic pathology suggests that tau-dependent processes normally buffer the severity of the NPC phenotype. Our studies have identified a critical role for tau in the regulation of autophagy in NPC1-deficient cells and suggest that other disorders, where autophagy contributes to pathogenesis, may be similarly sensitive to affects of tau deletion. Furthermore, we suggest that alterations in tau mediated by its hyperphosphorylation may similarly act through a loss-of-function mechanism to exacerbate the phenotype of NPC patients.
Figure 7.

Model of the relationship between NPC1 deficiency, autophagy and tau. Functional deficiency of NPC1 impairs intracellular lipid trafficking and leads to the accumulation of unesterified cholesterol, sphingolipids and complex gangliosides in the late endosomal/lysosomal network. Pathological changes in NPC include progressive neurodegeneration associated with the accumulation of hyperphosphorylated tau (depicted at right). In NPC model systems and patient fibroblasts, defects in lipid transport stimulate a Beclin-1-dependent increase of basal autophagy, which we propose exerts a protective effect by facilitating the re-use of critical cellular components. The deletion of endogenous tau abrogates this response and exacerbates the NPC phenotype. We speculate that similar effects may be triggered by tau hyperphosphorylation, as occurs in NPC patients.
Our findings point toward the intriguing notion that tau loss-of-function may be more deleterious to the progression of NPC pathology than to the accumulation of hyperphosphorylated tau species. This novel mechanism for modulating the severity of the disease phenotype indicates that new treatment approaches aimed at restoring normal tau function could ameliorate or delay NPC disease progression. Other tauopathies may be similarly affected by tau loss-of-function, suggesting that this approach could prove beneficial in multiple disorders characterized by tau abnormalities. Additionally, our data raise the possibility that allelic differences in tau expression levels may modify the NPC phenotype. There is no established correlation between specific NPC1 mutations and disease age of onset or rate of progression (63). Furthermore, the severity of the NPC phenotype varies considerably even among siblings sharing the same mutation (64,65). These clinical observations suggest the existence of genetic modifiers (66,67). The work presented here identifies MAPT as a candidate modifier, a possibility that will be explored in future studies.
MATERIALS AND METHODS
Materials
Human fibroblasts from age- and sex-matched donors were from Coriell Institute for Medical Research (control cells, GM00038C and NPC1-deficient, GM03123A). Mouse samples were from age- and sex-matched BALB/cNctr-Npc1m1N/J (Jackson Laboratories stock no. 003092), Mapttm1(GFP)Kit//J (Jackson Laboratories stock number 004779) and wild-type littermates. Mice were backcrossed onto C57BL/6J for five generations; all experimental and control mice were on this same genetic background. LC3 antibody was from Novus Biologicals. GAPDH antibody was from Abcam. Hsp90 antibody was from Santa Cruz. Hsp40 and stress inducible hsp70 antibodies (SPA-812) were from Stressgen. Anti-20S proteasome was from Calbiochem. The p62 (C-terminal) antibody was from American Research Products. Horse radish peroxidase-conjugated secondary antibodies were from Biorad (goat anti-rabbit and goat anti-mouse) and Zymed (rabbit anti-guinea pig). MG132 was from Sigma. All procedures involving mice were approved by the University of Michigan Committee on Use and Care of Animals, in accord with the NIH Guidelines for the Care and Use of Experimental Animals.
Cell culture
Fibroblasts were maintained at 37°C, 5% CO2 in MEM with Earle's salts and non-essential amino acids (Gibco), supplemented with 15% fetal bovine serum (Atlanta Biologicals) and 10 µg/ml penicillin, 10 µg/ml streptomycin and 2 mm glutamine (referred to as complete MEM).
SiRNA knockdown of MAPT
Fibroblasts growing in complete MEM were collected using the Reagent Pack Subculture Kit (BioWhitaker), resuspended in Nucleofector solution (human dermal fibroblast Nucleofector Kit, Amaxa) and mixed with 1.5 µg ON-TARGETplus SMART pool human MAPT (NM_016841) siRNA (L-012488-00-0005) or siCONTROL-plus non-targeting pool (D-001810-10-05) (Dharmacon). The suspension was electroporated using the program U-23 on the Amaxa Nucleofector. Cells were plated in six-well dishes, and total RNA was collected 72 h later to measure MAPT mRNA expression by quantitative RT–PCR. Alternatively, cell lysates were collected 72 h after electroporation for western blot.
