

Abstract
The N-acyl-l-homoserine lactone hydrolases (AHL lactonases) have attracted considerable attention because of their ability to quench AHL-mediated quorum-sensing pathways in Gram-negative bacteria and because of their relation to other enzymes in the metallo-β-lactamase superfamily. To elucidate the detailed catalytic mechanism of AHL lactonase, mutations are made on residues that presumably contribute to substrate binding and catalysis. Steady-state kinetic studies are carried out on both the wild-type and mutant enzymes using a spectrum of substrates. Two mutations, Y194F and D108N, present significant effects on the overall catalysis. On the basis of a high-resolution structural model of the enzyme−product complex, a hybrid quantum mechanical/molecular mechanical method is used to model the substrate binding orientation and to probe the effect of the Y194F mutation. Combining all experimental and computational results, we propose a detailed mechanism for the ring-opening hydrolysis of AHL substrates as catalyzed by the AHL lactonase from Bacillus thuringiensis. Several features of the mechanism that are also found in related enzymes are discussed and may help to define an evolutionary thread that connects the hydrolytic enzymes of this mechanistically diverse superfamily.
Proteins in the metallo-β-lactamase superfamily span all three domains of life and are quite diverse, encompassing noncatalytic proteins as well as a diverse set of enzymes (1). At least 17 different activities have been reported within this superfamily, including nitric oxide and oxygen reduction as well as cleavage of C−O, C−N, C−S, S−O, P−O, and possibly P−N bonds (2). Many of these proteins are important for understanding clinically important processes, including anticancer drug detoxification pathways, mRNA processing, and antibacterial resistance mechanisms (2). The metal centers at the active sites of these enzymes are considerably diverse, with examples of mononuclear proteins, mononuclear proteins with the metal ion bound at a different site, dinuclear proteins with both metals bound in the proximity of each other in a cocatalytic site, and two-metal proteins where the second metal-binding site is distant but allosterically linked to the active site (3,4). Both zinc(II) and iron(II) can confer activity to various superfamily members, and some enzymes appear to incorporate (and display activity with) a wide variety of different divalent metal ions found in their host organism’s growth medium (5–7). Even within a single family of enzymes, there is considerable diversity in metal stoichiometry, metal placement, and catalytic mechanism (3). The rich functionality and high diversity of this superfamily, as well as the clinical importance of some members, make it an attractive area for studying the divergence and evolution of enzyme activities.
Recognition of a signature sequence motif, HxHxDH∼H∼D∼H, in the quorum-quenching N-acyl-l-homoserine lactone hydrolases (AHL1 lactonases) led to the adoption of this family of enzymes into the metallo-β-lactamase superfamily (8). Although initial reports suggested that the AHL lactonases were not metal-dependent, subsequent work on at least two different isoforms has shown that they are in fact dinuclear metalloproteins with two zinc(II) ions bound in the proximity of each other at the active site, and that these metal ions are essential for catalytic activity (9–13). The first reported structure of AHL lactonase did not have a bound substrate or inhibitor, but a subsequent structure was reported containing l-homoserine lactone, a competitive inhibitor that lacks the N-acyl substitution usually found in substrates (11,12). On the basis of the structure of this complex, a catalytic mechanism was proposed using a substrate binding orientation similar to that of the l-homoserine lactone inhibitor, but the opposite of that observed in enzyme−substrate and enzyme−product complex structures of related metallo-β-lactamase superfamily members (3,12,14,15). This hypothesis sparked us to further investigate the catalytic mechanism of AHL lactonases. The preceding paper in this series describes structural determination of three product-bound complexes and their implications for both substrate orientation and the ring-opening mechanism (16). Here, we use computational modeling, site-directed mutagenesis, synthesis of alternative substrates, and steady-state kinetics to test the proposed substrate binding model and the importance of several active site residues. In conclusion, a detailed catalytic mechanism is proposed for the AHL lactonase from Bacillus thuringiensis.
Materials and Methods
Unless otherwise noted, all chemicals were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO) and all restriction enzymes were purchased from New England BioLabs (Beverly, MA). Metal analysis of purified proteins was performed using inductively coupled plasma mass spectrometry (ICP-MS) as previously described (Departments of Geological Sciences and Chemistry and Biochemistry, The University of Texas) (10). The substrates N-hexanoyl-l-homoserine lactone (C6-HSL) and N-hexanoyl-dl-homocysteine thiolactone (C6-HCTL) were synthesized as reported previously (7). The catalytic activities of dizinc and dicobalt AHL lactonases were quantified using a continuous spectrophotometric phenol red-based assay as described previously (10). Wild-type and mutant AHL lactonase enzymes were expressed in M9 minimal medium with either ZnSO4 or CoCl2 supplements and purified as previously described (7).
Model Computational Method
The initial geometry in our simulations was adopted from a 1.4 Å X-ray structure of the AHL lactonase from B. thuringiensis complexed with a hydrolyzed N-hexanoyl-l-homoserine lactone (C6-HSL), namely the enzyme−product (EP) complex (PDB entry 3DHB) (16). The active site contains two zinc ions, customarily called Zn1 and Zn2. Zn1 is coordinated by His104, His106, and His169 (along with a weaker bridging interaction to Asp191, not shown), while Zn2 is coordinated by His109, His235, Asp108, and Asp191 (atomic definition given in Scheme 1). In our model, all histidine ligands were assumed to be neutral and the two aspartate ligands ionized. The bridging oxygen moiety is assumed to be a deprotonated water, namely a hydroxide anion, and its remaining proton is oriented toward Oδ2 of Asp108. To reconstruct the enzyme−substrate (ES) complex, we manually closed the opened lactone ring by connecting O2 and C1. In addition, the redundant C1−Ow bond was cleaved and the resulting Ow moiety was placed between the two zinc ions. All other heavy atoms in the enzyme were unaltered. Hydrogen atoms were then added by the HBUILD module of CHARMM (17). This was followed by a short minimization of the substrate with all other atoms fixed in their crystallographic positions.
Scheme 1. Atom Definition and Interaction Pattern in the Active Site of the AHL Lactonase.

The resulting ES complex was then solvated repeatedly with pre-equilibrated TIP3P (18) water spheres centered at the Zn1 atom with a radius of 25 Å. Any solvent water molecule that came within a 2.8 Å radius of a non-hydrogen atom was deleted. The solvent waters were then relaxed with a short MD run with all protein and crystal waters fixed. Using the stochastic boundary (SB) protocol (19), the solvated system was then divided into the reaction zone (r < 22 Å) and buffer zone (22 Å < r < 25 Å), with atoms outside the 25 Å radius deleted.
QM/MM Computational Method
In the QM/MM approach (20,21), the enzymatic system is partitioned into a QM region and a MM region. The former is treated with a quantum mechanical model for the electrons, whereas the latter is approximated by a molecular mechanics force field. For the dizinc ES complex model studied here, the QM region includes the two metal ions, side chains of their protein ligands, the hydroxide nucleophile, and the entire substrate. As a result, the QM model has to be very efficient to handle the large number of electrons. In this work, we chose to employ a semiempirical density functional method, namely the self-consistent charge density functional tight binding (SCC-DFTB) method (22). SCC-DFTB is very efficient, and reasonably accurate as demonstrated by several recent studies (23,24). Using a new parametrization of the zinc ion in the biological environment (25), the same type of computational calculation has been successfully conducted on metallo-β-lactamases (26–30), which bear many similarities with AHL lactonases.
The simulations reported here employed a SCC-DFTB/CHARMM implementation (23), in which the QM region was described by the SCC-DFTB method, whereas the MM region was described by the CHARMM all atom force field (31). At the boundary, the link atom approach (32) was used to saturate the dangling bonds. For the seven protein ligands, a link atom was placed between Cα and Cβ atoms in each case. There were 109 atoms in the QM region, while the total number of atoms in both QM and MM regions was 7227 for the wild type or 7223 for the Y194F mutant.
