

Abstract
The reaction center (RC) from Rhodobacter sphaeroides converts light into chemical energy through the reduction and protonation of a bound quinone molecule QB (the secondary quinone electron acceptor). We investigated the proton transfer pathway by measuring the proton-coupled electron transfer, kAB(2) [QA⨪QB⨪ + H+ → QA(QBH)−] in native and mutant RCs in the absence and presence of Cd2+. Previous work has shown that the binding of Cd2+ decreases kAB(2) in native RCs ≈100-fold. The preceding paper shows that bound Cd2+ binds to Asp-H124, His-H126, and His-H128. This region represents the entry point for protons. In this work we investigated the proton transfer pathway connecting the entry point with QB⨪ by searching for mutations that greatly affect kAB(2) (≳10-fold) in the presence of Cd2+, where kAB(2) is limited by the proton transfer rate (kH). Upon mutation of Asp-L210 or Asp-M17 to Asn, kH decreased from ≈60 s−1 to ≈7 s−1, which shows the important role that Asp-L210 and Asp-M17 play in the proton transfer chain. By comparing the rate of proton transfer in the mutants (kH ≈ 7 s−1) with that in native RCs in the absence of Cd2+ (kH ≥ 104 s−1), we conclude that alternate proton transfer pathways, which have been postulated, are at least 103-fold less effective.
Keywords: bacterial photosynthesis, Rhodobacter sphaeroides, metal binding, proton-coupled electron transfer
The conversion of light into chemical energy in photosynthetic bacteria is initiated within a membrane-bound pigment–protein complex called the reaction center (RC). The isolated RC from Rhodobacter (Rb.) sphaeroides is composed of three polypeptide subunits (L, M, and H); four bacteriochlorophylls; two bacteriopheophytins; one internally bound nonheme Fe2+; and two ubiquinone (UQ10) molecules (reviewed in refs. 1 and 2). Light induces electron transfer from the primary donor (a bacteriochlorophyll dimer) through a series of electron donor and acceptor molecules (a bacteriopheophytin and a quinone molecule QA) to a loosely bound secondary quinone QB. QB accepts two electrons, sequentially transferred through the electron transfer chain, and two protons, through the proton transfer pathway(s), to form quinol. The first electron transfer to QB (kAB(1)) does not involve direct protonation of the quinone (Eq. 1).
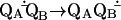 |
1 |
However, the second electron transfer (kAB(2)) is coupled to the direct protonation of the quinone (Eq. 2). The mechanism of the proton-coupled electron transfer reaction kAB(2) (Eq. 2) was shown to be a two-step process in which fast protonation precedes rate-limiting electron transfer (3). Subsequent protonation (Eq. 3) leads to the formation of quinol.
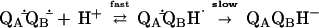 |
2 |
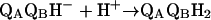 |
3 |
The quinol, QBH2, serves as a mobile electron and proton carrier (4–6) transferring electrons and protons from the RC to other components of the bioenergetic cycle.
In the RC embedded in the bacterial membrane, the protons, taken up to form quinol at the QB site, come from the cytoplasm. The pathways for these proton transfer events have been studied in isolated RCs by a number of groups (7–12). Large decreases (≥103-fold) in kAB(2) and/or the rate of proton uptake in RCs with mutations at Glu-L212, Ser-L223, and Asp-L213, located near QB⨪ (≤5 Å), had shown that these amino acid residues are important for the proton transfer reactions (Eqs. 2 and 3) (reviewed in refs. 13 and 14). The effect of mutations at sites located further from QB⨪ (≥10 Å) are much smaller. Because kAB(2) (Eq. 2) is in most mutant RCs not a direct measure of the rate of proton transfer (15–17), the observed decrease in kAB(2) does not necessarily represent a reduction in the rate of proton transfer. The lack of identification of a single amino acid that is clearly more important than any other led to the proposal of many possible pathways for proton transfer (7–12). Furthermore, the crystal structure revealed that Glu-L212, Asp-L213, and Ser-L223 could be connected to the surface through a number of routes (18–22), supporting the idea of several possible proton transfer pathways, each of them having a distinct surface entry point. The recent finding that binding of a single Zn2+ (23, 24) or Cd2+ metal ion to the RC surface (24) slows the rate of proton transfer ≥100-fold shows that there is a unique proton entry point—i.e., that not all possible pathways are equally effective (24).
An important consequence of the decrease in the rate of proton transfer is a change in the mechanism of the proton-coupled electron transfer kAB(2). In the presence of a bound metal ion, the first step becomes rate limiting (Eq. 4) (24).
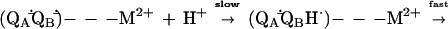 |
4 |
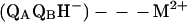 |
where M2+ is either Zn2+ or Cd2+. Thus, measurement of kAB(2) in the presence of a bound M2+ provides us with an assay to determine directly the effect of a mutation on the rate of proton transfer as opposed to inferring it from a change in the observed rate (Eq. 2).
