

Abstract
Molecular chaperones of the heat shock protein 70 (Hsp70) family counteract protein misfolding in a variety of neurodegenerative disease models. To determine whether human Hsp70 exerts similar effects on the aggregation of alpha-synuclein (α-Syn), the key component of insoluble fibrils present in Parkinson’s disease, we investigated α-Syn fibril assembly in the presence of Hsp70. We found in vitro assembly was efficiently inhibited by substoichiometric concentrations of purified Hsp70 in the absence of co-factors. Experiments using α-Syn deletion mutants indicated that interactions between the Hsp70 substrate binding domain and the α-Syn core hydrophobic region underlie assembly inhibition. This assembly process was inhibited prior to the elongation stage as we failed to detect any fibrils by electron microscopy. In addition, fluorescence polarization and binding assays suggest that Hsp70 recognizes soluble α-Syn species in a highly dynamic and reversible manner. Together, these results provide novel insights into how Hsp70 suppresses α-Syn aggregation. Furthermore, our findings suggest that this critical step in Parkinson’s disease pathogenesis may be subject to modulation by a common molecular chaperone.
The 140 amino acid protein alpha synuclein (α-Syn) is the principal component of intracellular inclusions characteristic of a family of neurological disorders known as synucleinopathies, which includes Parkinson’s disease (PD), dementia with Lewy bodies, and multiple systems atrophy (for review, see ref. 1). Although highly soluble under physiological conditions, pathological α–Syn forms insoluble amyloid fibrils that aggregate as Lewy bodies, Lewy neurites, and glial cytoplasmic inclusions (2-4). The correlation between α–Syn misfolding and pathogenesis is further strengthened by the observation that amplification or mutation of the α–Syn gene give rise to early onset PD (5-7). Similarly, overexpression in animal models also leads to fibrillar inclusions accompanied by neurotoxicity (8-10).
The highly conserved heat shock protein 70 (Hsp70) family of molecular chaperones interact with misfolded proteins, preventing their subsequent aggregation, and are instrumental in targeting them for degradation when this fails (11). In a number of animal models of protein misfolding, Hsp70 overexpression attenuates toxicity and ameliorates disease pathology (12-14). These findings also extend to models of α–Syn overexpression where elevated Hsp70 levels reduce α–Syn accumulation and toxicity in mice and Drosophila (8;15). Intracellular α–Syn inclusions are intimately associated with chaperones, likely reflecting their integral role in the cellular response to misfolded α–Syn (8;16;17). However, given the pleiotropic functions of Hsp70 in vivo (18) it is unclear whether or not the observed decreases in toxicity result from Hsp70 directly interacting with misfolded α–Syn. For example, although elevating Hsp70 levels increased survival in transgenic α–Syn flies, insoluble aggregates remained relatively unchanged (8). Therefore, Hsp70 may reduce toxicity by facilitating clearance of α–Syn and/or destabilizing toxic (e.g. non-fibrillar) species in cells.
The direct actions of Hsp70 on α–Syn have been explored in two recent in vitro studies demonstrating that Hsp70 can prevent α–Syn fibril assembly (19;20). Inhibition appears to be mediated by the selective binding of Hsp70 to soluble prefibrillar species and fibrils which may neutralize their ability to act as sites for nucleation and elongation, respectively. However, the precise mechanism underlying this selective recognition, including the actual α–Syn domain interacting with Hsp70, remains unclear.
Here, we identify the region of α–Syn recognized by Hsp70-1A, an inducible Hsp70 isoform expressed in multiple tissues including the brain (21-23), using a series of deletion mutants and demonstrate that the core hydrophobic region of α–Syn is responsible for interaction with Hsp70. We also provide additional evidence that, contrary to previous reports, the bulk of α–Syn retains its monomeric form following exposure to Hsp70. Our data suggest that inhibition of fibril assembly occurs as a result of transient and reversible interactions between Hsp70 and soluble α–Syn.
EXPERIMENTAL PROCEDURES
Protein purification
Recombinant full-length and truncated forms of wildtype (WT) human α-Synuclein (α-Syn) were expressed in Escherichia coli as previously described (24) with minor modifications. With the exception of full-length α-Syn and α-Syn21-140, proteins were eluted over a 0.1-1.0 M NaCl gradient from HiTrap SP-HP columns (GE Healthcare) equilibrated with 0.1 M MES (pH 6.5). All α-Syn proteins were dialyzed into 50 mM Tris-HCl, 100 mM NaCl pH 7.0, concentrated, and stored at -80°C in aliquots until used.