Molecular Dynamics Computational Method
In the molecular dynamics (MD) simulations, atoms in the reaction region were governed by Newtonian dynamics on the QM/MM potential. Atoms in the buffer zone were simulated with Langevin dynamics with friction and random forces stemming from the bulk solvent that were not explicitly included in the simulation (19). Nonbonded interactions were cut off at 12 Å, but the electrostatic interactions were treated with the group-based extended electrostatics model (33), which has been shown to provide a balanced treatment of the QM−MM interactions (28).
The MD simulations were performed with a time step of 1.0 fs, and the SHAKE algorithm (34) was applied to maintain the covalent bonds involving H atoms. All the calculations reported in this work were carried out using the CHARMM suite of simulation codes (17). The temperature was increased slowly, and the system was allowed to equilibrate for 200 ps. Data collection was carried out for an additional 800 ps.
Site-Directed Mutagenesis of AHL Lactonase
Construction of the expression vector for the maltose binding protein (MBP)−D108N AHL lactonase fusion protein was completed by using the overlap extension PCR method (35). Two primers were designed to amplify the N-terminal region of the gene, an outside primer 5′-CGGGAAGGATTTCAATGACAGTAAAGAAGCTTTAT-3′ and a mutagenic primer 5′-AATAATCAAGAGTGAATGTAAAAttgGTACGTCCTCCTTTGCCACG-3′ (the mutant codon is designated in lowercase). Similarly, two primers were designed to amplify the C-terminal end of the gene, an outside primer 5′-GTCGAATTCCTCAACAAGATACTCCTAATG-3′ and a mutagenic primer 5′-TTATTAGTTCTCACTTACATTTTaacCATGCAGGAGGAAACGGTGC-3′. Each set of primers (final concentration of 0.4 μM) was combined with template (1 ng of pMal-c2x-AiiA) (10), dNTPs (each at a final concentration of 0.8 mM), and Triplemaster polymerase (2 units, Eppendorf, Westbury, NY) and brought to a final volume of 20 μL. Each mixture was then subjected to the following thermocycler procedure: 94 °C for 2 min, 30 cycles at 94 °C for 20 s, 52 °C for 20 s, and 72 °C for 45 s, followed by 72 °C for 2 min. The size of the N-terminal and C-terminal fragments was verified on a 1% agarose gel, and the fragments were purified using the Qiaquick PCR product purification kit (Qiagen, Valencia, CA). A second round of PCR was performed using the N- and C-terminal fragments as a template with the two outside primers previously mentioned (each at a final concentration of 1 μM). The resulting product was purified using the Qiaquick kit and digested with EcoRI and XmnI restriction enzymes. The pMal-c2x plasmid (New England BioLabs) was digested with these enzymes as well, and the resulting linear DNA fragments were ligated overnight at 14 °C. The ligation mixture was transformed into competent Escherichia coli DH5α. Colonies were selected on LB plates supplemented with ampicillin (50 μg/mL) and subsequently screened for the presence of the D108N AHL lactonase gene by colony PCR.
Expression vectors expressing Y194F, A206W, G207W, and G207D mutants were obtained using the Quikchange kit (Stratagene, La Jolla, CA). Mutagenic primers used to obtain each mutant are given in Table 1. Briefly, both forward and reverse primers (125 ng) were combined with template (100 ng of pMal-t-AiiA plasmid) (10), dNTPs (each at a final concentration of 200 μM), and Pfu polymerase (2.5 units). The reaction mixture was brought to a volume of 50 μL by the addition of reaction buffer and subjected to the following thermocycler program: 95 °C for 30 s, 17 cycles at 95 °C for 30 s, 55 °C for 1 min, and 68 °C for 7 min and 30 s, followed by 68 °C for 10 min. DpnI restriction enzyme was added and allowed to react for 2 h at 37 °C. The digested mixture was transformed into competent E. coli Top10 (5 μL into 50 μL of cells). Transformed cells were selected by plating onto LB supplemented with ampicillin (50 μg/mL). The plasmid expressing the G207D mutant gene was obtained in a similar manner as described above, utilizing the appropriate primers described in Table 1, and the following thermocycler parameters: 95 °C for 2 min, 15 cycles at 95 °C for 30 s, 73 °C for 30 s, and 72 °C for 9 min, followed by 72 °C for 10 min. All mutations described herein were verified by fully sequencing the gene inserts (DNA Core Facility, The University of Texas).
Table 1. Primers Used for Quikchange Mutagenesisa.
| primer | sequence |
|---|---|
| Y194F forward | 5′-CGATTGATGCATCGttcACGAAAGAGAATTTTGAATGAAGTGTGC-3′ |
| Y194F reverse | 5′-GCACACTTCATTCAAAATTCTCTTTCGTgaaCGATGCATCAATCG-3′ |
| A206W forward | 5′-GAAGTGCCGTTCtggGGATTTGATCCAGAATTAGCTTTATCT-3′ |
| A206W reverse | 5′-AGATAAAGCTAATTCTGGATCAAATCCccaGAACGGCACTTC-3′ |
| G207W forward | 5′-GAAGTGCCGTTCGCAtggTTTGATCCAGAATTAGCTTTATCT-3′ |
| G207W reverse | 5′-AGATAAAGCTAATTCTGGATCAAAccaTGCGAACGGCACTTC-3′ |
| G207D forward | 5′-GATGAAGTGCCGTTCGCAgatTTTGATCCAGAATTAGCT-3′ |
| G207D reverse | 5′-AGCTAATTCTGGATCAAAatcTGCGAACGGCACTTCATC-3′ |
Mutagenic codons are represented in lowercase.
Synthesis of N-Pentanoyl-(S)-homoserine Lactone (C5-HSL)
In a procedure similar to those previously described (7,10), triethylamine (12 mmol) was added to a stirred suspension of (S)-α-amino-γ-butyrolactone hydrobromide (6.5 mmol) in dimethylformamide (50 mL) at 0 °C. Valeroyl chloride (7 mmol) was added dropwise. The reaction mixture was allowed to come to room temperature and stirred for 2 h. Solvent was removed by rotary evaporation with heating at ≤55 °C. The residue was dissolved in dichloromethane and washed sequentially with sodium sulfate (1 M, 2 × 50 mL), saturated sodium bicarbonate (1 × 50 mL), and saturated sodium chloride (1 × 50 mL). The organic layer was dried over anhydrous magnesium sulfate, and solvent was removed by rotary evaporation. The final compound was further purified by recrystallization from ethyl acetate and petroleum ether (50:50, v/v): calculated yield 30%; Rf = 0.49 in ethyl acetate; mp 125−128 °C (uncorrected); 1H NMR (300 MHz, CDCl3) δ 0.88 (t, 3H), 1.29−1.36 (m, 2H), 1.55−1.63 (m, 2H), 2.08−2.15 (m, 1H), 2.22 (m, 2H), 2.75−2.82 (m, 1H), 4.21−4.30 (m, 1H), 4.40 (t, 1H), 4.46−4.59 (m, 1H), 6.18 (broad, N−H); 13C NMR (75 MHz, CdCl3) δ 13.72, 22.27, 27.46, 30.41, 35.83, 49.11, 66.09, 173.80, 175.70; EI-HRMS MH+calc = 186.1130, MH+obs = 186.1133; [α]D,methanol20 °C−23°.