In this study, we investigated the pathway for proton transfer from solution to the bound semiquinone QB⨪. We measured kAB(2) in the presence of Cd2+, where proton transfer is the rate-limiting step (24), to determine the effect of a mutation on the rate of proton transfer. We searched for mutant RCs that decreased the rate of proton transfer without affecting other kinetic rates to eliminate mutations that affect the properties (e.g., the redox potential and pKa) of the proton acceptor QBH⋅. This approach ensures that the mutations affect the proton pathway between the entry point (25) and QB⨪. Two mutant RCs, DN(L210) [Asp-L210 → Asn] and DN(M17) [Asp-M17 → Asn], satisfied the above criteria and are the focus of this work. The effects of these mutations on the transfer rate of the first electron, kAB(1) (Eq. 1), and on the rate of the proton-coupled second electron transfer, kAB(2) (Eq. 2), in the absence and presence of Cd2+ were measured. In addition, the rates of charge recombination kAD (D+QA⨪ → DQA) and kBD (D+QAQB⨪ → DQAQB), which are sensitive probes of the electrostatic and structural changes near QB, were determined.
Materials and Methods
Reagents and Quinones.
Coenzyme Q10 (2,3-dimethoxy-5-methyl-6-decaisoprenyl-1,4-benzoquinone) was obtained from Sigma, and the QB site inhibitor stigmatellin (prepared in ethanol) was from Fluka. Horse heart cytochrome c (Cyt c) was obtained from Sigma, reduced (>95%) by hydrogen gas on platinum black (Aldrich), and filtered (0.2-μm pore size acetate filter). All other reagents were of analytical grade.
Modification, Isolation, and Preparation of RCs.
The site-directed mutations Asp-L210 → Asn [DN(L210)] and Asp-M17 → Asn [DN(M17)] were constructed as previously described (7). RCs from Rb. sphaeroides R26 and the mutant strains were isolated in 15 mM Tris⋅HCl, pH 8/0.025% lauryldimethylamine N-oxide (LDAO)/0.1 mM EDTA by following published procedures (26). The final ratio of absorbance, A280/A800, was ≤1.25. Reconstitution of the QB site was achieved by incubating the RC solution with Q10 (≈5 Q10 per RC) solubilized in 1% LDAO followed by dialysis against TL buffer (10 mM Tris⋅HCl, pH 8.0/0.025% LDAO). Occupancy of the QB sites was 70% to 80%.
Transient Optical Spectroscopy.
Charge recombination rates were measured by monitoring the recovery of the donor band at 865 nm after bleaching with a single laser flash (PhaseR DL2100c, 590 nm, ≈0.2 J per pulse, 0.4-μs full-width-half-maximum) using a single-beam spectrophotometer (27). All measurements were performed at 21°C. To determine the recombination rate, kBD (D+QAQB⨪ → DQAQB), the observed absorption decays were fitted to two exponentials by using procedures previously described (28). The recombination rate, kAD (D+QA⨪ → DQA), was measured in the presence of 10 μM stigamatellin, which blocks electron transfer to QB.
The rate constant, kAB(1), for the transfer of the first electron to QB (Eq. 1) was measured by monitoring the bacteriopheophytin bandshift at 750 nm, which is differentially sensitive to the reduction state of the quinones QA and QB (27, 29). To improve the signal-to-noise ratios, 9 to 36 traces were averaged.
The proton-coupled electron transfer kAB(2) (Eq. 2) was determined by monitoring the decay of the semiquinone absorption at 450 nm after a second saturating laser flash in the presence of an external reductant (10 μM horse heart Cyt c) (30). The kinetic decay was fitted to the sum of two exponentials. The observed Cd2+ concentration dependence of kAB(2) was fitted to the kinetic model (Eq. A4) by using origin 6.0 (Microcal).
Results
Charge Recombination Rates.
The charge recombination rates for the reactions D+QA⨪ → DQA (kAD) and D+QAQB⨪ → DQAQB (kBD) were measured at 865 nm. In the absence of CdSO4, the measured values of kAD (≈9 s−1) and kBD (≈ 1 s−1) were approximately the same for native and mutant DN(M17) [Asp-M17 → Asn] and DN(L210) [Asp-L210 → Asn] RCs at pH 7.7 (Table 1). The addition of 1 mM Cd2+ produced only small changes in kAD and kBD for native and mutant RCs (Table 1).
Table 1.