Gene expression analysis
Total RNA isolated from cells with Trizol (Invitrogen) served as a template for cDNA synthesis using the High Capacity cDNA Archive Kit from Applied Biosystems. Gene-specific primers and probes labeled with a fluorescent reporter dye and quencher were purchased from Applied Biosystems. Quantitative RT–PCR (qPCR) assays were performed using 5 ng aliquots of cDNA. Replicate tubes were analyzed for the expression of 18S ribosomal RNA (rRNA) using a VIC-labeled probe. Threshold cycle (Ct) values were determined by an ABI Prism 7900HT Sequence Detection System, and relative expression levels normalized to rRNA were calculated using the standard curve method of analysis.
Measurement of long-lived protein degradation
Degradation of long-lived proteins was determined as described previously (40) with minor modifications. Cells were pooled after electroporation, seeded in six-well plates in complete MEM for 4 h, washed with HBSS and labeled with 2 µCi/ml 3H-leucine (Amersham) in complete MEM. After 48 h of labeling, cells were washed with HBSS and incubated in complete MEM supplemented with 2.8 mm leucine. Aliquots of medium were collected at the indicated times and added to 20% TCA, and then BSA was added to a final concentration of 3 mg/ml. All samples were stored at 4°C for at least 1 h, and then centrifuged at 10 000g for 5 min at 4°C. Supernatants were collected, and pellets were washed twice with cold 20% TCA, and then all supernatants for each sample were pooled and the total radioactivity was measured by scintillation counting. Pellets were dissolved in cell lysis buffer (0.1 N NaOH, 0.1% Na deoxycholate), and radioactivity was measured by scintillation counting. Cells were washed with phosphate-buffered saline (PBS) three times and incubated for 1 h at 37°C in cell lysis buffer. An aliquot of the solubilized cells was used to determine the total protein concentration (BioRad), and another aliquot was used to measure the total radioactivity in the cells by scintillation counting. Relative proteolysis was determined by calculating TCA soluble radioactivity in the medium relative to total radioactivity in the well. Relative proteolysis was normalized to protein concentration from the solubilized cells.
Western blot analysis
Cells were harvested, washed with PBS, lysed in RIPA buffer containing cOmplete Protease Inhibitor Cocktail (Roche Diagnostics) and 0.1% β-mercaptoethanol and sonicated. Liver, cerebellar and telencephalon samples were homogenized with the same buffer using a motor homogenizer. Lysates were pre-cleared by centrifugation at 15 000g for 10 min at 4°C. For analysis of p62 levels in the pellet, protein concentrations were determined for crude lysates and sample concentrations were normalized. Samples were centrifuged at 15 000g for 10 min at 4°C, and pellets were resuspended in 1× loading buffer. All samples were electrophoresed through either 10% SDS–PAGE or 4–20% Tris–glycine gradient gels (Cambrex) and then transferred to nitrocellulose membranes (BioRad) using a semidry transfer apparatus. Immunoreactive proteins were detected by chemiluminescence (Perkin-Elmer).
Footprint analysis
Mice were trained to walk across an 83 cm long platform (7 cm wide), until they were able to consistently complete the task by walking down the center of the path. Forepaws and hindpaws were painted maize and blue, respectively, with non-toxic paint and animals walked over white paper to record footprints.
20S proteasome assay
Proteasome activity in mouse liver lysates was determined using the 20S Proteasome Assay Kit, SDS-Activation Format (K-900) from Boston Biochem. Liver was homogenized in sample buffer (2.5 mm Tris–HCl, pH 7.5, 0.1 mM EDTA, pH 8.0, 0.1 mM NaN3), and supernatants were collected after spinning at 15 000g for 30 min at 4°C. About 100 µg of soluble protein was added to each well of a 96-well plate and brought to a final volume of 60 µl with sample buffer. SDS-activation buffer was added to all samples and allowed to equilibrate for 5 min at room temperature. Substrate solution (Suc-LLVY-AMC) was then added, and fluorescent AMC counts released every 5 min for 1 h at 37°C were measured (excitation wavelength 390, emission filter 510) by an Ascent Fluroskan microplate reader. Each sample had a companion well with the same amount of protein, pre-treated with 1 mM MG132 (a proteasome inhibitor) for 10 min at room temperature to control for non-specific degradation of the substrate. Data represent AMC release per minute in samples minus AMC release per minute in companion MG132-treated wells.