Synthesis of N-Decanoyl-(S)-homoserine Lactone (C10-HSL)
In a procedure similar to those previously described (7,10), triethylamine (12 mmol) was added to a stirred suspension of (S)-α-amino-γ-butyrolactone hydrobromide (5.7 mmol) in dimethylformamide (50 mL) at 0 °C. Decanoyl chloride (7 mmole) was added dropwise. The reaction mixture was allowed to come to room temperature and stirred for 75 min. Solvent was removed by rotary evaporation with heating at ≤55 °C. The residue was dissolved in dichloromethane and washed sequentially with sodium sulfate (1 M, 2 × 50 mL), saturated sodium bicarbonate (1 × 50 mL), and saturated sodium chloride (1 × 50 mL). The organic layer was dried over anhydrous magnesium sulfate, and solvent was removed by rotary evaporation. The final compound was further purified by recrystallization from ethyl acetate and petroleum ether (50:50, v/v): calculated yield 46%; Rf = 0.55 in ethyl acetate; mp 127−130 °C (uncorrected); 1H NMR (300 MHz, CDCl3) δ 0.86 (t, 3H), 1.24 (m, 10H), 1.57 (m, 2H), 1.64 (m, 2H), 2.06−2.13 (m, 1H), 2.20−2.25 (m, 1H), 2.81−2.90 (m, 1H), 4.22−4.31 (m, 1H), 4.42−4.48 (m, 1H), 4.49−4.53 (m, 1H), 5.95 (broad, N−H); 13C NMR (75 MHz, CdCl3) δ 14.10, 22.65, 25.41, 29.19, 29.24, 29.29, 29.39, 30.74, 31.84, 36.20, 49.29, 66.11, 175.50, 173.76; EI-HRMS MH+calc = 256.1913, MH+obs = 256.1915; [α]D,methanol20 °C−23.5°.
Results
Computational Molecular Dynamics of Wild-Type AHL Lactonase
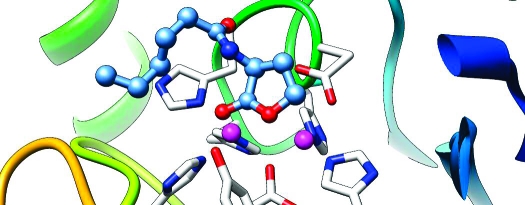
During the 1.0 ns MD simulation, the ES complex was fairly stable. The root-mean-square deviation (rmsd) of backbone atoms from the X-ray coordinates was calculated and is displayed in Figure 1. The averaged rmsd excluding the first 200 ps of the equilibration period was 1.05 ± 0.05 Å. The mode of substrate binding in the enzyme active site is sketched in Scheme 1, and a snapshot is given in Figure 2.
Figure 1.

rmsd of backbone atoms in 1.0 ns MD simulations of wild-type (black) and Y194F (red) AHL lactonase complexed with C6-HSL.
Figure 2.

Snapshot of the active site of the wild-type AHL lactonase complexed with C6-HSL. Zinc ions are shown as purple spheres. Carbon atoms are colored green, oxygens red, nitrogens blue, and hydrogens white. For clarity, the C6-HSL substrate is shown as a thicker stick model than protein residues. Key hydrogen bonds and strong coordination bonds in the active site are indicated with dashed lines.
The experimentally observed Zn coordination at the active site of AHL lactonase was satisfactorily reproduced by the MD simulation. In particular, Zn1 is tetracoordinated by His104, His106, His169, and the bridging hydroxide ion. Zn2 has five ligands, including Asp191, His235, His109, Asp108, and the bridging hydroxide ion. The metal−ligand distances listed in Table 2 are mostly in agreement with the experimental data. The only exception is that of Oδ2 of Asp191, which seems to form a stronger interaction with Zn1 in the X-ray structures than in the simulations.
Table 2. Comparison of Calculated Geometric Parameters of the ES Complex for the Wild-Type Enzyme and Y194F Mutant with Available X-ray Structures of AHL Lactonases.
| bond length (Å) |
||||||
|---|---|---|---|---|---|---|
| wild type (ES) | Y194F (ES) | 2A7M (11) (apo) | 2BTN (12) (apo) | 2BR6 (12) (EI) | wild type (16) (EP) | |
| Zn1−Zn2 | 3.49 ± 0.11 | 3.51 ± 0.11 | 3.3 | 3.4 | 3.4 | 3.7 |
| Zn2−Oδ1(Asp108) | 2.34 ± 0.13 | 2.36 ± 0.14 | 2.3 | 2.3 | 2.3 | 2.8 |
| Zn1−Oδ2(Asp191) | 2.90 ± 0.18 | 3.08 ± 0.29 | 2.6 | 2.6 | 2.6 | 2.4 |
| Zn2−Oδ2(Asp191) | 2.22 ± 0.08 | 2.21 ± 0.08 | 2.0 | 2.1 | 2.1 | 2.1 |
| Zn2−Oδ1(Asp191) | 3.04 ± 0.15 | 3.07 ± 0.15 | 3.3 | 3.2 | 3.2 | 2.7 |
| Zn1−Nε2(His104) | 2.05 ± 0.06 | 2.04 ± 0.06 | 2.1 | 2.3 | 2.3 | 2.1 |
| Zn1−Nδ1(His106) | 2.09 ± 0.08 | 2.07 ± 0.07 | 2.2 | 2.3 | 2.3 | 2.1 |
| Zn1−Nε2(His169) | 2.02 ± 0.06 | 2.02 ± 0.06 | 2.1 | 2.3 | 2.2 | 2.1 |
| Zn2−Nε2(His109) | 2.01 ± 0.06 | 2.03 ± 0.06 | 2.0 | 2.2 | 2.1 | 2.1 |
| Zn2−Nε2(His235) | 2.01 ± 0.06 | 2.02 ± 0.06 | 2.1 | 2.2 | 2.2 | 2.0 |
| Zn1−Ow | 1.99 ± 0.06 | 1.98 ± 0.05 | 2.0 | 2.1 | 2.0 | —a |
| Zn2−Ow | 2.14 ± 0.08 | 2.12 ± 0.07 | 2.0 | 2.2 | 2.2 | —a |
| O2−Zn2 | 3.79 ± 0.32 | 4.83 ± 0.54 | —a | —a | —a | —a |
| Ow−Oδ2(Asp108) | 2.77 ± 0.09 | 2.78 ± 0.10 | 2.9 | 2.6 | 2.5 | —a |
| Hw−Oδ2(Asp108) | 1.84 ± 0.10 | 1.84 ± 0.11 | —a | —a | —a | —a |
| C1−Ow | 2.58 ± 0.36 | 3.74 ± 0.63 | —a | —a | —a | —a |
| O1−HO(Tyr194) | 1.82 ± 0.20 | —a | —a | —a | —a | —a |
| O3−Ob | 2.73 ± 0.15 | 2.76 ± 0.23 | —a | —a | —a | 2.6 |
| O3−Hb1 | 1.79 ± 0.17 | 1.81 ± 0.26 | —a | —a | —a | —a |
| O3−N(Phe107) | 4.05 ± 0.73 | 4.05 ± 0.74 | —a | —a | —a | 3.8 |
| O3−HN(Phe107) | 3.11 ± 0.27 | 3.16 ± 0.74 | —a | —a | —a | —a |
| Ob−N(Phe107) | 3.12 ± 0.27 | 3.12 ± 0.37 | —a | —a | —a | 3.0 |
| Ob−HN(Phe107) | 2.24 ± 0.33 | 2.25 ± 0.46 | —a | —a | —a | —a |
Value not applicable.
The calculated Zn1−Zn2 distance of 3.49 ± 0.11 Å is close to those observed in the known AHL lactonase structures, as shown in Table 2. The much longer Zn1−Zn2 distance in the EP complex of the AHL lactonase from B. thuringiensis(16), which is used here to reconstruct the ES complex, can be attributed to the replacement of the bridging hydroxide ion with the carboxylate group of the product. The bridging hydroxide ion was found to be slightly closer to Zn1 (1.99 ± 0.06 Å) than to Zn2 (2.14 ± 0.08 Å), due apparently to the large negative charge of the Zn2 coordination sphere.
As expected, the bridging hydroxide ion forms a hydrogen bond with Oδ2 of Asp108, as evidenced by the Hw−Oδ2 distance of 1.84 ± 0.10 Å. As shown in Table 2, the Ow−Oδ2 distance is in accord with that in several known structures of AHL lactonases. The hydrogen bond from Asp108 is partially responsible for orienting the hydroxide nucleophile for attack at the carbonyl carbon in the substrate and is similar to at least one of the multiple roles assigned to the corresponding aspartate residue in dizinc metallo-β-lactamases (3,36–40).
