

Abstract
Recent studies have demonstrated key roles for several membrane guanylyl cyclase receptors in the regulation of cell hyperplasia, hypertrophy, migration and extracellular matrix production, all of which having an impact on clinically relevant diseases, including tissue remodeling after injury. Additionally, cell differentiation, and even tumor progression, can be profoundly influenced by one or more of these receptors. Some of these receptors also mediate important communication between the heart and intestine, and the kidney to regulate blood volume and Na+ balance.
Introduction
In rodents, there appear to be seven plasma membrane forms of the guanylyl cyclase (GC) receptor, three of which possess identified ligands (Table 1). Two of the GCs (GCD and GCG) appear to be pseudogenes in the human [1]. GCA, also referred to as natriuretic peptide receptor A (NPR-A) and GCB, also referred to as natriuretic peptide receptor B (NPR-B), have served as prototypes for the other membrane forms. These two receptors contain a module (Figure 1) whose phosphorylation state clearly regulates sensitivity to ligand. Although early work suggested that ATP was a requisite factor for ligand activation of cyclase activity, recent studies have suggested otherwise [2]. This review concentrates on the most recent studies on regulation of the various membrane GCs and the medical and biological implications of such regulation.
Table 1.
Members of the transmembrane GC family
| Ligand | Sites of synthesis and release | Membrane GC receptor | Sites of receptor |
|---|---|---|---|
| ANP BNP | Hearta, brain, testis, other tissues | GCA; NPR-A | Heart, kidney, adrenal gland, vasculature |
| CNP | Endothelial cellsa, brain, bone, epithelium, other tissues | GCB; NPR-B | Adipose, fibroblasts, dentate gyrus, bone, hippocampus |
| STa | Intestinea, kidney, other tissues | GCC | Intestine, kidney, liver |
| Guanylin | |||
| Uroguanylin | |||
| Lymphoguanylin | |||
| Presumed orphan receptor | GCD | Olfactory neurons | |
| Presumed orphan receptor | GCE | Retina, pineal gland | |
| Presumed orphan receptor | GCF | Retina | |
| Presumed orphan receptor | GCG | Lung, intestine, skeletal muscle |
Abbreviation: STa, heat-stable enterotoxin.
Major sites of expression.
Figure 1.

The conserved regulatory phosphorylation module of GCA and GCB. Despite the high similarity between the regulatory regions, GCB can be desensitized specifically by S1P and various growth factors. Abbreviations: PPi, inorganic pyrophosphate; TM, transmembrane segment.
The physiology and medical significance of the GC receptors for natriuretic peptides
Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are both produced predominantly in the heart, and both seem to bind to the same signaling receptor (GCA; NPR-A). A so-called clearance receptor (NPR-C) also binds each of the natriuretic peptides and is known to alter circulating levels of these peptides [3]; however, it remains possible that this receptor also specifically signals in response to ligand binding [4].
GCA (NPR-A) gene disruption
Disruption of the GCA gene first suggested that ANP or BNP might have local effects within the heart [5,6]. Blood pressure was elevated by disruption of the gene, and although this should lead to cardiac hypertrophy, the size of the heart in mice lacking the GCA gene product was found to be considerably larger than that seen in other mouse models exhibiting similar elevations of blood pressure. Subsequently, GCA was introduced back into the cardiac myocytes of otherwise GCA-null mice, and cardiac myocyte hypertrophy was reduced, despite continued elevations of blood pressure [7]. Recent reports from two groups support significant paracrine or autocrine functions of ANP and/or BNP within the heart. In these studies, GCA expression was either interrupted in the mouse through Cre–Lox recombination [8] or inhibited through the over-expression of a dominant negative form of GCA in transgenic animals [9]. In the first study [8], an increase in cardiac hypertrophy and elevation of known hypertrophy-marker genes was evident; this occurred despite a significant drop in blood pressure in the mice, presumably owing to increased production of ANP as a result of eliminating GCA in the heart. In the second study [9], transgenic animals overproducing the dominant negative construct were now susceptible to cardiac fibrosis in response to aortic clamping. Although attenuation of the cardiac myocyte GCA with the dominant negative GCA transgene resulted in modest accentuation of the hypertrophic response to pressure overload, as found in the previous work [8], the novel findings were the dramatic effects on the fibrotic response to pressure overload and increased mortality observed in banded mutant mice. These findings both confirmed and extended the earlier study of Holtwick et al. [8], who examined interstitial fibrosis ten days after transverse aortic banding; in the latter study, hearts were examined at 21 days, thereby possibly accounting for the greater fibrosis. In addition, the high mortality reported in the later study [9] might have been related to the exclusive use of males compared with the use of both male and female mice in earlier studies by Holtwick et al. [8]. As reported by Tamura et al. [10], male mice with BNP gene disruption exhibited greater fibrosis and higher death rates than did female mice. Most importantly, these studies taken together support an important role for the ANP–BNP–GCA signaling pathway in the control of extra-cellular matrix production and the subsequent development of cardiac fibrosis. This is not surprising, given that the most robust increases in ANP and BNP gene expression occur at the time of cardiogenesis in the developing heart [11].