Statistics
Statistical significance was assessed by unpaired Student's t-test, analysis of variance (ANOVA) with the Newman–Keul's multiple comparison test or log-rank test, using the software package Prism 4 (GraphPad Software). P-values less than 0.05 were considered significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by grants from the NIH (F31 NS51143 to C.D.P and T32 GM 07863 to M.J.E.).
ACKNOWLEDGEMENTS
We thank Diane Robins for helpful discussions and Elizabeth Walker for preparation of the figures.
Conflict of Interest statement. The authors have no financial conflicts of interest to report.
REFERENCES
- 1.Higgins J.J., Patterson M.C., Dambrosia J.M., Pikus A.T., Pentchev P.G., Sato S., Brady R.O., Barton N.W. A clinical staging classification for type C Niemann–Pick disease. Neurology. 1992;42:2286–2290. doi: 10.1212/wnl.42.12.2286. [DOI] [PubMed] [Google Scholar]
- 2.Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B., et al. Niemann–Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 3.Ioannou Y.A. Multidrug permeases and subcellular cholesterol transport. Nat. Rev. Mol. Cell Biol. 2001;2:657–668. doi: 10.1038/35089558. [DOI] [PubMed] [Google Scholar]
- 4.Davies J.P., Chen F.W., Ioannou Y.A. Transmembrane molecular pump activity of Niemann–Pick C1 protein. Science. 2000;290:2295–2298. doi: 10.1126/science.290.5500.2295. [DOI] [PubMed] [Google Scholar]
- 5.Garver W.S., Heidenreich R.A., Erickson R.P., Thomas M.A., Wilson J.M. Localization of the murine Niemann–Pick C1 protein to two distinct intracellular compartments. J. Lipid Res. 2000;41:673–687. [PubMed] [Google Scholar]
- 6.Higgins M.E., Davies J.P., Chen F.W., Ioannou Y.A. Niemann–Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol. Genet. Metab. 1999;68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- 7.Neufeld E.B., Wastney M., Patel S., Suresh S., Cooney A.M., Dwyer N.K., Roff C.F., Ohno K., Morris J.A., Carstea E.D., et al. The Niemann–Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999;274:9627–9635. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- 8.Wojtanik K.M., Liscum L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003;278:14850–14856. doi: 10.1074/jbc.M300488200. [DOI] [PubMed] [Google Scholar]
- 9.Liscum L., Ruggiero R.M., Faust J.R. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann–Pick type C fibroblasts. J. Cell Biol. 1989;108:1625–1636. doi: 10.1083/jcb.108.5.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sokol J., Blanchette-Mackie J., Kruth H.S., Dwyer N.K., Amende L.M., Butler J.D., Robinson E., Patel S., Brady R.O., Comly M.E., et al. Type C Niemann–Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J. Biol. Chem. 1988;263:3411–3417. [PubMed] [Google Scholar]
- 11.Loftus S.K., Morris J.A., Carstea E.D., Gu J.Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M.A., Tagle D.A., et al. Murine model of Niemann–Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- 12.Walkley S.U. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience. 1995;68:1027–1035. doi: 10.1016/0306-4522(95)00208-z. [DOI] [PubMed] [Google Scholar]
- 13.Xie C., Burns D.K., Turley S.D., Dietschy J.M. Cholesterol is sequestered in the brains of mice with Niemann–Pick type C disease but turnover is increased. J. Neuropathol. Exp. Neurol. 2000;59:1106–1117. doi: 10.1093/jnen/59.12.1106. [DOI] [PubMed] [Google Scholar]
- 14.Zervas M., Dobrenis K., Walkley S.U. Neurons in Niemann–Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 15.Takikita S., Fukuda T., Mohri I., Yagi T., Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann–Pick type C mouse. J. Neuropathol. Exp. Neurol. 2004;63:660–673. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- 16.Bu B., Li J., Davies P., Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann–Pick type C murine model. J. Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.German D.C., Quintero E.M., Liang C.L., Ng B., Punia S., Xie C., Dietschy J.M. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann–Pick C disease. J. Comp. Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashi Y., Murayama S., Pentchev P.G., Suzuki K. Cerebellar degeneration in the Niemann–Pick type C mouse. Acta Neuropathol. (Berl.) 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka J., Nakamura H., Miyawaki S. Cerebellar involvement in murine sphingomyelinosis: a new model of Niemann–Pick disease. J. Neuropathol. Exp. Neurol. 1988;47:291–300. doi: 10.1097/00005072-198805000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Voikar V., Rauvala H., Ikonen E. Cognitive deficit and development of motor impairment in a mouse model of Niemann–Pick type C disease. Behav. Brain Res. 2002;132:1–10. doi: 10.1016/s0166-4328(01)00380-1. [DOI] [PubMed] [Google Scholar]