Hydrogen bonding interactions between the substrate and active site residues exist. The two carbonyl groups in the substrate serve as hydrogen bond acceptors and contribute to substrate binding. In the X-ray structure of the EP complex (16), the substrate O3 interacts with the backbone amide group of Phe107 via a water molecule. This water-bridged hydrogen bond network is preserved in the MD simulation of the ES complex, as shown in Scheme 1 and Figure 2. The other carbonyl group on the lactone ring is hydrogen bonded with the side chain of the conserved residue Tyr194, as evidenced by the O1···H distances of 1.82 ± 0.20 Å. This confirms the importance of Tyr194 in substrate binding and is suggestive of its role in helping in formation of an oxyanion hole in catalysis (11).
Due largely to the two aforementioned hydrogen bonding interactions between the substrate and the enzyme active site, the lactone ring is held in place close to the nucleophile. Indeed, the Ow−C1 distance of 2.58 ± 0.36 Å indicates a well-organized near-attack configuration. On the other hand, the alkyl tail of the substrate is very floppy and experiences extensive conformational changes during the MD simulation.
Computational Molecular Dynamics of Y194F AHL Lactonase
It has been shown that Tyr194 plays an important role in both substrate binding and catalysis (12,41). To understand its impact, we have carried out MD simulation of the Y194F mutant using the same QM/MM protocol. The initial structure of the mutant was constructed by replacement of the OH moiety in the tyrosine side chain by H, transforming it into phenylalanine. The MD result shows that the Y194F mutant complexed with the same C6-HSL substrate is stable during the 1.0 ns of MD simulation. The rmsd is shown in Figure 1 and has an average of 0.97 ± 0.11 Å.
The Y194F mutant removes the hydrogen bond between Tyr194 and the lactone carbonyl of the substrate. As shown in Table 2, the zinc binding sites are not significantly affected by the mutation. Even the water-bridged hydrogen bonds connecting O3 with the backbone NH group of Phe107 are preserved. However, the lactone ring becomes quite floppy due apparently to the loss of the important anchoring point with Tyr194. As a result, the distance between the carbonyl carbon (C1) and the nucleophilic Ow is significantly elongated (3.74 ± 0.63 Å) when compared with that in the wild-type enzyme (2.58 ± 0.36 Å). Although a quantitative assessment of its impact on the catalysis awaits the calculation of the free energy barriers, it is clear that Tyr194 plays an important role in arranging the near-attack configuration in the active site.
Protein Purification and Characterization
Wild-type and Y194F AHL lactonase were expressed, purified, and characterized for purity and metal content as described previously (7). When protein expression was completed from medium supplemented with ZnSO4, wild-type AHL lactonase was purified with 2.0 equiv of zinc, and Y194F with 1.9 equiv of zinc, with negligible amounts of other bound metal ions. When expression cultures were supplemented with CoCl2, the wild-type protein contained 2.1 equiv of cobalt and 0.1 equiv of zinc, and Y194F contained 2.2 equiv of cobalt and 0.1 equiv of zinc with negligible amounts of other metal ions. The D108N variant of AHL lactonase was cloned without a tobacco etch virus protease cleavage site, and it was expressed, purified, and characterized with its N-terminal MBP fusion tag intact. Previous experiments have shown that the rates of hydrolysis exhibited by the fusion protein do not significantly differ from those of the untagged version (10). When expressed in medium supplemented with ZnSO4, the D108N MBP−AHL lactonase complex was found to contain 1.9 equiv of zinc. When expressed in medium containing a CoCl2 supplement, the mutant contained 1.3 equiv of cobalt and 0.1 equiv of zinc. Several residues lining the putative binding acyl chain binding site were mutated to purturb substrate binding. These mutations, A206W (2.0 equiv of cobalt and 0.2 equiv of zinc), G207W (2.1 equiv of cobalt and 0.1 equiv of zinc), and G207D (1.3 equiv of cobalt and 0.1 equiv of zinc), were all well expressed in CoCl2-supplemented medium and were purified to give untagged proteins. Circular dicroism spectroscopy was used to show there were no major structural perturbations induced by the mutations (Supporting Information).
Steady-State Hydrolysis Kinetics
Substrate turnover of C6-HSL by the Y194F and D108N variants of dizinc AHL lactonase was quite slow (Table 3), with kcat/KM values 2 orders of magnitude lower than that of the wild-type protein. Earlier work has shown that dicobalt-substituted AHL lactonase from B. thuringiensis exhibits hyperactivity (7). Therefore, to obtain more reliable kinetic parameters from these slower variants, dicobalt-substituted AHL lactonases were used. X-ray absorption spectroscopy (7) and X-ray crystallography (data not shown) have shown very similar metal coordination environments for the dicobalt- and dizinc-substituted lactonases. Using dicobalt-substituted AHL lactonase, a 2−3 order of magnitude decrease in kcat/KM values for the Y194F and D108N mutations (Table 3) was observed, as compared to the values of the wild type. The majority of this effect stemmed from a decrease in kcat values (30−150-fold) rather than changes in KM values (4−6-fold), consistent with an important role for these residues in the lactone hydrolysis mechanism.
Table 3. Steady-State Kinetic Constants of C6-HSL Hydrolysis by Dizinc and Dicobalt AHL Lactonase Variants.
| enzyme | metala | KM (mM) | kcat (s−1) | kcat/KM (M−1 s−1) |
|---|---|---|---|---|
| wild typeb | ZnSO4 | 5.6 ± 0.6 | 91 ± 3 | 1.6 × 104 |
| Y194F | ZnSO4 | 2.1 ± 0.5 | 0.31 ± 0.02 | 1.5 × 102 |
| D108N | ZnSO4 | 1.6 ± 0.6 | 1.4 ± 0.1 | 8.8 × 102 |
| wild typeb | CoCl2 | 0.36 ± 0.04 | 510 ± 10 | 1.4 × 106 |
| Y194F | CoCl2 | 2.0 ± 0.4 | 16.6 ± 0.8 | 8.3 × 103 |
| D108N | CoCl2 | 0.09 ± 0.04 | 3.5 ± 0.1 | 3.9 × 104 |
Metal supplements used during protein expression.
Values taken from ref (7).
Hydrolysis of the synthetic thiolactone substrate C6-HCTL was used to probe the effects of sulfur substitution in the substrate’s leaving group position on the relative changes induced by these mutations. Because of an observed kinetic thio effect ((7)), it was suggested that breaking a metal−thiol bond could be partially rate-limiting in the wild-type lactonase. Compared to the dicobalt wild-type enzyme, the dicobalt D108N mutant showed some interesting differences. KM and kcat values for C6-HCTL were decreased (5- and 180-fold, respectively) to an extent similar to that observed for the oxygen-containing C6-HSL substrate [4- and 150-fold, respectively (Tables 3 and 4)]. In the same comparison with the dicobalt wild-type enzyme, the Y194F mutant showed decreased values for KM and kcat (11- and 37-fold, respectively), but the oxygen congener showed an increase in KM and a decrease in kcat [6- and 30-fold, respectively (Tables 3 and 4)]. Changes in KM upon mutation may arise from changes in the substrate dissociation constant and/or from changes in other microscopic rate constants. The kcat values for thiolactone hydrolysis by the wild-type and mutant enzymes are generally 3-fold lower than for the oxygen congener, suggesting that both the oxygen-for-sulfur substitution in the substrate and the D108N or Y194F mutations of the enzyme can contribute to rate-limiting steps during steady-state hydrolysis.
Table 4. Steady-State Kinetic Constants for Hydrolysis of C6-HCTL by Dicobalt AHL Lactonase Variants.
| enzyme | KM (mM) | kcat (s−1) | kcat/KM (M−1 s−1) |
|---|---|---|---|
| wild typea | 6.7 ± 0.7 | 198 ± 8 | 3.0 × 104 |
| Y194F | 0.6 ± 0.1 | 5.4 ± 0.2 | 9.0 × 103 |
| D108N | 1.3 ± 0.2 | 1.11 ± 0.04 | 8.5 × 102 |
Values taken from ref (7).