Measured rate constants for native and mutant RCs in the presence and absence of Cd2+ (pH 7.7; 21°C)
| RC* | kAD, s−1 | kBD, s−1 | kAB(1)† s−1 | kAB(2), s−1 |
|---|---|---|---|---|
| Native | 8.8 | 0.8 | 7,000 | 1,200 |
| DN(M17) | 9.0 | 0.6 | 1,000 | 500 |
| DN(L210) | 8.9 | 1.0 | 900 | 600 |
| Native + Cd2+ | 9.0 | 0.8 | 700 | 60 |
| DN(M17) + Cd2+ | 8.9 | 0.8 | 300 | 8 |
| DN(L210) + Cd2+ | 9.0 | 2.2 | 150 | 8 |
Errors (statistical) in the rates are estimated to be ≈8% for the charge recombination rate constants kAD and kBD and ≈15% for the forward electron transfer rate constants kAB(1) and kAB(2). It should be noted that the forward rate constants are dependent on conditions, such as type and concentration of detergent, and can vary by factors of 2 between different measurement conditions.
Conditions for kinetic measurements as described in the text. The samples labeled “+ Cd2+” were measured in the presence of 1 mM CdSO4.
The First Electron Transfer Rate, kAB(1).
In the absence of CdSO4, the transfer rates for the first electron to QB (kAB(1), Eq. 1), measured at 750 nm, were reduced ≈7-fold in the DN(M17) and DN(L210) compared with native RCs at pH 7.7 (Table 1). Upon addition of 1 mM CdSO4, kAB(1) was decreased ≈10-fold in native RCs, ≈3-fold in the DN(M17), and ≈6-fold in the DN(L210) RCs (Table 1). The observed rate constant was essentially independent of the metal concentration above 10 μM.
The Proton-Coupled Electron Transfer Rate, kAB(2).
The rate of transfer of the second electron to QB (kAB(2), Eq. 2), after a second saturating laser flash, was measured at 450 nm in native RCs to be 1200 s−1 at pH 7.7 (Table 1). In the DN(M17) and DN(L210) RCs, kAB(2) was decreased ≈2-fold (Table 1). In addition, kAB(2) in the DN(L210) and DN(M17) mutant RCs displayed the same pH profile as native RCs (9, 10) with a shift of ≤0.5 pH unit to lower pH (data not shown).
The dependence of kAB(2) on the driving force for electron transfer was measured as described by Graige et al. (3), by replacing the native Q10 in the QA site with a series of naphthoquinones. As previously observed in native RCs, kAB(2) in the mutant RCs was also dependent on the driving force for electron transfer (data not shown).
Upon addition of low concentrations of CdSO4 (≤5 μM), a fraction of the sample exhibited a slower rate. From the concentration dependence of this fraction, we determined a dissociation constant (Kd) of 3.1 ± 0.6 μM and 1.2 ± 0.5 μM for the DN(M17) and DN(L210) RCs, respectively (Table 2); Kd for native RCs was previously determined to be ≈0.3 μM (24).
Table 2.
Parameters obtained from the fit of the CdSO4 concentration dependence on kAB(2) (see Fig. 2) to Eqs. 5 and A4
| RC | Kd, μM | kAB(2)(Cd2+), s−1 | kAB(2)(0), s−1 | kon, μM−1⋅s−1 | koff, s−1 |
|---|---|---|---|---|---|
| Native | ≈0.3 | 60 | ND | ND | ≪10 |
| DN(M17) | 3.1 | 7.1 | 630 | 30 | 100 |
| DN(L210) | 1.2 | 7.5 | 690 | 90 | 110 |
ND, not determinable (in view of the Cd2+ concentration independence). Errors are 10% for kAB(2) (Cd2+) and koff, 20% for Kd, kAB(2)(0), and kon.
Upon addition of 10 μM CdSO4, kAB(2) became essentially monophasic with rate constants of 60 s−1 in native RCs (data not shown) and ≈100 s−1 in DN(M17) and in DN(L210) RCs (Fig. 1). Upon further addition of CdSO4, kAB(2) did not change in native RCs but decreased to a limiting rate of ≈7 s−1 above 1 mM Cd2+ in DN(M17) and in DN(L210) RCs (Fig. 2). The limiting value for kAB(2) was the same for both mutants, being ≈10-fold smaller than in native RCs (Fig. 3).
Figure 1.

Absorbance decay of the semiquinones at 450 nm as a function of time after the second laser flash in the presence of 10 μM, 100 μM, or 1000 μM CdSO4 in the DN(M17) [Asp-M17 → Asn] RCs. From the decay, the rate constant kAB(2) was determined. Note the slowing of the kinetics with increasing CdSO4 concentrations. A similar behavior is observed in the DN(L210) [Asp-L210 → Asn] RCs (data not shown). This is in contrast to the behavior of native RCs, where no change in the kinetics is observed above CdSO4 concentrations of 10 μM. The pedestal at long times is due to the absorbance change of Cyt c used to reduce the primary donor. Conditions: 2 μM RCs in TL buffer, pH 7.7; concentrations of CdSO4 as indicated.
Figure 2.