GCA ligands
These studies raise the question of whether ANP, BNP or both are primarily responsible for the cardiac effects in GCA-knockout animals. With disruption of the ANP gene, it was first reported that null animals demonstrated a salt-sensitive hypertension [12]; in later studies, it was concluded that ANP lowered blood pressure in the absence of changes in renal function, and that ANP was not essential for normal salt balance [13]. O’Tierny et al. [14] suggested that because kidney function remained normal in ANP-null mice, the elevation of blood pressure in response to high salt levels was a compensatory mechanism to maintain natriuresis in these animals. The absence of ANP by itself does not appear to be sufficient to cause the dramatic cardiac hypertrophy or the fibrosis seen in GCA-null animals. The BNP gene lies very close to the ANP gene at chromosome locus p36.2 in the human, and genes encoding the peptides guanylin and uroguanylin are also located on the same chromosome. Tamura et al. [10] have extended the studies of ANP gene deletion to include disruption of the BNP gene. These studies provide compelling evidence of the antifibrotic properties of BNP in vivo. Importantly, in the absence of any increase in arterial pressure, the cardiac phenotype of mice lacking the BNP gene was fibrosis. Since the publication of this report in mice, two studies have demonstrated that, in cultured cardiac fibroblasts, BNP might activate selective matrix metalloproteinases that degrade collagen and suppress profibrotic extracellular matrix genes activated by transforming growth factor β (TGF-β ) [15,16]. Thus, BNP appears to possess important antifibrotic properties in addition to those of ANP.
Clinical implications of ANP and BNP
The clinical implications of GCA can be inferred from research on the use of ANP or BNP as either prognosticators or therapeutic agents in the treatment of heart disease [17,18]. Here, we concentrate our discussion on BNP, because concentrations of BNP and aminoterminal (NT)-proBNP in patients with known cardiac diseases are reliable predictors of clinical events such as heart failure and death [19,20] and can be utilized for the detection of ventricular systolic dysfunction in the general population [21]. For hospitalized patients with acute decompensated heart failure, BNP has a better prognostic value than any other serum marker or imaging modality in predicting short-term death and rehospitalization [22]. In recent years, clinical investigators have made similar observations in the general population. Elevated BNP or NT-proBNP levels are closely associated with incremental increases in risk for cardiovascular events such as myocardial infarction, stroke and heart failure [23–25].
Although the use of BNP and NT-proBNP as biomarkers for cardiovascular disease continues to increase, the failing heart can also release altered forms of BNP, together with rapid processing of mature biologically active BNP-32 to a smaller molecular form (BNP-30). It has been reported that high molecular weight BNP is present in human heart failure, whereas others, using state-of-the-art mass spectrometry, have reported the absence of BNP-32 in human plasma in advanced heart failure [26–28]. This latter clinical proteomics report used an immunoaffinity purification assay to isolate BNP-32 from New York Heart Association class IV patients with high levels of immunoreactive BNP for subsequent analysis by nano-liquid chromatography electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS), which provided unprecedented measurement accuracy. These MS studies provide molecularly specific evidence for the absence of circulating BNP-32 in advanced stage heart failure, also suggesting that the existence of altered forms of BNP contributes to increases in plasma BNP by conventional assays [28]. Two current theories are emerging. One is that the absence of BNP-32 might be explained by rapid processing of mature BNP by the enzyme dipeptidyl peptidase IV [29]. The other is that downregulation or saturation of the proBNP processing enzyme corin might result in the release of intact proBNP1–108. Taken together, the presence of altered forms of BNP suggests that one physiological problem of heart failure is the absence of biologically active BNP-32. Presumably, the altered BNP forms have reduced GCA-activating properties or possibly even act as antagonists. This will, no doubt, be an intense area of further investigation.
Although BNP and NT-proBNP are valuable prognosticators, clinicians have discovered some limitations in their ability to improve clinical outcomes. Nesiritide, a recombinant form of human BNP, was anticipated to serve as a modifying therapeutic agent, especially for high-risk patients with severe heart failure, owing to its ability to promote a more favorable hemodynamic profile [30–32]. Since its approval for the treatment of acute decompensated heart failure in 2001, there has been considerable controversy with respect to its efficacy and safety, from a renal perspective. Data from the three randomized, double-blind trials involving 862 patients with acute decompensated heart failure demonstrated a modest but significant improvement of dyspnea during the 3–6 hours of nesiritide infusion relative to placebo. Furthermore, favorable hemodynamic measurements, such as decreased pulmonary capillary wedge pressure and increased cardiac index, were seen with nesiritide infusion compared with placebo and nitroglycerin [33–35]. Although it did not reach statistical significance, owing to the limited number of patients that participated in each trial, there was a trend toward increased short-term mortality in the nesiritide-treated group. According to the recent meta-analysis of these trials, nesiritide therapy was associated with an augmented risk of death at 30 days after treatment [36]. Such observations might indicate that the current recommended doses for clinical uses are inappropriately high, and the means to identify those high-risk patients who would benefit from nesiritide therapy are still lacking. The ongoing European trial involving 1900 patients might soon provide the vital evidence needed to clarify the safety and role of nesiritide in the treatment of acute decompensated heart failure. The concept of efficacy of lower doses of BNP in human heart failure has received support from recent studies in heart failure patients with hypotension and preexisting renal insufficiency [37]. In these most recent studies, use of half the clinical dose of nesiritide (0.005 μ g kg−1 min−1) improved renal function, compared with the clinical dose. One could speculate that doses of nesiritide which decrease blood pressure excessively might do this by reducing renal perfusion pressure, negating the renal-enhancing actions of BNP, and even result in further renal impairment, as was originally reported with ANP in early experimental studies [38].