To probe the binding site for the N-acyl chain of the substrates, dicobalt variants of AHL lactonase were used to obtain steady-state kinetic constants for the hydrolysis of substrates with five substitutions: γ-butyrolactone (GBL), N-pentanoyl-l-homoserine lactone (C5-HSL), N-hexanoyl-l-homoserine lactone (C10-HSL), N-(tert-butyloxycarbonyl)-dl-HSL (BOC-HSL), and N-(benzyloxycarbonyl)-l-HSL (CBZ-HSL). See the Supporting Information for the structures of these substrates. Hydrophobic (A206W and G207W) or charged (G207D) side chains were introduced via site-directed mutations at positions predicted to interact with the N-acyl chain of the bound substrate molecule. These interactions are predicted to occur approximately near the C5 position of the N-acyl substitution, based on the observed product-bound structures (Figure 3) (16).
Figure 3.

Product-bound AHL lactonase. The broad substrate-binding cavity surface (in blue) is shown in relation to the product’s hydrophobic acyl chain with residues Gly207 and Ala206 colored red and yellow, respectively. This figure was prepared using coordinates for Complex II (16) using USCF Chimera (49).
Because of the suspected weak nonspecific interactions made by the substrate’s acyl chain, large nonconservative mutations of residues lining its binding site were made to maximize their effect. In general, hydrolysis of C10-HSL by all mutants displayed the highest kcat/KM values (106 M−1 s−1). The kcat/KM values for C5-HSL were approximately 10-fold lower, and GBL was the poorest substrate, with kcat/KM values 5 orders of magnitude lower than those of substrates containing N-pentanoyl substitutions (Table 5). The non-natural HSL compounds, BOC-HSL and CBZ-HSL, were processed efficiently with kcat/KM values comparable to those of naturally occurring AHLs. Despite their nonconservative nature, mutations in the N-acyl binding region (A206W, G207W, and G207D) exhibited only small changes in KM values (<2.6-fold) for hydrolysis of GBL, C5-HSL, or C10-HSL, with two modest exceptions: The KM value for C5-HSL hydrolysis by the charged mutant, G207D, was increased approximately 5-fold, and the KM for C10-HSL hydrolysis by the hydrophobic mutant, G207W, was decreased approximately 10-fold, compared to that of the wild type. Changes in the resulting kcat/KM values for all mutants were <3.6-fold. Apparent substrate discrimination energies were calculated by comparing the kcat/KM values for each enzyme−substrate pair to the value for wild-type lactonase hydrolysis of GBL using the equation ΔΔG = RT ln[(kcat/KM)WT:GBL/(kcat/KM)enzyme:substrate] in a manner similar to that described previously (42).
Table 5. Steady-State Kinetic Constants for Substrate Hydrolysis by Dicobalt AHL Lactonase Variants.
| enzyme | substrate | KM (mM) | kcat (s−1) | kcat/KM (M−1 s−1) | ΔΔGa (kcal/mol) |
|---|---|---|---|---|---|
| wild type | GBL | 303 ± 40 | 5.9 ± 0.5 | 20 | 0a |
| A206W | GBL | 200 ± 15 | 3.4 ± 0.1 | 17 | 0.1 |
| G207W | GBL | 115 ± 15 | 3.3 ± 0.1 | 30 | −0.2 |
| G207D | GBL | 210 ± 30 | 1.4 ± 0.1 | 6 | 0.7 |
| wild type | C5-HSL | 0.8 ± 0.1 | 490 ± 30 | 6.1 × 105 | −6.1 |
| A206W | C5-HSL | 0.32 ± 0.06 | 148 ± 3 | 4.6 × 105 | −6.0 |
| G207W | C5-HSL | 0.7 ± 0.1 | 263 ± 7 | 3.8 × 105 | −5.8 |
| G207D | C5-HSL | 3.5 ± 0.3 | 650 ± 20 | 1.7 × 105 | −5.4 |
| wild type | C10-HSL | 0.15 ± 0.02 | 650 ± 20 | 4.3 × 106 | −7.3 |
| A206W | C10-HSL | 0.08 ± 0.01 | 106 ± 5 | 1.3 × 106 | −6.6 |
| G207W | C10-HSL | 0.015 ± 0.002 | 123 ± 4 | 8.2 × 106 | −7.7 |
| G207D | C10-HSL | 0.24 ± 0.01 | 500 ± 10 | 2.1 × 106 | −6.9 |
| wild type | BOC-HSL | 3.8 ± 0.3 | 185 ± 4 | 4.9 × 104 | −4.6 |
| wild type | CBZ-HSL | 0.48 ± 0.04 | 790 ± 20 | 1.6 × 106 | −6.7 |
The differences in discrimination energy (ΔΔG) are calculated by comparing the kcat/KM values for GBL hydrolysis by wild-type dicobalt AHL lactonase to each of the dicobalt enzyme and substrate pairs (Results).
Discussion
The metallo-β-lactamase superfamily shows great diversity (1,2), even within enzyme families (3), making a priori prediction of catalytic mechanisms difficult. Understanding substrate specificities and the ring-opening mechanism(s) of the AHL lactonases holds particular interest because of their potential application in controlling intercellular signaling pathways and virulence factor production in bacteria that use AHL-based quorum sensing (43,44). Comparing and contrasting these quorum-quenching AHL lactonases with other superfamily members can also provide insight into how related enzymes operate and into the evolutionary history and potential of this group of catalysts.
The preceding paper describes three product-bound structures of the AHL lactonase from B. thuringiensis with respect to the structures of unliganded (11) and l-homoserine lactone-inhibited (12) enzyme (16). The observed product binding mode is fundamentally different from that of the l-homoserine lactone complex and suggests a substrate orientation in which the lactone ring is rotated nearly 180° from that depicted in an earlier proposed mechanism (12). Additionally, the placement of two active site residues, Asp108 and Tyr194, suggested possible roles for these groups in the catalytic mechanism. This structural evidence is reinforced here by computational molecular dynamics and functional studies and is used in combination with other studies to propose a catalytic mechanism of AHL lactonase.
In molecular dynamics simulations, the alternative substrate orientation depicted in a previously proposed mechanism did not result in a stable complex (data not shown). On the basis of the X-ray structure of the EP complex and the QM/MM simulation reported here, we believe that this substrate orientation represents a nonproductive, inhibitory complex rather than a species on the reaction pathway. Starting from the structure of the product complex, a different substrate model was constructed in which the carbonyl oxygen of the lactone ring (O1) coordinates Zn1 and the leaving group oxygen (O2) coordinates Zn2. The QM/MM simulation using this alternative substrate binding mode was, in contrast, quite stable and underscored several important binding interactions with the enzyme. In addition to the oxygen−zinc interactions noted above, the lactone ring’s carbonyl oxygen (O1) was further stabilized through an interaction with the phenol of Tyr194, and the amide carbonyl oxygen (O3) was stabilized by a through-water hydrogen bond to the backbone amide nitrogen of Phe107. Both of these contacts persisted throughout the entire 1 ns simulation, indicating stable interactions. The importance of the interaction with Tyr194 was illustrated with a molecular dynamics simulation of the Y194F variant that resulted in destabilization of the lactone ring binding mode and less optimal positioning for catalysis. Unlike the lactone ring, the hydrophobic N-hexanoyl chain of the bound substrate was much less stable and exhibited considerable motion throughout the simulation, making transient interactions at various sites along the surface of the wide binding cavity.