The slow phase of kAB(2) as a function of CdSO4 concentration for native (●), and mutant DN(M17) (■) and DN(L210) (○) RCs. In native RCs, kAB(2) is independent of CdSO4 concentration above 10 μM. In mutant RCs, kAB(2) displays a sigmoidal dependence on CdSO4 concentration. The solid curve represents a theoretical fit using the kinetic model described by Eq. 5 (see also Eq. A4 of Appendix) with the parameters shown in Table 2. The drastic difference in behavior between native and mutant RCs is because of the lack of competition of koff with kAB(2)(Cd2+) in native RCs (see Discussion). Conditions: same as in Fig. 1.
Figure 3.

The absorbance decay of the semiquinones at 450 nm as a function of time in native and the mutant DN(M17) and DN(L210) RCs in the presence of 1 mM CdSO4. This represents the rate of proton transfer in RCs with a bound Cd2+ metal ion. Note that kAB(2) is decreased ≈10-fold in the mutant RCs compared with native RCs. The observed decay rate for the two mutant RCs is the same within experimental error. Conditions: same as in Fig. 1.
Discussion
In this study, we investigated the pathway for proton transfer from solution to the bound semiquinone QB⨪ in isolated RCs from Rb. sphaeroides by measuring the proton-coupled electron transfer rate kAB(2) [QA⨪QB⨪ + H+ → QA(QBH)−] in the absence and presence of Cd2+. We searched for mutant RCs in which kAB(2) in the presence of Cd2+, where proton transfer is the rate-limiting step (24), was decreased significantly (≳10-fold). This occurred in two mutant RCs, DN(L210) and DN(M17). The effect of these mutations on the conformationally gated step of the first electron transfer, kAB(1) (Eq. 1), on the protonation step of the proton-coupled second electron transfer, kAB(2) (Eqs. 2 and 4), and the implications of these results for the identification of the proton transfer pathways are discussed below.
Effect of the DN(L210) and DN(M17) Mutations and of Cd2+ on the Conformationally Gated Step of kAB(1).
The rate of the first electron transfer reaction kAB(1) (Eq. 1) was reduced ≈8-fold in the DN(L210) and DN(M17) mutant RCs (Table 1) compared with the native rate. Upon addition of Cd2+, kAB(1) was decreased ≈10-fold in native RCs (20, 21), ≈3-fold in the DN(M17) RCs, and ≈6-fold in the DN(L210) RCs compared with their respective rate in the absence of Cd2+ (Table 1). Thus, mutant RCs with bound Cd2+ have a smaller kAB(1) than their native counterpart.
The reaction mechanism of kAB(1) in isolated RCs involves a slow rate-limiting gating step, which involves the movement of QB (20) before electron transfer (31, 32). Thus, the decreased rate observed in the mutant RCs and upon binding Cd2+ implies a slowing of the conformationally gated step (23, 24). The effects of the mutation and the binding of Cd2+ produce similar effects on kAB(1). Because the mutations and the binding of Cd2+ both result in a local charge change, the decrease in kAB(1) is attributed to a change in the electrostatic environment (more positive) near the mutation and Cd2+ binding sites. The further reduction in kAB(1), produced by the binding of Cd2+ to the mutant RCs, shows that the effects of the two changes in the electrostatic potential are partially additive.
A possible candidate for the involvement in the gating process is Glu-H173. It is located near the mutation sites (≈4–5 Å) and becomes more disordered when QB is reduced, indicating its sensitivity to nearby charges (20). In the presence of Cd2+ (or Zn2+) it becomes well resolved in the crystal structure (25), indicating a reduction in its dynamic mobility, which may account for the reduction in kAB(1). Thus, the mobility of Glu-H173 may be an important factor in determining the rate of the conformationally gated step, by affecting, for instance, the displacement of water molecules (19–21) or proton uptake and/or redistribution (33–38).
Effect of the DN(L210) and DN(M17) Mutations on kAB(2) in the Absence of Cd2+.
In the absence of Cd2+, kAB(2) (Eq. 2) decreased only ≈2-fold in both mutant RCs compared with native RCs (see Table 1). The pH dependence of kAB(2) in the mutant RCs displayed a small pH shift of ≤0.5 pH unit toward lower pH values. The mechanism of kAB(2), determined from the dependence of kAB(2) on the driving force for electron transfer (3), was found to be the same as in native RCs—i.e., electron transfer was the rate-limiting step. The similar values and behavior of kAB(2) in the native and mutant RCs shows that (i) neither mutation introduces significant changes that affect the redox potential or pKa of QBH⋅ and (ii) electron transfer remains the rate-limiting step in the mutant RCs. Consequently, because kAB(2) is a measure of the electron transfer rate, we cannot assess the effect of the mutation on proton transfer. Indeed, as we shall show in the next section, proton transfer in the mutant RCs in the presence of Cd2+ is reduced ≈10-fold (see Table 2) and is expected to be similarly slowed in the absence of Cd2+.