- 21.Auer I.A., Schmidt M.L., Lee V.M., Curry B., Suzuki K., Shin R.W., Pentchev P.G., Carstea E.D., Trojanowski J.Q. Paired helical filament tau (PHFtau) in Niemann–Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. (Berl.) 1995;90:547–551. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- 22.Love S., Bridges L.R., Case C.P. Neurofibrillary tangles in Niemann–Pick disease type C. Brain. 1995;118:119–129. doi: 10.1093/brain/118.1.119. [DOI] [PubMed] [Google Scholar]
- 23.Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 24.Hirokawa N., Shiomura Y., Okabe S. Tau proteins: the molecular structure and mode of binding on microtubules. J. Cell Biol. 1988;107:1449–1459. doi: 10.1083/jcb.107.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee G., Neve R.L., Kosik K.S. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 26.Alonso A.C., Grundke-Iqbal I., Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 27.Dixit R., Ross J.L., Goldman Y.E., Holzbaur E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnani E., Fan J., Gasparini L., Golding M., Williams M., Schiavo G., Goedert M., Amos L.A., Spillantini M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harada A., Oguchi K., Okabe S., Kuno J., Terada S., Ohshima T., Sato-Yoshitake R., Takei Y., Noda T., Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- 30.Bu B., Klunemann H., Suzuki K., Li J., Bird T., Jin L.W., Vincent I. Niemann–Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol. Dis. 2002;11:285–297. doi: 10.1006/nbdi.2002.0551. [DOI] [PubMed] [Google Scholar]
- 31.Sawamura N., Gong J.S., Garver W.S., Heidenreich R.A., Ninomiya H., Ohno K., Yanagisawa K., Michikawa M. Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann–Pick type C mice. J. Biol. Chem. 2001;276:10314–10319. doi: 10.1074/jbc.M009733200. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M., Li J., Chakrabarty P., Bu B., Vincent I. Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann–Pick Type C mice. Am. J. Pathol. 2004;165:843–853. doi: 10.1016/S0002-9440(10)63347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klionsky D.J., Emr S.D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Vicente M., Cuervo A.M. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 35.Rubinsztein D.C., Gestwicki J.E., Murphy L.O., Klionsky D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 36.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 37.Settembre C., Fraldi A., Rubinsztein D.C., Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy. 2008;4:113–114. doi: 10.4161/auto.5227. [DOI] [PubMed] [Google Scholar]
- 38.Ko D.C., Milenkovic L., Beier S.M., Manuel H., Buchanan J., Scott M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann–Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao G., Yao Y., Liu J., Yu Z., Cheung S., Xie A., Liang X., Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1−/− mouse brain. Am. J. Pathol. 2007;171:962–975. doi: 10.2353/ajpath.2007.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pacheco C.D., Kunkel R., Lieberman A.P. Autophagy in Niemann–Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007;16:1495–1503. doi: 10.1093/hmg/ddm100. [DOI] [PubMed] [Google Scholar]
- 41.Iwata A., Riley B.E., Johnston J.A., Kopito R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 42.Ravikumar B., Acevedo-Arozena A., Imarisio S., Berger Z., Vacher C., O'Kane C.J., Brown S.D., Rubinsztein D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 43.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 44.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 45.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanida I., Ueno T., Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 2004;279:47704–47710. doi: 10.1074/jbc.M407016200. [DOI] [PubMed] [Google Scholar]