The active site arrangement revealed by these QM/MM MD simulations has important implications for understanding the catalytic mechanism of AHL lactonase. The proximity of the zinc-bound nucleophilic hydroxide to the lactone carbonyl carbon (C1) indicates an optimized near-attack configuration. The addition of this hydroxide nucleophile to C1 would likely result in a tetrahedral intermediate and subsequent collapse via cleavage of the C1−O2 bond. Interaction of O1 with Zn1 and Tyr194 could polarize the carbonyl bond, making it more electrophilic for attack by the hydroxide ion, and may also serve to stabilize a tetrahedral intermediate. Although the contribution by Tyr194 has not yet been quantified, it is clear from these simulations that it plays an important role in arranging the near-attack configuration in the active site. Because of the relative proximity of Zn2 to O2 (3.79 ± 0.32 Å), it is conceivable that Zn2 could further polarize the lactone bond as well as stabilize the developing negative charge on the leaving group oxygen (O2) during ring opening.
Steady-state kinetics of substrate hydrolysis by the D108N and Y194F mutant enzymes, which display kcat/KM values at least 2 orders of magnitude lower than that of wild-type AHL lactonase, are consistent with the roles of these residues that are suggested by structural studies and by computational molecular dynamics. The Y194F mutation has a higher KM value (6-fold) and a 30-fold slower kcat value, indicating that disrupting the substrate’s interaction with the phenol group has a significant effect on catalysis and likely on binding, although KM may not directly reflect the substrate’s Kd value. The effect of the D108N mutation is even more drastic, resulting in a 150-fold decrease in kcat and a modest decrease (4-fold) in KM, consistent with a significant role of this residue in catalysis. Although the rate-limiting step in these mutants has not yet been defined, it is interesting to note that a sulfur-for-oxygen substitution in the lactone’s leaving group does not significantly change the relative effects on KM or kcat values for either mutation, as compared to wild-type hydrolysis of the respective lactone and thiolactone substrates. However, in general, turnover of the thiolactone is 3-fold slower than that of the oxygen congener. These observations suggest that both the mutations and the leaving group substitution can contribute to the overall rate-limiting steps during steady-state catalysis. However, further experiments will be required to better define the kinetic mechanism of AHL lactonase.
Several observations suggest that the N-acyl chain binding pocket of the B. thuringiensis AHL lactonase is not fully optimized for AHL substrates. Typical AHL substrates have millimolar KM values (9,10). This particular AHL lactonase (from B. thuringiensis) does not show much selectivity between acyl chains of varying length or substitutions (9). The product-bound structures exhibit alternative binding modes for the hexanoyl chains (16), and the molecular dynamics simulations show considerable motion and conformational flexibility in this part of the substrate. To improve our understanding of these interactions better, we compared the hydrolysis kinetics of an unsubstituted lactone, γ-butyrolactone (GBL), with substrates that had progressively longer acyl chains, C5-HSL and C10-HSL. The unsubstituted GBL was a very poor substrate with high KM values (ca. 300 mM) and is only slowly processed by the enzyme. C5-HSL was a much better substrate, exhibiting lower KM values (800 μM) and higher kcat values, leading to a 4 order of magnitude increase in kcat/KM values. The longer C10-HSL substrate displayed even lower KM values (ca. 150 μM), although kcat values did not show much improvement. Since these enzymes are expected to usually operate under second-order conditions, it is useful to compare the apparent substrate discrimination energies derived from their respective kcat/KM values (Table 5). Compared to the unsubstituted GBL, the five-carbon amide substitution in C5-HSL results in a 6 kcal/mol improvement in substrate discrimination, but five more carbon units in the C10-HSL substrate only translates into an additional 1.2 kcal/mol. Considering the relatively nonspecific binding surface for the acyl chain and the similar hydrolysis kinetics for substrates between 4 and 12 carbons in length (9), we can approximate that lengthening the substrate’s acyl chain from 5 to 10 carbons improves substrate discrimination by only 0.24 kcal/mol per carbon increment. In comparison, transfer of n-alkyl alcohols from water into n-octanol is favorable by approximately 0.38 kcal/mol per carbon increment, and binding hydrophobic groups to enzymes can be even more favorable due to the additional displacement of waters from binding sites (42). Therefore, AHL lactonase’s binding site for the substrate’s acyl chain is considerably less hydrophobic than n-octanol and does not appear to be fully optimized for acyl chain binding. These observations are consistent with the binding surface observed in the product-bound structures (16). To probe this binding cavity in more detail, we introduced large nonconservative mutations to increase (A206W and G207W) and to decrease (G207D) the hydrophobicity of the binding surface. As predicted, the G207W mutation resulted in a considerably lower KM value (ca. 15 μM) for the longer C10-HSL substrate, which presumably could take advantage of the increased hydrophobicity of the cavity. Likewise, the more polar G207D mutation resulted in an increased KM for C5-HSL, a shorter substrate that presumably cannot compensate for this lost hydrophobic interaction. However, in general, mutations in the shallow binding cavity resulted in only minor changes to kcat/KM values, consistent with the multivalent interactions predicted to occur between the substrates’ acyl chains and the wide binding surface. This relatively nonspecific nature of the binding surface is demonstrated by the facile hydrolysis of artificial substrates that carry non-natural N-acyl substitutions with hydrophobicity similar to that of AHLs but that have very different steric constraints, BOC-HSL and CBZ-HSL (Table 4). On the basis of this broad specificity, it is likely that the AHL lactonase can also hydrolyze many non-natural quorum-sensing agonists and antagonists that are N-substituted homoserine lactones (45) and may also reflect a selective pressure to hydrolyze a large variety of AHLs found in native B. thuringiensis growth environments. It should be noted that although the enzyme’s binding site does not appear to be fully optimized for a particular substrate’s acyl chain, the presence of this acyl substituent does appear to be required to prevent the nonproductive binding orientation observed with l-homoserine (12), which lacks this acyl substitution. Presumably, the unsubstituted GBL substrate can access both orientations.