Effect of Cd2+ on kAB(2) in Native and Mutant RCs.
The binding of Cd2+ affects kAB(2) in native and mutant RCs in drastically different ways. Fig. 2 shows the slow component of kAB(2), which monitors the decay of the semiquinone state with a bound metal ion, (QA⨪QB⨪)- - -Cd2+. In native RCs, equimolar concentrations of Cd2+ decrease kAB(2) ≈20-fold without further changes with increasing Cd2+ concentration. This observation shows that there is a single binding site for Cd2+, in agreement with the x-ray crystal structure (25).
In the mutant RCs, there is a pronounced dependence of kAB(2) on Cd2+ concentration (Figs. 1 and 2). At the concentration of Cd2+ (1 mM) above which no further changes in kAB(2) occur, kAB(2) was decreased ≈10-fold compared with native RCs (Fig. 3).† Because in RCs with a bound Cd2+, kAB(2) is a measure of proton transfer (24), this large decrease of kAB(2) in the mutant RCs points to the important role of Asp-L210 and Asp-M17 in the proton transfer.
To gain further insight into the binding and proton transfer rates, we analyzed the Cd2+ concentration dependence of the slow component of kAB(2) using the kinetic scheme given by Eq. 5.
 |
5 |
where the upper states represent RCs with a bound Cd2+ (left) and unbound Cd2+ (right). The proton-coupled electron transfer rate proceeds with a bound Cd2+ at a rate kAB(2)(Cd2+) (Eq. 4) and without a bound Cd2+ at a rate kAB(2)(0) (Eq. 2). The upper bound and unbound states equilibrate with a dissociation constant Kd = koff/kon.
For the mutant RCs kAB(2)(0) > koff > kAB(2)(Cd2+). Therefore, at low Cd2+ concentrations, (QA⨪QB⨪)- - -Cd2+ decays via the (QA⨪QB⨪) state. Because koff < kAB(2)(0), the rate-limiting step for this decay is koff, which is independent of Cd2+ concentration. It is responsible for the flat region of the slow phase of kAB(2) at low Cd2+ concentration (Fig. 2). Thus, the intercept of the ordinate corresponds to koff. As the Cd2+ concentration is increased, kon[Cd2+] starts to compete with kAB(2)(0), resulting in an increase in the steady-state concentration of the (QA⨪QB⨪)- - -Cd2+ state with a concomitant decrease in the observed rate. This is responsible for the region that depends on Cd2+ concentration. At high Cd2+ concentration (i.e., [Cd2+] ≫ Kd), essentially all RCs have a bound Cd2+ and the observed rate is kAB(2)(Cd2+), which is independent of Cd2+ concentration. It corresponds to the asymptotic value of kAB(2) at high Cd2+ concentration (Fig. 2).
In native RCs, the more negative potential due to the Asp residues increases kAB(2)(Cd2+) and decreases koff with respect to the mutant RCs such that kAB(2)(Cd2+) > koff. Consequently, (QA⨪QB⨪)- - -Cd2+ does not decay via the (QA⨪QB⨪) state and no concentration dependence is observed (Fig. 2).
The qualitative considerations discussed above are borne out by the exact mathematical solution of Eq. 5 presented in the Appendix (Eq. A4). The parameters used to fit the experimental results are summarized in Table 2.
Proton Transfer Pathway to the QB Site.
We have previously shown (25) that the binding of Cd2+ to the surface accessible region on the H subunit (His-H126, His-H128, and Asp-H124) resulted in a ≥102-fold reduction in the rate of proton transfer to QB⨪ (Eq. 4) (24). This result establishes the location of the entry point of the protons. In this work we address the question of the proton transfer pathway connecting the entry point with the QB⨪ site.
The x-ray crystal structure of the RC shows that the two residues Asp-L210 and Asp-M17 are located in the intervening region between the bound Cd2+ and QB⨪ (Fig. 4) (25). These residues are, therefore, logical candidates as members of the chain of residues and water molecules that constitute the proton transfer pathway. To test their involvement in proton transfer, we replaced the protonatable Asp residues with Asn and measured the effect of the mutations on the proton-coupled electron transfer rate, kAB(2).
Figure 4.

Part of the RC structure with a bound Cd2+ showing the region between the bound metal ion and QB⨪ [modified from Axelrod et al. (25)]. The metal (large sphere) is coordinated (solid lines) to Asp-H124, His-H126, His-H128, and three water molecules. Located in this region are Asp-L210, Asp-M17, Asp-L213, Ser-L223 (as indicated), and several water molecules (small spheres). The kinetic results presented in this work show that Asp-L210 and Asp-M17 are involved in proton transfer to QB⨪ in the presence of a bound Cd2+. The most likely proton transfer pathways connecting the surface near Cd2+ to QB⨪ are indicated by the dashed lines. In the absence of a bound Cd2+, the same pathways are expected to predominate with an even greater effectiveness.