It has long been an unresolved therapeutic challenge to develop biologically active but orally available peptides. Based upon favorable cardiovascular and renal properties of BNP, an orally active form would have major therapeutic implications beyond severe heart failure. Such a form could be employed for the treatment of hypertension with fibrosis-inhibiting properties and also used early in the natural history of evolving heart failure so as to delay disease progression. Most recently, Cataliotti and co-workers [39] have used proprietary technology relying on short, amphiphilic oligomers covalently attached to BNP. Such advanced conjugation, as used with conjugated BNP, enhances pharmacokinetic and pharmacodynamic profiles, enabling oral administration. These studies reported for the first time that novel conjugated oral BNP in conscious dogs was absorbed and detected in plasma, elevated plasma cGMP concentrations and significantly reduced arterial pressure, thus implying an efficacious coupling of conjugated BNP to GCA. These data advance an innovative concept of orally administered chronic BNP therapy for cardiovascular disease.
Clinical implications of C-type natriuretic peptide
Over the past few years, the possible clinical implications of targeting the C-type natriuretic peptide (CNP)–GCB signaling pathway has also become evident. Inflammatory proliferative responses occur after arterial injury, and the atherosclerosis and restenosis that occurs following angioplasty has been a significant therapeutic challenge [40,41]. The central events in these processes are believed to be migration of smooth muscle cells from the media to the intima, followed by growth factor- and cytokine-induced unregulated proliferation coincident with extracellular matrix formation. CNP is expressed in the vascular endothelium and is a potent inhibitor of smooth muscle [42–44] and fibroblast [45,46] proliferation; it therefore acts as an antagonist to a variety of growth factors. Additionally, CNP production is stimulated in cultured aortic endothelial cells by addition of TGF-β , a polypeptide also expressed at the site of vascular injury [42,43]. In support of the potential importance of CNP, it is known that infusion of the peptide inhibits the intimal proliferation induced in rats after arterial injury [47–49]. Furthermore, GCB is expressed at the site of vascular injury. These results and that of others [50] support the concept that CNP and GCB undergo changes in expression in relation to the progression of an atherosclerotic lesion in humans. The prominence of CNP is particularly evident in endothelial cells and in plaque microvessels and adventitial vasa vasorum, and therefore it is reasonable to speculate a role for CNP–GCB signaling in vascular remodeling and in the progression or regression of plaques. Given the strong adversarial relationship between CNP–GCB signaling and mitogens, such as sphingosine 1-phosphate (S1P), a failure of CNP to inhibit proliferative responses could result in overproliferation and overproduction of extracellular matrix; indeed, in the heart, natriuretic peptides have been shown to inhibit fibrosis [17,18]. The significant effect of CNP–GCB in tissue remodeling in the human liver has also been reported. Here, hepatocytes were shown to be a major source of CNP in the human liver, and peptide expression was increased or decreased during chronic liver diseases [51,52]. Thus, during chronic liver injury, CNP could also have a key role in counteracting liver fibrosis and resultant portal hypertension through inhibition of human stellate cell proliferation and inhibition of contractile responses. Our understanding and subsequent intervention in the regulation of the CNP–GCB signaling pathway is therefore likely to result in significant effects on tissue remodeling.
Given that CNP appears to inhibit collagen synthesis by cardiac fibroblasts more potently than does ANP or BNP [53,54], that CNP is a more potent antihypertrophic agent than is ANP or BNP on cultured fibroblasts [54] and that CNP is even produced locally by the failing heart [55], it is reasonable to conclude that CNP might also be a primary regulatory natriuretic peptide locally within the heart. Given that exogenous CNP is much less potent than ANP or BNP in terms of altering blood pressure or causing diuresis or natriuresis, it is possible that the CNP–GCB signaling pathway could prove to be more selective, in terms of altering tissue remodeling, than the GCA or soluble GC signaling pathways.
Furthermore, recent research has shown in both cultured smooth muscle and fibroblast cells that serum (Figure 2) and S1P at low concentrations (not shown) specifically desensitize GCB as opposed to GCA or the nitric oxide-sensitive forms of GC [46] [also shown by Chrisman et al. (unpublished)]. Some of this work is summarized in Figure 2. Although we show only data using NIH–3T3 cells overproducing GCA or GCB, the same results are obtained using primary cultures of fibroblasts from mouse or human, or primary cultures of mouse smooth muscle cells. Given that the regulatory phosphorylation modules in GCA and GCB are very similar in primary sequence (Figure 1), this was surprising and suggests compartmentalization of the GCA and GCB regulatory complexes, at least within some cells. In previous studies, Chrisman et al. [45] demonstrated that serum contained a factor(s) capable of causing desensitization of GCB. The factor was purified and shown to be S1P [46]. The work of Abbey-Hosch et al. [56] has also shown that S1P can induce desensitization of GCB.
Figure 2.