All available results can be combined in proposing a catalytic mechanism for the AHL lactonase from B. thuringiensis (Scheme 2). On the basis of metal analysis, reconstitution studies, 1H NMR, EXAFS, and X-ray crystallography, the resting enzyme contains two zinc(II) ions bound closely together in a dinuclear site and these metal ions are essential for both enzyme activity and protein folding (7,10–12). As suggested by X-ray crystallography of product complexes (16) and observed in molecular dynamics simulations, AHL substrates bind with the less hindered re face of the lactone ring facing the dinuclear metal center with the ring’s carbonyl oxygen poised over Zn1 and the leaving group oxygen over Zn2. It is interesting to note that the related metallo-β-lactamases also position the less hindered exo face of their β-lactam substrates toward the metal center. As observed in a product complex, the lactonase’s active site appears to somewhat tighten around the bound ligand, although not around the hydrophobic acyl chain (16). Stabilizing interactions are also made between the lactone’s carbonyl oxygen and the phenol group of Tyr194, as demonstrated by molecular dynamics experiments, site-directed mutagenesis, and steady-state kinetics (12,41). The substrate’s amide carbonyl oxygen makes a through-water hydrogen bond to the backbone amide nitrogen of Phe107 [observed in the product complexes (16) and MD simulations], and the hydrophobic acyl chain makes weak multivalent surface interactions along a wide shallow cavity [consistent with product structures (16), MD simulations, site-directed mutagenesis, alternative substrates, and steady-state kinetics]. In this proposed substrate orientation, the metal ions can provide further polarization of the lactone bond, increasing the electrophilicity of the ring’s carbonyl carbon. Isotopic labeling studies have indicated that ring opening occurs via an addition elimination reaction with water addition at this same carbonyl carbon, consistent with this proposal (7). The substrate is positioned well for attack by the nucleophilic hydroxide ion, which bridges both metal ions and coordinates to Asp108. As proposed for other dinuclear metallo hydrolases (46), when substrate binds the bridging hydroxide may release one of the zinc ions and become more nucleophilic. However, other than a slightly shorter Zn1−Ow distance in the calculated ES complex (Table 2), there is currently no experimental evidence for this rearrangement in AHL lactonase. Both Zn1 and the phenol of Tyr194 are well placed to stabilize the formation of a tetrahedral adduct. This proposed role for Tyr194 is consistent with the observation of significantly decreased kcat values (Table 3) and destabilization of the bound lactone (MD simulations) upon mutation of this residue to Phe. Although deprotonation of a similar tetrahedral intermediate to the dianion appears to be required for β-lactam ring opening by one related metallo-β-lactamase (47), the dianion may not be required to cleave the intrinsically less stable lactone substrates. Collapse of the tetrahedral adduct (either before or after deprotonation) and cleavage of the carbon oxygen bond could be facilitated by Zn2, which is well placed to help stabilize the developing negative charge on the leaving group oxygen. An observed kinetic thio effect on kcat is consistent with a kinetically significant metal−leaving group interaction during catalysis (7). Structural and spectroscopic evidence for such an intermediate in AHL lactonase has not been published, but similar intermediates have been detected in related metallo-β-lactamase enzymes (3). Coordination of the anionic leaving group to Zn2 may weaken the Zn2−Asp108 interaction, freeing Asp108 to release Zn2 and to deprotonate the intermediate. Asp108, observed to coordinate the bridging hydroxide in the X-ray structure of the unliganded enzyme (11), is well placed to serve as the general base for this deprotonation. The loss of coordination of Asp108 to Zn2 is observed in the product complex (16), in a phosphate-bound structure of a related AHL lactonase (13), and in a low-pH structure of a related metallo-β-lactamase (48). Also, mutation of this position in AHL lactonase to an asparagine results in a significant decrease in kcat values. All of these observations support the role of Asp108 as the proton acceptor. After deprotonation by Asp108, the product’s carboxylate would be fully available to coordinate both metal sites, bridging these metal ions in a bidentate fashon as observed in the product complexes (16) and possibly leading to the longer Zn1−Zn2 distances observed in the product structures, MD simulations, and the phosphate-bound structure of a related AHL lactonase (13). This rearrangement could reposition the leaving group oxygen, allowing it to release its transient coordination to Zn2 and to receive a proton from the protonated Asp108. Consistent with this proposal, the leaving group alcohol is found to be within hydrogen bonding distance of Asp108 in one product complex (16). Transfer of the proton back to the leaving group would result in a deprotonated Asp108, which could then recoordinate Zn2, facilitate product release, and return the enzyme to its original resting state. This proposed shuttling by Asp108 would provide a favorable pathway for the transferred proton to move between groups with successively higher pKa values, from the zinc-bound product carboxyl, to Asp108, to the zinc-bound hydroxyl leaving group. In metallo-β-lactamase, a homologous Asp has been proposed to serve primarily as a Zn2 anchor (among other roles) (37–40). However, in AHL lactonase, this burden appears to be shared by Asp191, which makes a weak compensating aniso-bidentate interaction with Zn2 when Asp108 releases this metal ion (16). Also in contrast to the case with metallo-β-lactamases (3), Asp191 serves as a bridging ligand between the two active site metal ions and may help mediate the placticity of this dinuclear active site.
Scheme 2. Proposed Catalytic Mechanism of the Dizinc AHL Lactonase from B. thuringiensis.

Despite these differences, it is interesting to note that there are three aspects of the proposed mechanism, substrate orientation, a cocatalytic metal site, and leaving group stabilization by Zn2, that are also conserved in the catalytic mechanisms of metallo-β-lactamase and glyoxalase II (3,14). The common aspects of these chemical mechanisms may help to define an evolutionary thread that connects the hydrolytic enzymes of the mechanistically diverse metallo-β-lactamase superfamily.
Acknowledgments
We thank Terezie H. Schaller for her assistance in constructing D108N AHL lactonase and Paul Enemark for his assistance in the synthesis of C5-HSL.
Supporting Information Available
Steady-state kinetic studies, circular dichroism spectroscopy of AHL lactonase, and supplemental references. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: AHL, N-acyl-l-homoserine lactone; HSL, homoserine lactone; HCTL, homocysteine thiolactone; MBP, maltose binding protein; PCR, polymerase chain reaction; ICP-MS, inductively coupled plasma mass spectrometry; aiiA, gene for AHL lactonase from B. thuringiensis; ESI-HRMS, electrospray ionization high-resolution mass spectrometry; TLC, thin layer chromatography; mp, melting point; NMR, nuclear magnetic resonance; Rf, retention factor; rmsd, root-mean-square deviation; MD, molecular dynamics; QM, quantum mechanical; MM, molecular mechanical; EP, enzyme−product; ES, enzyme−substrate; EI, enzyme−inhibitor; SCC-DFTB, self-consistent charge-density functional tight binding.
Supplementary Material
References
- Aravind L. (1999) An evolutionary classification of the metallo-β-lactamase fold proteins. In Silico Biol. 1, 69–91. [PubMed] [Google Scholar]
- Daiyasu H.; Osaka K.; Ishino Y.; Toh H. (2001) Expansion of the zinc metallo-hydrolase family of the β-lactamase fold. FEBS Lett. 503, 1–6. [DOI] [PubMed] [Google Scholar]
- Crowder M. W.; Spencer J.; Vila A. J. (2006) Metallo-β-lactamases: Novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 39, 721–728. [DOI] [PubMed] [Google Scholar]
- Costello A. L.; Sharma N. P.; Yang K. W.; Crowder M. W.; Tierney D. L. (2006) X-ray absorption spectroscopy of the zinc-binding sites in the class B2 metallo-β-lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry 45, 13650–13658. [DOI] [PubMed] [Google Scholar]
- Zang T. M.; Hollman D. A.; Crawford P. A.; Crowder M. W.; Makaroff C. A. (2001) Arabidopsis glyoxalase II contains a zinc/iron binuclear metal center that is essential for substrate binding and catalysis. J. Biol. Chem. 276, 4788–4795. [DOI] [PubMed] [Google Scholar]