Of particular interest is the value of kAB(2) in the presence of Cd2+, because in this system (unlike RCs devoid of Cd2+), kAB(2) is a measure of the rate-limiting proton transfer step (24). We found that in both mutants kAB(2)(Cd2+) ≅ 10 s−1, which is close to an order of magnitude smaller than in native RCs (Table 1). The simplest explanation of these results is that the removal of a negative charge in the Asp-M17/Asp-L210 region introduces a barrier for proton transfer,‡ rendering the direct pathway between the Cd2+ binding region and QB⨪ ineffective, thereby allowing an alternate pathway to take over.§ The maximum rate of this alternate pathway, which does not involve Asp-L210 or Asp-M17, such as P1 (11) or P2 (20) (see figure 1 of ref. 21 or figure 4 of ref. 24), cannot be larger than the observed rate of ≈10 s−1. This rate is at least 103-fold smaller than the physiological transfer rate in native RCs (kH ≥ 104 s−1) (3).
The results presented above address the pathways for the transfer of the first proton to QB⨪ (Eq. 2). Although likely, it has not been established whether these pathways are also dominant for the transfer of the second proton (Eq. 3) that ends up at a spatially different oxygen (O4) of QBH− located near His-L190 (Fig. 4) (13, 14).
The greater rate of proton transfer (≥103-fold) through the pathways near Asp-L210 and Asp-M17 shows that the activation barrier for proton transfer is smaller than for the other possible pathways, such as P1 and P2 (see, e.g., refs. 21 and 24). This smaller activation energy could be a consequence of the relatively short length of the pathway and the high density of carboxylic acid groups (Asp-L210, Asp-M17, and Asp-L213), which electrostatically stabilize the proton in the interior of the protein (see, e.g., ref. 41). The involvement of carboxylic acid groups in proton conduction has also been suggested in several other membrane-bound proteins. These include bacteriorhodopsin (reviewed in refs. 42 and 43), terminal oxidases (reviewed in refs. 44 and 45), lactose permease (reviewed in ref. 46), and the ubiquinol:Cyt c oxidoreductases (47–49). Thus, the involvement of carboxylic acid residues may represent a general strategy used to lower the activation energy to facilitate fast proton conduction through proteins.
Acknowledgments
We thank Michael Graige, Andrea Juth, Herbert Axelrod, and Edward Abresch for technical assistance. This work was supported by the National Science Foundation (Grant MCB94–16652) and the National Institutes of Health (Grants GM 41637 and GM 13191).
Abbreviations
- D
primary donor
- QA
primary quinone electron acceptor
- QB
secondary quinone electron acceptor
- Q
quinone molecule
- Q10
coenzyme Q10 (2,3-dimethoxy-5-methyl-6-decaisoprenyl-1,4-benzoquinone)
- RC
reaction center
- Cyt c
cytochrome c
Appendix
To obtain the kinetic parameters of Eq. 5 we rewrite it in the form:
 |
A1 |
where A = (QA⨪QB⨪), B = (QAQBH−), k1 = kAB(2)(Cd2+) and k2 = kAB(2)(0). The differential equations describing the decay of [A- - -Cd2+] and [A] are obtained from Eq. A1:
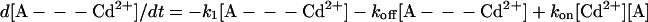 |
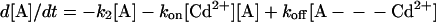 |
A2 |
We are interested in the slow component of the decay of the semiquinones—i.e., the decay of [A- - -Cd2+], after the second laser flash. For this process, the initial conditions at time t = 0 are [A- - -Cd2+(0)] = 1 and [A(0)] = 0 (this state quickly disappears after the second flash because k2 ≫ koff, k1). The solution for the time dependence of [A- - -Cd2+] is given by:
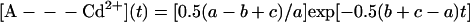 |
A3 |
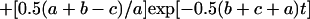 |
where a = {k12 + 2k1koff − 2k1k2 − 2k1kon[Cd2+] + koff2 − 2koffk2 + 2kon[Cd2+]koff + k22 + 2kon[Cd2+]k2 + (kon[Cd2+])2}1/2, b = k1 + koff, and c = k2 + kon[Cd2+]. For our situation, k2 > k1, koff (see Tables 1 and 2). This makes the first term in Eq. A3 the dominant term and the observed rate constant is
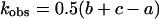 |
A4 |
We fitted the observed Cd2+ concentration dependence for the two mutants with Eq. A4 by leaving k1, k2, koff, and kon as free parameters subject to the constraint Kd = koff/kon, where Kd is the measured dissociation constant (Table 2). The values of the intrinsic rate constants are summarized in Table 2.