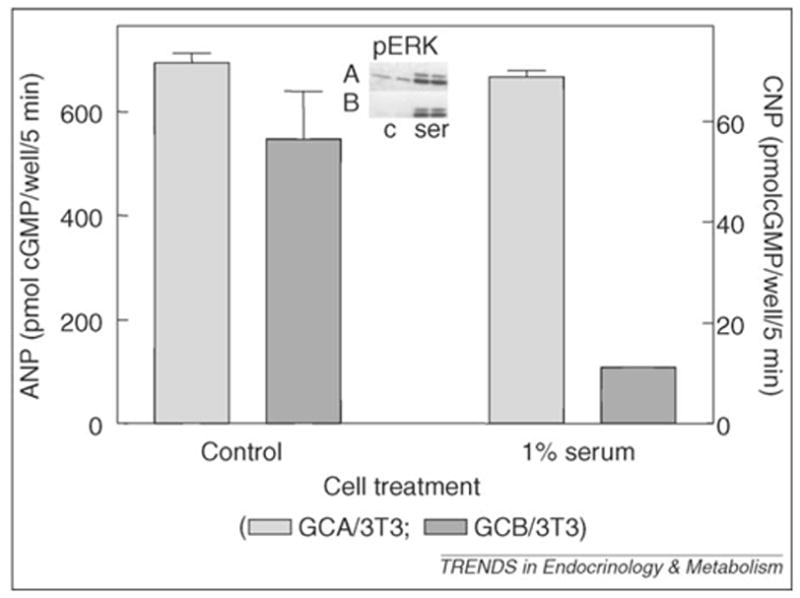
Selective desensitization of GCB by serum. NIH–3T3 cell lines over-expressing rat GCA (GCA/3T3) or rat GCB (GCB/3T3) were serum starved for 24 -hours and then incubated in the absence (control) or presence of 1% fetal bovine serum (serum) for 30 minutes, followed by the addition of 20 nM ANP (GCA/3T3) or 20 nM CNP (GCB/3T3) for 5 minutes. Cyclic GMP (cGMP) contents were determined by radioimmunoassay. Inset: pERK in companion wells (A, GCA/3T3; B, GCB/3T3) were analyzed by western blotting after 5 minutes in the absence (c) or presence of 1% fetal bovine serum (ser). The results show that GCB is specifically desensitized to ligand stimulation by the addition of serum.
S1P is bound principally to high-density lipoprotein (HDL) in serum, where its concentrations (if all considered unbound to protein) range between 500–1000 pM. Highly purified preparations of HDL are also effective in causing desensitization of GCB. The significance of S1P being carried principally by HDL remains unclear. Some reports have suggested that S1P promotes the positive effects of HDL, and yet S1P clearly promotes smooth muscle hyperplasia, and thus it remains unclear whether S1P is pro- or anti-atherogenic.
S1P signaling pathways
A second area of intrigue with respect to S1P-induced desensitization of GCB is which of the S1P receptors are mediating these effects, and also whether this signaling pathway is unique. There are five putative S1P receptors, all apparently coupled to G proteins (Figure 3). Importantly, the desensitization of GCB caused by either serum or S1P is pertussis toxin insensitive [45,46,56]. The most widely distributed receptors, S1PR1–S1PR3, have been suggested to couple to pertussis toxin-sensitive Gi or Go, or insensitive Gq and G12 or G13 [57–61]. Adult and mouse embryonic fibroblasts (MEFs) have been important models for defining the role(s) of the S1P receptors. MEFs express the lysophosphatidic acid receptors LPA1 and LPA2, and also the three S1P receptors S1P1, S1P2 and S1P3 [60]. Presumably, therefore, one or more of these receptors mediate the specific desensitization of GCB.
Figure 3.
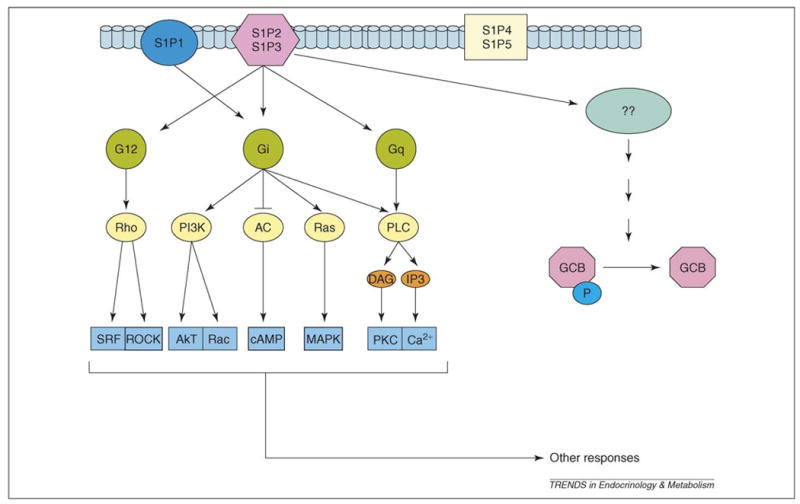
A model for the specific desensitization of GCB by S1P. The five putative receptors for S1P include three (S1P1, S1P2, S1P3) that are expressed in fibroblasts. The desensitization of GCB by S1P is insensitive to pertussis toxin, apparently eliminating Giα as a component of the desensitization pathway. Given that S1P1 has been suggested to signal through Gi, this receptor would then be eliminated as an element within the GCB desensitization pathway. This suggests that either the S1P2 or S1P3 receptor, or both, mediate S1P-specific desensitization of GCB. Abbreviations: AC, adenylyl cyclase; DAG, diacylglycerol; IP3, inositol trisphosphate; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; PKC, protein kinase C; PLC, phospholipase C; SRF, serum response factor.
In terms of mouse genetic models, S1P1-null mice show embryonic hemorrhage, resulting in intrauterine death between E12.5 and E14.5 [57]. Recruitment of vascular smooth muscle cells to blood vessel walls is severely impaired in these animals. S1P2 and S1P3 receptors have also been implicated in processes relevant to vascular maturation and angiogenesis, such as cell migration and the formation of adherence junctions. S1P2-null mice have been generated independently by two groups [59,60]. S1P2-null mouse pups are born with no obvious anatomical or physiological defects at birth, but show spontaneous and sporadic seizures between 3–7 weeks of age [59]. S1P3-null mice seem to be healthy at birth.