Expression plasmids for Hsp70s were kindly provided by Dr. R. Morimoto (Northwestern University). Full-length human Hsp70 (Hsp70-1A) and truncated forms lacking the N-terminal ATPase domain (Hsp70386-640) or extreme C-terminal portion (Hsp701-611) were purified as previously published following expression in BL21(DE3)RIL cells (25). GST-tag was cleaved from Hsp70386-640 with thrombin before use, except for in chaperone depletion experiments where the tag was retained. Protein concentrations were determined using a BCA protein quantification kit (Pierce) with bovine serum albumin (BSA) as a standard.
Fibril assembly reactions
WT or mutant α-Syn was incubated in assembly buffer (50 mM Tris, 50 mM KCl, 5 mM MgCl2, pH 7.0) at a final concentration of 72 μM (~1 mg/ml wildtype protein). Reactions were performed either in low-volume 384-well polycarbonate plates (Nunc) or 200 μL polypropylene microcentrifuge tubes. For chaperone experiments, Hsp70 was added at the final concentrations indicated with an equal concentration of BSA included in control samples. For reactions requiring ATP, 2 mM ATP was added to the assembly buffer along with a regeneration system consisting of pyruvate kinase (20 ug/ml), phosphoenolpyruvate (4 mM), and DTT (1 mM). All plates and tubes were incubated at 37°C with continuous agitation on an orbital shaker at 1000 rpm.
Thioflavin T and K114 readings
At various timepoints, 10 μL aliquots were removed from reactions and added to 100 μL of 20 μM Thioflavin T (ThT; Sigma), a dye recognizing amyloid in 100 mM glycine pH 8.0. Fluorescence was read using a Spectramax M5 plate reader (Molecular Devices) at 490 nm/510 nm (ex/em; cutoff 475 nm). Parallel aliquots were also added to 100 μM (trans,trans)-1-bromo-2,5-bis-(4-hydroxy)styrylbenzene (K114) in 100 mM glycine (pH 8.5) and fluorimetry performed at 380 nm/550 nm (530 nm cutoff) as previously described (26).
Synthetic peptide and assembly
The peptide corresponding to residues 61-95 of wildtype human α-Syn (referred to as non-amyloid component or NAC) was synthesized by solid-phase coupling at the Biotechnology Resource Center at Yale University (New Haven, CT). After dissolving in 35% acetonitrile/0.01% trifluoroacetic acid the peptide was diluted into assembly buffer containing 0.01% Triton-X 100, and freeze-dried. For assembly experiments, lyophilized peptide was first resuspended in ultrapure water, and filtered (0.22 micron) to remove any oligomers or aggregates before further dilution into assembly buffer and addition of BSA or chaperone.
Sedimentation and immunoblot analysis
Reactions were subjected to ultracentrifugation (100,000 × g) for 30 min at 4°C. Both the pellet and supernatant were recovered, resuspended in SDS sample buffer and boiled for 10 min. Samples were separated by SDS-PAGE (15%) and stained with Coomassie Blue. NAC samples were separated on precast 16.5% Tris-tricine gels (Biorad, Hercules, CA). For specific immunodetection of α-Syn, Hsp70, or the NAC peptide, samples were transferred onto nitrocellulose membrane following electrophoresis, blocked with 5% milk in TBS, and incubated with anti-α-Syn mouse monoclonal antibody (Mab) LB509 (1:5,000), anti-Hsp70 Mab 3a3 (1:10,000), and a rabbit polyclonal to the NAC1 domain of α-Syn (1:500), respectively (27-29). Blots were developed and scanned after incubation with the appropriate horseradish peroxidase-conjugated secondary antibody.
Electron microscopy
Assembly reactions were adsorbed onto 300-mesh carbon coated copper grids and stained with 1% uranyl acetate for transmission electron microscopy (EM). Immuno-EM was performed by incubating grids with SNL-1, a rabbit polyclonal antibody to α-Syn (1:100), anti-Hsp70 Mab 3a3 (1:200) or anti-Oregon-Green 488 Mab (Molecular Probes, 1:200). Grids were visualized after incubation with colloidal gold-conjugated (6, 10 or 16 nm) secondary antibodies against mouse or rabbit immunoglobulins (1:20) (Aurion, Wageningen, The Netherlands) and negative staining with 1% uranyl acetate on a JEOL 1010 transmission electron microscope. Images were captured with a Hamamatsu digital camera using the AMT acquisition software.