- Schilling O.; Wenzel N.; Naylor M.; Vogel A.; Crowder M.; Makaroff C.; Meyer-Klaucke W. (2003) Flexible metal binding of the metallo-β-lactamase domain: Glyoxalase II incorporates iron, manganese, and zinc in vivo. Biochemistry 42, 11777–11786. [DOI] [PubMed] [Google Scholar]
- Momb J.; Thomas P. W.; Breece R. M.; Tierney D. L.; Fast W. (2006) The quorum-quenching metallo-γ-lactonase from Bacillus thuringiensis exhibits a leaving group thio effect. Biochemistry 45, 13385–13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y. H.; Xu J. L.; Li X. Z.; Zhang L. H. (2000) AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. U.S.A. 97, 3526–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. H.; Weng L. X.; Dong Y. H.; Zhang L. H. (2004) Specificity and enzyme kinetics of the quorum-quenching N-acyl homoserine lactone lactonase (AHL-lactonase). J. Biol. Chem. 279, 13645–13651. [DOI] [PubMed] [Google Scholar]
- Thomas P. W.; Stone E. M.; Costello A. L.; Tierney D. L.; Fast W. (2005) The quorum-quenching lactonase from Bacillus thuringiensis is a metalloprotein. Biochemistry 44, 7559–7569. [DOI] [PubMed] [Google Scholar]
- Liu D.; Lepore B. W.; Petsko G. A.; Thomas P. W.; Stone E. M.; Fast W.; Ringe D. (2005) Three-dimensional structure of the quorum-quenching N-acyl homoserine lactone hydrolase from Bacillus thuringiensis. Proc. Natl. Acad. Sci. U.S.A. 102, 11882–11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. H.; Choi W. C.; Kang H. O.; Lee J. S.; Kang B. S.; Kim K. J.; Derewenda Z. S.; Oh T. K.; Lee C. H.; Lee J. K. (2005) The molecular structure and catalytic mechanism of a quorum-quenching N-acyl-l-homoserine lactone hydrolase. Proc. Natl. Acad. Sci. U.S.A. 102, 17606–17611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Thomas P. W.; Momb J.; Hoang Q. Q.; Petsko G. A.; Ringe D.; Fast W. (2007) Structure and specificity of a quorum-quenching lactonase (AiiB) from Agrobacterium tumefaciens. Biochemistry 46, 11789–11799. [DOI] [PubMed] [Google Scholar]
- Cameron A. D.; Ridderstrom M.; Olin B.; Mannervik B. (1999) Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Struct. Folding Des. 7, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Spencer J.; Read J.; Sessions R. B.; Howell S.; Blackburn G. M.; Gamblin S. J. (2005) Antibiotic recognition by binuclear metallo-β-lactamases revealed by X-ray crystallography. J. Am. Chem. Soc. 127, 14439–14444. [DOI] [PubMed] [Google Scholar]
- Liu D.; Momb J.; Thomas P. W.; Moulin A.; Petsko G. A.; Fast W.; Ringe D. (2008) Mechanism of the quorum-quenching lactonase (AiiA) from Bacillus thuringiensis. 1. Product-bound structures. Biochemistry 47, 7706–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. (1983) CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Brooks C. L. III; Karplus M. (1989) Solvent effects on protein motion and protein effects on solvent motion. J. Mol. Biol. 208, 159–181. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Levitt M. (1976) Theoretical studies of enzymatic reactions: Dielectric, electrostatic and steric stabilization of carbonium ion in the reaction of lysozyme. J. Mol. Biol. 103, 227–249. [DOI] [PubMed] [Google Scholar]
- Gao J. (1996) Methods and applications of combined quantum mechanical and molecular mechanical potentials, in Reviews in Computational Chemistry (Lipkowitz K. B., and Boyd D. B., Eds.) VCH, New York. [Google Scholar]
- Elstner M.; Porezag D.; Jungnickel G.; Elsner J.; Haugk M.; Frauenheim T.; Suhai S.; Seigert G. (1998) Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268. [Google Scholar]
- Cui Q.; Elstner M.; Kaxiras E.; Frauenheim T.; Karplus M. (2001) A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J. Phys. Chem. B 105, 569–585. [DOI] [PubMed] [Google Scholar]
- Riccardi D.; Schaefer P.; Yang Y.; Yu H.; Ghosh N.; Prat-Resina X.; Konig P.; Li G.; Xu D.; Guo H.; Elstener M.; Cui Q. (2006) Development of effective quantum mechanical/molecular mechanical (QM/MM) methods for complex biological processes. J. Phys. Chem. B 110, 6458–6469. [DOI] [PubMed] [Google Scholar]
- Elstner M.; Cui Q.; Munih P.; Kaxiras E.; Frauenheim T.; Karplus M. (2003) Modeling zinc in biomolecules with the self consistent charge density functional tight binding (SCC-DFTB) method: Applications to structure and energetic analysis. J. Comput. Chem. 24, 565–581. [DOI] [PubMed] [Google Scholar]
- Xu D.; Zhou Y.; Xie D.; Guo H. (2005) Antibiotic binding to monozinc CphA β-lactamase from Aeromonas hydropila: Quantum mechanical/molecular mechanical and density functional theory studies. J. Med. Chem. 48, 6679–6689. [DOI] [PubMed] [Google Scholar]
- Xu D.; Xie D.; Guo H. (2006) Catalytic mechanism of class B2 metallo-β-lactamase. J. Biol. Chem. 281, 8740–8747. [DOI] [PubMed] [Google Scholar]
- Xu D.; Guo H.; Cui Q. (2007) Antibiotic binding to dizinc β-lactamase L1 from Stenotrophomonas maltophilia: SCC-DFTB/CHARMM and DFT studies. J. Phys. Chem. A 111, 5630–5636. [DOI] [PubMed] [Google Scholar]
- Wang C.; Guo H. (2007) Inhibitor binding by metallo-β-lactamase IMP-1 from Pseudomonas aeruginosa: Quantum mechanical/molecular mechanical simulations. J. Phys. Chem. B 111, 9986–9992. [DOI] [PubMed] [Google Scholar]
- Xu D.; Guo H.; Cui Q. (2007) Deactivation of penicillin by dizinc β-lactamase: Mechanistic insights from QM/MM and DFT studies. J. Am. Chem. Soc. 129, 10814. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Bashford D.; Bellott M.; Dunbrack R. L. Jr.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E. III; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Field M. J.; Bash P. A.; Karplus M. (1990) A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11, 700–733. [Google Scholar]
- Steinbach P. J.; Brooks B. R. (1994) New spherical-cutoff methods for long-range forces in macromolecular simulations. J. Comput. Chem. 15, 667. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Ho S. N.; Hunt H. D.; Horton R. M.; Pullen J. K.; Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59. [DOI] [PubMed] [Google Scholar]
- Xu D.; Guo H.; Cui Q. (2007) Antibiotic binding to dizinc β-lactamase L1 from Stenotrophomonas maltophilia: SCC-DFTB/CHARMM and DFT studies. J. Phys. Chem. A 111, 5630–5636. [DOI] [PubMed] [Google Scholar]
- Crisp J.; Conners R.; Garrity J. D.; Carenbauer A. L.; Crowder M. W.; Spencer J. (2007) Structural basis for the role of Asp-120 in metallo-β-lactamases. Biochemistry 46, 10664–10674. [DOI] [PubMed] [Google Scholar]
- Garrity J. D.; Carenbauer A. L.; Herron L. R.; Crowder M. W. (2004) Metal binding Asp-120 in metallo-β-lactamase L1 from Stenotrophomonas maltophilia plays a crucial role in catalysis. J. Biol. Chem. 279, 920–927. [DOI] [PubMed] [Google Scholar]
- Llarrull L. I.; Fabiane S. M.; Kowalski J. M.; Bennett B.; Sutton B. J.; Vila A. J. (2007) Asp120 locates Zn2 for optimal metallo-β-lactamase activity. J. Biol. Chem. 282, 18276–18285. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y.; Kuroki T.; Yasuzawa H.; Higashi T.; Jin W.; Kawanami A.; Yamagata Y.; Arakawa Y.; Goto M.; Kurosaki H. (2005) Probing the role of Asp-120(81) of metallo-β-lactamase (IMP-1) by site-directed mutagenesis, kinetic studies, and X-ray crystallography. J. Biol. Chem. 280, 20824–20832. [DOI] [PubMed] [Google Scholar]
- Lu X.; Yuan Y.; Xue X. L.; Zhang G. P.; Zhou S. N. (2006) Identification of the critical role of Tyr-194 in the catalytic activity of a novel N-acyl-homoserine lactonase from marine Bacillus cereus strain Y2. Curr. Microbiol. 53, 346–350. [DOI] [PubMed] [Google Scholar]
- Fersht A. (1999) Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding, W. H. Freeman, New York. [Google Scholar]
- Dong Y. H.; Wang L. H.; Zhang L. H. (2007) Quorum-quenching microbial infections: Mechanisms and implications. Philos. Trans. R. Soc. London, Ser. B 362, 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y. H.; Zhang L. H. (2005) Quorum sensing and quorum-quenching enzymes. J. Microbiol. 43, 101–109. [PubMed] [Google Scholar]
- Geske G. D.; O’Neill J. C.; Blackwell H. E. (2007) N-Phenylacetanoyl-l-homoserine lactones can strongly antagonize or superagonize quorum sensing in Vibrio fischeri. ACS Chem. Biol. 2, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz R. C.; Bzymek K. P.; Swierczek S. I. (2003) Co-catalytic metallopeptidases as pharmaceutical targets. Curr. Opin. Chem. Biol. 7, 197–206. [DOI] [PubMed] [Google Scholar]
- Bounaga S.; Laws A. P.; Galleni M.; Page M. I. (1998) The mechanism of catalysis and the inhibition of the Bacillus cereus zinc-dependent β-lactamase. Biochem. J. 331, Part 3703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A. M.; Rasia R. M.; Vila A. J.; Sutton B. J.; Fabiane S. M. (2005) Effect of pH on the active site of an Arg121Cys mutant of the metallo-β-lactamase from Bacillus cereus: Implications for the enzyme mechanism. Biochemistry 44, 4841–4849. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.