- 47.Corcelle E., Nebout M., Bekri S., Gauthier N., Hofman P., Poujeol P., Fenichel P., Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006;66:6861–6870. doi: 10.1158/0008-5472.CAN-05-3557. [DOI] [PubMed] [Google Scholar]
- 48.Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 49.Cao Y., Espinola J.A., Fossale E., Massey A.C., Cuervo A.M., MacDonald M.E., Cotman S.L. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006;281:20483–20493. doi: 10.1074/jbc.M602180200. [DOI] [PubMed] [Google Scholar]
- 50.Vergarajauregui S., Connelly P.S., Daniels M.P., Puertollano R. Autophagic dysfunction in mucolipidosis type Iv patients. Hum. Mol. Genet. 2008;17:2723–2737. doi: 10.1093/hmg/ddn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Komatsu M., Waguri S., Koike M., Sou Y.S., Ueno T., Hara T., Mizushima N., Iwata J.I., Ezaki J., Murata S., et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 52.Gozuacik D., Kimchi A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007;78:217–2145. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]
- 53.Ingelson M., Vanmechelen E., Lannfelt L. Microtubule-associated protein tau in human fibroblasts with the Swedish Alzheimer mutation. Neurosci. Lett. 1996;220:9–12. doi: 10.1016/s0304-3940(96)13218-3. [DOI] [PubMed] [Google Scholar]
- 54.Auteri J.S., Okada A., Bochaki V., Dice J.F. Regulation of intracellular protein degradation in IMR-90 human diploid fibroblasts. J. Cell. Physiol. 1983;115:167–174. doi: 10.1002/jcp.1041150210. [DOI] [PubMed] [Google Scholar]
- 55.Cuervo A.M., Dice J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 56.Fuertes G., Martin De Llano J.J., Villarroya A., Rivett A.J., Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 2003;375:75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Tallec B., Barrault M.B., Courbeyrette R., Guerois R., Marsolier-Kergoat M.C., Peyroche A. 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol. Cell. 2007;27:660–674. doi: 10.1016/j.molcel.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 58.Hasegawa M. Biochemistry and molecular biology of tauopathies. Neuropathology. 2006;26:484–490. doi: 10.1111/j.1440-1789.2006.00666.x. [DOI] [PubMed] [Google Scholar]
- 59.Goedert M., Crowther R.A., Spillantini M.G. Tau mutations cause frontotemporal dementias. Neuron. 1998;21:955–958. doi: 10.1016/s0896-6273(00)80615-7. [DOI] [PubMed] [Google Scholar]
- 60.Heutink P. Untangling tau-related dementia. Hum. Mol. Genet. 2000;9:979–986. doi: 10.1093/hmg/9.6.979. [DOI] [PubMed] [Google Scholar]
- 61.Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 62.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 63.Runz H., Dolle D., Schlitter A.M., Zschocke J. NPC-db, a Niemann–Pick type C disease gene variation database. Hum. Mutat. 2008;29:345–350. doi: 10.1002/humu.20636. [DOI] [PubMed] [Google Scholar]
- 64.Fink J.K., Filling-Katz M.R., Sokol J., Cogan D.G., Pikus A., Sonies B., Soong B., Pentchev P.G., Comly M.E., Brady R.O., et al. Clinical spectrum of Niemann–Pick disease type C. Neurology. 1989;39:1040–1049. doi: 10.1212/wnl.39.8.1040. [DOI] [PubMed] [Google Scholar]
- 65.Yatziv S., Leibovitz-Ben Gershon Z., Ornoy A., Bach G. Clinical heterogeneity in a sibship with Niemann–Pick disease type C. Clin. Genet. 1983;23:125–131. doi: 10.1111/j.1399-0004.1983.tb01860.x. [DOI] [PubMed] [Google Scholar]
- 66.Vanier M.T., Duthel S., Rodriguez-Lafrasse C., Pentchev P., Carstea E.D. Genetic heterogeneity in Niemann–Pick C disease: a study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 1996;58:118–125. [PMC free article] [PubMed] [Google Scholar]
- 67.Vanier M.T., Rodriguez-Lafrasse C., Rousson R., Duthel S., Harzer K., Pentchev P.G., Revol A., Louisot P. Type C Niemann–Pick disease: biochemical aspects and phenotypic heterogeneity. Dev. Neurosci. 1991;13:307–314. doi: 10.1159/000112178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.