Fluorescence polarization
Full-length α-Syn was dialyzed into PBS and labeled using the Oregon Green-488 or Alexa fluor-594 labeling kit (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions to achieve a dye to protein ratio of 1:8. Unbound dye was removed by extensive dialysis into assembly buffer. Fluorescence polarization (FP) was performed to monitor α-Syn oligomerization and fibrillization as described (30). In these experiments, fibril assembly reactions were performed as described above with Oregon Green-labeled α-Syn (α-Syn488) added at a final dye to protein ratio of 1:250. Unlabeled α-Syn was adjusted to maintain total α-Syn concentrations constant at 72 μM. Fluorescence in perpendicular and parallel planes at 490/525 nm (ex/em) were recorded (average of 100 reads per sample) throughout the course of reactions using a Spectramax M5 reader. After subtraction of background fluorescence, FP values were calculated and expressed in milli-polarization units (mP) relative to freshly prepared monomeric α–Syn (30).
Chaperone depletion assay
GST-tagged Hsp70386-640 (7.2 μM) was added to assembly reactions containing 72 μM α-Syn. Glutathione sepharose 4B beads (GE Healthcare) was used to deplete Hsp70386-640 after 72 h incubation. Following depletion, reactions were resumed and monitored using ThT fluorescence and sedimentation analysis.
Dual-fluorescence assembly assay
Assembly reactions containing 72 μM α-Syn were spiked with α-Syn594 (1:250 dye to protein ratio) and incubated for 72 h in the presence of either GST-Hsp70386-640 (7.2 μM) or GST alone. An equal volume of fresh monomeric α-Syn containing α-Syn488 was then added and the soluble fraction of each fluorescent protein monitored by quantifying fluorescence intensity following centrifugation at 100,000 × g.
RESULTS
Hsp70 inhibits α-Syn fibril assembly in the absence of co-factors
We first examined the effects of Hsp70 on α-Syn fibril formation by monitoring changes in ThT and K114 fluorescences in assembly reactions. Monomeric α-Syn readily converted to fibrils as indicated by a 100-200 fold increase in ThT (Fig. 1A) and K114 (Fig. 1B) fluorescence in control reactions containing BSA, a protein with no chaperone activity but is similar to Hsp70 in molecular weight. Consistent with nucleation-dependent assembly reactions involving amyloid proteins (31), an increase in ThT and K114 fluorescence was observed in control reactions following an initial lag phase of 25-30 h, in agreement with values previously reported for α-Syn assembly under similar conditions (30;32;33). In contrast to BSA-containing assembly reactions, those containing Hsp70 exhibited baseline levels of ThT and K114 fluorescence during the course of incubation, indicating that α-Syn does not fibrillize in the presence of this chaperone in agreement with recent reports (19;20). Since styryl benzenes and benzothiazoles recognize separate binding sites in amyloids (34), these results are unlikely to arise Hsp70 displacing either dye from formed α-Syn fibrils. This was further confirmed by sedimentation and EM analyses of Hsp70-treated and–untreated samples (see below).
FIG. 1. Hsp70 inhibits α-Syn fibril assembly in vitro.