Membrane GC receptors and bone development
Gene disruption of the GC receptors GCA, GCC or GCE does not appear to influence bone development; however, disruption of the GCB (NPR2) gene results in the same general bone phenotype as that seen with disruption of the CNP gene [62,63]. In humans with acromesomelic dysplasia (type Maroteaux), which results in significant decreases in stature, mutations in the NPR2 gene have been reported to account for the phenotype; likewise, the cn/cn dwarf mouse appears to result from mutations in the GCB gene [64,65]. More recently, humans heterozygous for the GCB gene mutation were also shown to be shorter, on average, in stature [66]. CNP or GCB mRNA expression is evident in the columnar proliferating and prehypertrophic chondrocyte layers of the murine growth plate, suggesting that signaling is of a paracrine or autocrine nature. It has been concluded as a result of these various studies that CNP–GCB signaling is essential for chondrocyte differentiation.
Membrane GC receptors and the intestine
The GCC receptor is predominately expressed in the intestine of most animals but is also found in the kidney and other organs of the body. Likewise, two peptides (guanylin and uroguanylin) that bind to and stimulate the receptor are produced predominantly in the intestine, although forms of both peptides can be detected in the blood. Several studies have concentrated on the relationship between the GCC receptor and colon cancer [67–72]. Stimulation of GCC promotes colon cancer cell cytostasis, thereby acting as a tumor suppressor. It has also been shown that calcium entry through store-operated calcium channels promotes colon cancer cell proliferation, and it has been suggested that a combination of GCC agonists and store-operated calcium inhibitors would offer a novel means for therapy and prevention of colorectal tumors. However, mice lacking GCC in the multiple intestinal neoplasia mouse model actually demonstrated a decrease in polyp number, with no change in polyp size. The authors suggested that an increase in apoptosis represented a compensatory mechanism for the loss of GCC [73]. In summary, the most notable effects of GCC seem to be consistent with the general model, which suggests that elevations of cGMP normally block cell proliferation. Given that the intestine is remarkably proliferative, the regulation of cell production is crucial, particularly under conditions where food is scarce. Therefore, it is reasonable to suggest that the effects on tumor cytostasis reflect a natural role for GCC in the regulation of intestinal cell proliferation. One might then expect starvation to lead to activation of the receptor and subsequent inhibition of intestinal mucosal cell proliferation. Further support for such a model is the disruption of the gene for guanylin; here, null animals showed an expansion of the crypt compartment of the colon [74].
Box 1. Some questions for the future
Is there a unifying model for regulation of the various GCs at the molecular level?
Is cGMP the only means by which the cyclases signal?
Have we identified the most important targets for cGMP?
What is the explanation for a differential regulation of desensitization of GCA and GCB in tissue remodeling?
What are the mechanisms by which the cyclase receptors appear to protect the heart from hypertrophy?
What are the mechanisms by which the cyclase receptors appear to protect the heart from fibrosis?
Mechanistically, what is the explanation of the positive effects of low-dose administration of BNP in the face of high apparent endogenous concentrations of BNP during heart failure?
Will development of oral BNP markedly expand the therapeutic utility of the natriuretic peptide class for treatment of cardiovascular disease?
Will targeting GCB significantly alter bone development?
Will targeting GCC significantly change the progression of certain intestinal cancers?
Although GCC is stimulated by pathogenic bacteria that produce heat-stable enterotoxins, resulting in an acute secretory diarrhea [75,76], and thus suggesting a role for the receptor in intestinal fluid secretion, mice null for the GCC gene do not demonstrate adverse intestinal effects when on normal diets [77]. However, from genetic models and other studies, it seems that uroguanylin provides a means for the intestine to communicate with the kidney to regulate sodium balance [78–81]. Disruption of the gene encoding uroguanylin, for example, leads to elevations in blood pressure and decreased kidney responses to an intestinal sodium load [82].
Conclusions
Some of the important, as yet unanswered, questions that relate to the GC receptors and their cognate ligands are presented in Box 1. That two natriuretic peptide receptors with similar phosphorylation-regulatory modules are independently modified with respect to activity within the same cell is an important observation to explain, and one that will probably have translational implications in medicine. Additionally, the regulation of cell proliferation, extracellular matrix production and cardiac hypertrophy raises the question of whether tissue remodeling in general is crucially regulated by the GC receptors. From a clinical perspective, a significant advance in the application of natriuretic peptides to clinical cardiovascular medicine has taken place over the past several years. Novel therapies employing intravenous BNP, and now in experimental laboratory-based studies with oral BNP, are leading to efficacious strategies that should reduce the burden of human disease. These advances in the cardiovascular clinical setting might soon be mirrored within the intestine and bone.
Acknowledgments
The authors acknowledge the support of grants from the National Institutes of Health (HL36634 (J.C.B.), HL63282 (M.M.R.), HL776611 (J.C.B.) and DK73120 (D.L.G.)), and support from the Howard Hughes Medical Institute and the Cecil H. and Ida Green Center for Reproductive Biology Sciences.