Footnotes
At 1 mM Cd2+, kAB(2) was decreased in several other mutant RCs (e.g., Glu-H173 → Gln, Asn-M44 → Asp) (data not shown). However, in contrast to the DN(L210) and DN(M17) mutant RCs, the observed decrease was no greater than the change in other kinetic rates, indicating possible changes in the protein interaction with QB⨪. Thus, the observed change in kAB(2) in these mutant RCs might not be caused by alteration of the proton transfer pathway.
We exclude the possibility that the effects are due to a change in the properties of the proton acceptor QB⨪, which could also reduce the rate of proton transfer (39, 40). This conclusion is based on the similarity of the charge recombination rates kAD, kBD, and kAB(2) in the absence of Cd2+ between the native and mutant RCs (Table 1).
An alternate explanation can, at present, not be excluded. A mutation at one Asp eliminates the pathway involving the mutated residue, but allows proton transfer through the pathway involving the second (unchanged) Asp (Fig. 4), albeit at a diminished rate. The fact that both mutations result in the same transfer rates makes this explanation unlikely. This point can be settled by constructing a double mutant in which both Asp-L210 and Asp-M17 are changed to Asn.
References
- 1.Feher G, Allen J P, Okamura M Y, Rees D C. Nature (London) 1989;339:111–116. [Google Scholar]
- 2.Blankenship R E, Madigan M T, Bauer C E, editors. Anoxygenic Photosynthetic Bacteria. Dordrecht, the Netherlands: Kluwer; 1995. [Google Scholar]
- 3.Graige M S, Paddock M L, Bruce J M, Feher G, Okamura M Y. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- 4.Crofts A R, Wraight C A. Biochim Biophys Acta. 1983;726:149–185. [Google Scholar]
- 5.McPherson P H, Okamura M Y, Feher G. Biochim Biophys Acta. 1990;1016:289–292. doi: 10.1016/0005-2728(93)90116-w. [DOI] [PubMed] [Google Scholar]
- 6.Gunner M. Curr Topics Bioenerg. 1991;16:319–367. [Google Scholar]
- 7.Paddock M L, Rongey S H, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1989;86:6602–6606. doi: 10.1073/pnas.86.17.6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi E, Wraight C A. Biochim Biophys Acta. 1990;1020:107–111. [Google Scholar]
- 9.Takahashi E, Wraight C A. Biochemistry. 1992;31:855–866. doi: 10.1021/bi00118a031. [DOI] [PubMed] [Google Scholar]
- 10.Paddock M L, Rongey S H, McPherson P H, Juth A, Feher G, Okamura M Y. Biochemistry. 1994;33:734–745. doi: 10.1021/bi00169a015. [DOI] [PubMed] [Google Scholar]
- 11.Baciou L, Michel H. Biochemistry. 1995;34:7967–7972. doi: 10.1021/bi00025a001. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi E, Wraight C A. Proc Natl Acad Sci USA. 1996;93:2640–2645. doi: 10.1073/pnas.93.7.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi E, Wraight C A. In: Advances in Molecular and Cell Biology. Barber J, editor. Greenwich, CT: JAI Press; 1994. pp. 197–251. [Google Scholar]
- 14.Okamura M Y, Feher G. In: Anoxygenic Photosynthetic Bacteria. Blankenship R E, Madigan M T, Bauer C E, editors. the Netherlands: Kluwer; 1995. pp. 577–594. [Google Scholar]
- 15.Paddock M L, Feher G, Okamura M Y. Biochemistry. 1997;36:14238–14249. doi: 10.1021/bi971192m. [DOI] [PubMed] [Google Scholar]
- 16.Graige M S, Paddock M L, Feher G, Okamura M Y. Biophys J. 1998;74:A135. [Google Scholar]
- 17.Paddock M L, Senft M E, Graige M S, Rongey S H, Turanchik T, Feher G, Okamura M Y. Photosynth Res. 1998;55:281–291. [Google Scholar]
- 18.Ermler U, Fritzsch G, Buchanan S K, Michel H. Structure. 1994;2:925–936. doi: 10.1016/s0969-2126(94)00094-8. [DOI] [PubMed] [Google Scholar]
- 19.Lancaster C R D, Michel H, Honig B, Gunner M R. Biophys J. 1996;70:2469–2492. doi: 10.1016/S0006-3495(96)79820-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stowell M H B, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 21.Abresch E C, Paddock M L, Stowell M H B, McPhillips T M, Axelrod H L, Soltis S M, Rees D C, Okamura M Y, Feher G. Photosynth Res. 1998;55:119–125. [Google Scholar]