References
- 1.Potter LR. Domain analysis of human transmembrane guanylyl cyclase receptors: implications for regulation. Front Biosci. 2005;10:1205–1220. doi: 10.2741/1613. [DOI] [PubMed] [Google Scholar]
- 2.Antos LK, et al. ATP-independent activation of natriuretic peptide receptors. J Biol Chem. 2005;280:26928–26932. doi: 10.1074/jbc.M505648200. [DOI] [PubMed] [Google Scholar]
- 3.Sarzani R, et al. Natriuretic peptide clearance receptor alleles and susceptibility to abdominal adiposity. Obes Res. 2004;12:351–356. doi: 10.1038/oby.2004.44. [DOI] [PubMed] [Google Scholar]
- 4.Murthy KS, et al. G(i-1)/G(i-2)-dependent signaling by single-transmembrane natriuretic peptide clearance receptor. Am J Physiol Gastrointest Liver Physiol. 2000;278:G974–G980. doi: 10.1152/ajpgi.2000.278.6.G974. [DOI] [PubMed] [Google Scholar]
- 5.Lopez MJ, et al. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995;378:65–68. doi: 10.1038/378065a0. [DOI] [PubMed] [Google Scholar]
- 6.Oliver PM, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishimoto I, et al. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci U S A. 2001;98:2703–2706. doi: 10.1073/pnas.051625598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtwick R, et al. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003;111:1399–1407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel JB, et al. Cardiac-specific attenuation of natriuretic peptide A receptor activity accentuates adverse cardiac remodeling and mortality in response to pressure overload. Am J Physiol Heart Circ Physiol. 2005;289:H777–H784. doi: 10.1152/ajpheart.00117.2005. [DOI] [PubMed] [Google Scholar]
- 10.Tamura N, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A. 2000;97:4239–4244. doi: 10.1073/pnas.070371497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cameron V, Ellmers L. Minireview: Natriuretic peptides during development of the fetal heart and circulation. Endocrinology. 2003;144:2192–2194. doi: 10.1210/en.2003-0127. [DOI] [PubMed] [Google Scholar]
- 12.John SW, et al. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679–681. doi: 10.1126/science.7839143. [DOI] [PubMed] [Google Scholar]
- 13.Melo LG, et al. Chronic hypertension in ANP knockout mice: contribution of peripheral resistance. Regul Pept. 1999;79:109–115. doi: 10.1016/s0167-0115(98)00149-9. [DOI] [PubMed] [Google Scholar]
- 14.O’Tierny PF, et al. A potential role for the endothelin ETA receptor in salt-sensitive hypertension of the proANP gene-disrupted mouse. Mol Cell Biochem. 2005;275:57–66. doi: 10.1007/s11010-005-7716-3. [DOI] [PubMed] [Google Scholar]
- 15.Tsuruda T, et al. Brain natriuretic peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ Res. 2002;91:1127–1134. doi: 10.1161/01.res.0000046234.73401.70. [DOI] [PubMed] [Google Scholar]
- 16.Kapoun AM, et al. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res. 2004;94:453–461. doi: 10.1161/01.RES.0000117070.86556.9F. [DOI] [PubMed] [Google Scholar]
- 17.Gardner DG. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metab. 2003;14:411–416. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- 18.Munagala VK, et al. The natriuretic peptides in cardiovascular medicine. Curr Probl Cardiol. 2004;29:707–769. doi: 10.1016/j.cpcardiol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 19.de Lemos JA, et al. The prognostic value of B-type natriuretic peptide in patients with acute coronary syndromes. N Engl J Med. 2001;345:1014–1021. doi: 10.1056/NEJMoa011053. [DOI] [PubMed] [Google Scholar]
- 20.Maisel AS, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med. 2002;347:161–167. doi: 10.1056/NEJMoa020233. [DOI] [PubMed] [Google Scholar]
- 21.Bergler-Klein J, et al. Natriuretic peptides predict symptom-free survival and postoperative outcome in severe aortic stenosis. Circulation. 2004;109:2302–2308. doi: 10.1161/01.CIR.0000126825.50903.18. [DOI] [PubMed] [Google Scholar]
- 22.Logeart D, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J AmColl Cardiol. 2004;43:635–641. doi: 10.1016/j.jacc.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 23.Costello-Boerrigter LC, et al. Amino-terminal proB-type natriuretic peptide and B-type natriuretic peptide in the general population. J Am Coll Cardiol. 2006;47:345–353. doi: 10.1016/j.jacc.2005.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galasko GI, et al. What is the normal range for N-terminal probrain natriuretic peptide? How well does this normal range screen for cardiovascular disease? Eur Heart J. 2005;26:2269–2276. doi: 10.1093/eurheartj/ehi410. [DOI] [PubMed] [Google Scholar]
- 25.Kistorp C, et al. N-terminal probrain natriuretic peptide, C-reactive protein, and urinary albumin levels as predictors of mortality and cardiovascular events in older adults. JAMA. 2005;293:1609–1616. doi: 10.1001/jama.293.13.1609. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu H, et al. Molecular forms of human brain natriuretic peptide in plasma. Clin Chim Acta. 2002;316:129–135. doi: 10.1016/s0009-8981(01)00745-8. [DOI] [PubMed] [Google Scholar]
- 27.Yandle T, Richards AM. High molecular weight forms of BNP in human heart failure. J Clin Endocrinol Metab. 1993;76:832–838. doi: 10.1210/jcem.76.4.8473392. [DOI] [PubMed] [Google Scholar]
- 28.Hawkridge AM, et al. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc Natl Acad Sci U S A. 2005;102:17442–17447. doi: 10.1073/pnas.0508782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brandt I, Labmeir A. Dipeptidyl-peptidase IV converts intact B-type natriuretic peptide into its des-SerPro form. Clin Chem. 2006;52:82–87. doi: 10.1373/clinchem.2005.057638. [DOI] [PubMed] [Google Scholar]
- 30.Protter AA, et al. Relaxant effect of human brain natriuretic peptide on human artery and vein tissue. Am J Hypertens. 1996;9:432–436. doi: 10.1016/0895-7061(95)00435-1. [DOI] [PubMed] [Google Scholar]
- 31.Marcus LS, et al. Hemodynamic and renal excretory effects of human brain natriuretic peptide infusion in patients with congestive heart failure. A double-blind, placebo-controlled, randomized crossover trial. Circulation. 1996;94:3184–3189. doi: 10.1161/01.cir.94.12.3184. [DOI] [PubMed] [Google Scholar]
- 32.Boerrigter G, Burnett JC., Jr Recent advances in natriuretic peptides in congestive heart failure. Expert Opin Investig Drugs. 2004;13:643–652. doi: 10.1517/13543784.13.6.643. [DOI] [PubMed] [Google Scholar]
- 33.Colucci WS, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343:246–253. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- 34.Silver MA, et al. BNP Consensus Panel 2004: A clinical approach for the diagnostic, prognostic, screening, treatment monitoring, and therapeutic roles of natriuretic peptides in cardiovascular diseases. Congest Heart Fail. 2004;10 (5 Suppl 3):1–30. doi: 10.1111/j.1527-5299.2004.03271.x. [DOI] [PubMed] [Google Scholar]
- 35.Publication Committee for the VMAC Investigators (Vasodilatation in the Management of Acute CHF) Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540. doi: 10.1001/jama.287.12.1531. [DOI] [PubMed] [Google Scholar]
- 36.Sackner-Bernstein JD, et al. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293:1900–1905. doi: 10.1001/jama.293.15.1900. [DOI] [PubMed] [Google Scholar]
- 37.Riter HG, et al. Nonhypotensive low-dose nesiritide has differential renal effects compared with standard-dose nesiritide in patients with acute decompensated heart failure and renal dysfunction. J Am Coll Caridol. 2006;47:2334–2335. doi: 10.1016/j.jacc.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Burnett JC, et al. The renal action of atrial natriuretic peptide during control of glomerular filtration. Kidney Int. 1986;30:16–19. doi: 10.1038/ki.1986.144. [DOI] [PubMed] [Google Scholar]
- 39.Cataliotti A, et al. Oral human brain natriuretic peptide activates cyclic guanosine 3′,5′-monophosphate and decreases mean arterial pressure. Circulation. 2005;112:836–840. doi: 10.1161/CIRCULATIONAHA.105.538520. [DOI] [PubMed] [Google Scholar]
- 40.Landau C, et al. Percutaneous transluminal coronary angioplasty. N Engl J Med. 1994;330:981–993. doi: 10.1056/NEJM199404073301407. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz SM, et al. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 42.Suga S, et al. Endothelial production of C-type natriuretic peptide and its marked augmentation by transforming growth factor-beta. Possible existence of ‘vascular natriuretic peptide system’. J Clin Invest. 1992;90:1145–1149. doi: 10.1172/JCI115933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suga S, et al. Cytokine-induced C-type natriuretic peptide (CNP) secretion from vascular endothelial cells – evidence for CNP as a novel autocrine/paracrine regulator from endothelial cells. Endocrinology. 1993;133:3038–3041. doi: 10.1210/endo.133.6.8243333. [DOI] [PubMed] [Google Scholar]
- 44.Suga S, et al. Regulation of endothelial production of C-type natriuretic peptide by interaction between endothelial cells and macrophages. Endocrinology. 1998;139:1920–1926. doi: 10.1210/endo.139.4.5918. [DOI] [PubMed] [Google Scholar]
- 45.Chrisman TD, Garbers DL. Reciprocal antagonism coordinates C-type natriuretic peptide and mitogen-signaling pathways in fibroblasts. J Biol Chem. 1999;274:4293–4299. doi: 10.1074/jbc.274.7.4293. [DOI] [PubMed] [Google Scholar]
- 46.Chrisman TD, et al. Identification of a potent serum factor that causes desensitization of the receptor for C-type natriuretic peptide. Cell Commun Signal. 2003;1:4. doi: 10.1186/1478-811X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueno H, et al. Local expression of C-type natriuretic peptide markedly suppresses neointimal formation in rat injured arteries through an autocrine/paracrine loop. Circulation. 1997;96:2272–2279. doi: 10.1161/01.cir.96.7.2272. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi H, et al. Potentiation of C-type natriuretic peptide with ultrasound and microbubbles to prevent neointimal formation after vascular injury in rats. Cardiovasc Res. 2003;58:231–238. doi: 10.1016/s0008-6363(02)00833-7. [DOI] [PubMed] [Google Scholar]
- 49.Brown J, et al. An autocrine system for C-type natriuretic peptide within rat carotid neointima during arterial repair. Am J Physiol. 1997;272:H2919–H2931. doi: 10.1152/ajpheart.1997.272.6.H2919. [DOI] [PubMed] [Google Scholar]
- 50.Casco VH, et al. Natriuretic peptide system gene expression in human coronary arteries. J Histochem Cytochem. 2002;50:799–809. doi: 10.1177/002215540205000606. [DOI] [PubMed] [Google Scholar]
- 51.Tao J, et al. Biological effects of C-type natriuretic peptide in human myofibroblastic hepatic stellate cells. J Biol Chem. 1999;274:23761–23769. doi: 10.1074/jbc.274.34.23761. [DOI] [PubMed] [Google Scholar]
- 52.Henriksen JH, et al. Increased arterial compliance in cirrhosis is related to decreased arterial C-type natriuretic peptide, but not to atrial natriuretic peptide. Scand J Gastroenterol. 2003;38:559–564. doi: 10.1080/00365520310000393. [DOI] [PubMed] [Google Scholar]
- 53.Horio T, et al. Gene expression, secretion, and autocrine action of C-type natriuretic peptide in cultured adult rat cardiac fibroblasts. Endocrinology. 2003;144:2279–2284. doi: 10.1210/en.2003-0128. [DOI] [PubMed] [Google Scholar]