- 22.Fritzsch G, Kampmann L, Kapaun G, Michel H. In: Photosynthesis: Mechanisms and Effects. Garab G, editor. Vol. 2. the Netherlands: Kluwer; 1998. pp. 861–864. [Google Scholar]
- 23.Utschig L M, Ohigashi Y, Thurnauer M C, Tiede D M. Biochemistry. 1998;37:8278–8281. doi: 10.1021/bi980395n. [DOI] [PubMed] [Google Scholar]
- 24.Paddock M L, Graige M S, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1999;96:6183–6188. doi: 10.1073/pnas.96.11.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Axelrod H L, Abresch E C, Paddock M L, Okamura M Y, Feher G. Proc Natl Acad Sci USA. 2000;97:1542–1547. doi: 10.1073/pnas.97.4.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isaacson R A, Lendzian F, Abresch E C, Lubitz W, Feher G. Biophys J. 1995;69:311–322. doi: 10.1016/S0006-3495(95)79936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1984;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 28.Labahn A, Bruce J M, Okamura M Y, Feher G. Chem Phys. 1995;197:355–366. [Google Scholar]
- 29.Vermeglio A, Clayton R K. Biochim Biophys Acta. 1977;461:159–165. doi: 10.1016/0005-2728(77)90078-0. [DOI] [PubMed] [Google Scholar]
- 30.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1985;809:291–310. doi: 10.1016/0005-2728(85)90179-3. [DOI] [PubMed] [Google Scholar]
- 31.Tiede D M, Vázquez J, Córdova J, Marone P A. Biochemistry. 1996;35:10763–10775. doi: 10.1021/bi9605907. [DOI] [PubMed] [Google Scholar]
- 32.Graige M S, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1998;95:11679–11684. doi: 10.1073/pnas.95.20.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nabedryk E, Breton J, Okamura M Y, Paddock M L. Biochemistry. 1998;37:14457–14462. doi: 10.1021/bi981139d. [DOI] [PubMed] [Google Scholar]
- 34.Miksovska J, Kálmán L, Schiffer M, Maróti P, Sebban P, Hanson D K. Biochemistry. 1997;36:12216–12226. doi: 10.1021/bi970442w. [DOI] [PubMed] [Google Scholar]
- 35.McPherson P H, Okamura M Y, Feher G. Biochim Biophys Acta. 1988;934:348–368. doi: 10.1016/0005-2728(93)90116-w. [DOI] [PubMed] [Google Scholar]
- 36.Maróti P, Wraight C A. Biochim Biophys Acta. 1988;934:314–328. [Google Scholar]
- 37.Nabedryk E, Breton J, Hienerwadel R, Fogel C, Mäntele W, Paddock M L, Okamura M Y. Biochemistry. 1995;34:14722–14732. doi: 10.1021/bi00045a013. [DOI] [PubMed] [Google Scholar]
- 38.Sebban P, Maróti P, Hanson D K. Biochimie. 1995;77:677–694. doi: 10.1016/0300-9084(96)88183-1. [DOI] [PubMed] [Google Scholar]
- 39.Marcus R A. J Phys Chem. 1968;72:891–899. [Google Scholar]
- 40.Tu C, Qian M, Earnhardt J N, Laipis P J, Silverman D N. Biophys J. 1998;74:3182–3189. doi: 10.1016/S0006-3495(98)78024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutman M, Nachliel E. Biochim Biophys Acta. 1995;1231:123–138. [Google Scholar]
- 42.Lanyi J K. J Struct Biol. 1998;124:164–178. doi: 10.1006/jsbi.1998.4044. [DOI] [PubMed] [Google Scholar]
- 43.Haupts U, Tittor J, Oesterhelt D. Annu Rev Biophys Biomol Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 44.Brzezinski P, Adelroth P. J Bioenerg Biomembr. 1998;30:99–107. doi: 10.1023/a:1020567729941. [DOI] [PubMed] [Google Scholar]
- 45.Mills D A, Ferguson-Miller S. Biochim Biophys Acta. 1998;1365:46–52. doi: 10.1016/s0005-2728(98)00040-1. [DOI] [PubMed] [Google Scholar]
- 46.Kaback H R, Wu J. Q Rev Biophys. 1997;30:333–364. doi: 10.1017/s0033583597003387. [DOI] [PubMed] [Google Scholar]
- 47.Gennis R B, Barquera B, Hacker B, Van Doren S R, Arnaud S, Crofts A R, Davidson E, Gray K A, Daldal F. J Bioenerg Biomembr. 1993;25:195–209. doi: 10.1007/BF00762582. [DOI] [PubMed] [Google Scholar]
- 48.Zito F, Finazzi G, Joliot P, Wollman F-A. Biochemistry. 1998;37:10395–10403. doi: 10.1021/bi980238o. [DOI] [PubMed] [Google Scholar]
- 49.Crofts A R, Hong S, Ugulava N, Barquera B, Gennis R, Guergova-Kuras M, Berry E A. Proc Natl Acad Sci USA. 1999;96:10021–10026. doi: 10.1073/pnas.96.18.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]