- 54.Tokudome T, et al. Inhibitory effect of C-type natriuretic peptide (CNP) on cultured cardiac myocyte hypertrophy: interference between CNP and endothelin-1 signaling pathways. Endocrinology. 2004;145:2131–2140. doi: 10.1210/en.2003-1260. [DOI] [PubMed] [Google Scholar]
- 55.Kalra PR, et al. Myocardial production of C-type natriuretic peptide in chronic heart failure. Circulation. 2003;107:571–573. doi: 10.1161/01.cir.0000047280.15244.eb. [DOI] [PubMed] [Google Scholar]
- 56.Abbey-Hosch SE, et al. Sphingosine-1-phosphate inhibits C-type natriuretic peptide activation of guanylyl cyclase B (GC-B/NPR-B) Hypertension. 2004;43:1103–1109. doi: 10.1161/01.HYP.0000124668.80811.d3. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Payne SG, et al. Sphingosine-1-phosphate: dual messenger functions. FEBS Lett. 2002;531:54–57. doi: 10.1016/s0014-5793(02)03480-4. [DOI] [PubMed] [Google Scholar]
- 59.Kono M, et al. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 60.Ishii I, et al. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/Edg-3. J Biol Chem. 2002;277:25152–25159. doi: 10.1074/jbc.M200137200. [DOI] [PubMed] [Google Scholar]
- 61.Contos JJ, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2) Mol Cell Biol. 2002;22:6921–6929. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chusho H, et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A. 2001;98:4016–4021. doi: 10.1073/pnas.071389098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamura N, et al. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc Natl Acad Sci U S A. 2004;101:17300–17305. doi: 10.1073/pnas.0407894101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartels CF, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet. 2004;75:27–34. doi: 10.1086/422013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsuji T, Kunieda T. A loss-of-function mutation in natriuretic peptide receptor 2 (Npr2) gene is responsible for disproportionate dwarfism in cn/cn mouse. J Biol Chem. 2005;280:14288–14292. doi: 10.1074/jbc.C500024200. [DOI] [PubMed] [Google Scholar]
- 66.Olney RC, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J Clin Endocrinol Metab. 2006;91:1229–1232. doi: 10.1210/jc.2005-1949. [DOI] [PubMed] [Google Scholar]
- 67.Carrithers SL, et al. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci U S A. 1996;93:14827–14832. doi: 10.1073/pnas.93.25.14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carrithers SL, et al. Escherichia coli heat-stable enterotoxin receptors. A novel marker for colorectal tumors. Dis Colon Rectum. 1996;39:171–181. doi: 10.1007/BF02068072. [DOI] [PubMed] [Google Scholar]
- 69.Cohen MB, et al. Guanylin mRNA expression in human intestine and colorectal adenocarcinoma. Lab Invest. 1998;78:101–108. [PubMed] [Google Scholar]
- 70.Waldman SA, et al. Heterogeneity of guanylyl cyclase C expressed by human colorectal cancer cell lines in vitro. Cancer Epidemiol Biomarkers Prev. 1998;7:505–514. [PubMed] [Google Scholar]
- 71.Birbe R, et al. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005;36:170–179. doi: 10.1016/j.humpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 72.Kazerounian S, et al. Proliferative signaling bysStore-operated calcium channels opposes colon cancer cell cytostasis induced by bacterial enterotoxins. J Pharmacol Exp Ther. 2005;314:1013–1022. doi: 10.1124/jpet.105.089052. [DOI] [PubMed] [Google Scholar]
- 73.Mann EA, et al. Lack of guanylyl cyclase C, the receptor for Escherichia coli heat-stable enterotoxin, results in reduced polyp formation and increased apoptosis in the multiple intestinal neoplasia (Min) mouse model. Int J Cancer. 2005;116:500–505. doi: 10.1002/ijc.21119. [DOI] [PubMed] [Google Scholar]
- 74.Steinbrecher KA, et al. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am J Pathol. 2002;161:2169–2178. doi: 10.1016/S0002-9440(10)64494-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giannella RA, Mann EA. E. coli heat-stable enterotoxin and guanylyl cyclase C: new functions and unsuspected actions. Trans Am Clin Climatol Assoc. 2003;114:67–85. [PMC free article] [PubMed] [Google Scholar]
- 76.Schulz S, et al. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63:941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 77.Schulz S, et al. Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J Clin Invest. 1997;100:1590–1595. doi: 10.1172/JCI119683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Forte LR., Jr Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther. 2004;104:137–162. doi: 10.1016/j.pharmthera.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 79.Kikuchi M, et al. Role of uroguanylin, a peptide with natriuretic activity, in rats with experimental nephrotic syndrome. J Am Soc Nephrol. 2005;16:392–397. doi: 10.1681/ASN.2004040324. [DOI] [PubMed] [Google Scholar]
- 80.Kuhn M. Cardiac and intestinal natriuretic peptides: insights from genetically modified mice. Peptides. 2005;26:1078–1085. doi: 10.1016/j.peptides.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 81.Sindic A, et al. Guanylin and uroguanylin regulate electrolyte transport in isolated human cortical collecting ducts. Kidney Int. 2005;67:1420–1427. doi: 10.1111/j.1523-1755.2005.00219.x. [DOI] [PubMed] [Google Scholar]
- 82.Lorenz JN, et al. Uroguanylin knockout mice have increased blood pressure and impaired natriuretic response to enteral NaCl load. J Clin Invest. 2003;112:1244–1254. doi: 10.1172/JCI18743. [DOI] [PMC free article] [PubMed] [Google Scholar]