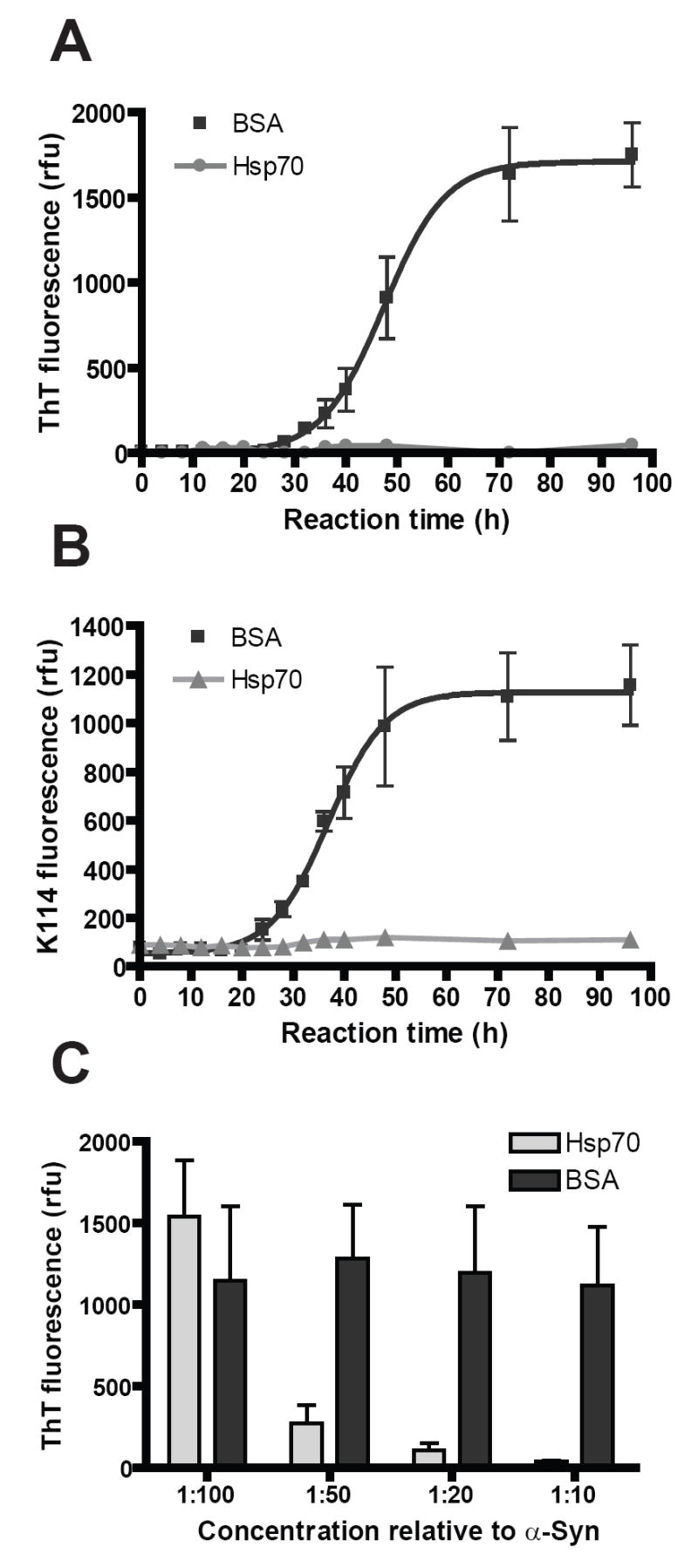
Monomeric recombinant α-Syn (72 μM) in assembly buffer was incubated in 384-well plates (20 μL per reaction) in the presence of either purified Hsp70 or BSA (7.2 μM). Parallel samples (10 μL each) were removed at various time points and α–Syn fibrillization are monitored with ThT (A) or K114 (B) fluorescence. Substoichiometric quantities of Hsp70 (1:10 relative to monomeric α-Syn) prevented increase of α–Syn amyloid fibrils as indicated by these two dyes. C, Reactions containing α-Syn (72 μM) combined with various concentrations of Hsp70 or BSA (0.72-7.2 μM) were measured for ThT fluorescence after 96 h incubation. Inhibition of α–Syn fibril assembly was observed in a concentration-dependent manner with Hsp70 but not BSA. Data are expressed as mean ± s.e.m. (n = 3-4 per concentration, 2 independent experiments).
We determined the minimal relative concentration of Hsp70 required for inhibiting α-Syn assembly by incubating decreasing quantities of Hsp70 (72 nM-7.2 μM) with a constant concentration of α-Syn monomer (72 μM). Complete inhibition was observed at Hsp70 concentrations as low as one-tenth of that of α-Syn (i.e. 7.2 μM). Inhibition efficiency decreased to 91% and 71%, when Hsp70 concentration was added at 1:20 and 1:50, respectively, while no inhibition occurred at 1:100 (Fig. 1C). Neither nucleotides nor co-factors (e.g. Hsp40 co-chaperone) were required for assembly inhibition as reactions were performed in their absence. Similarly, inhibition was comparable to full-length Hsp70 when using the Hsp70 substrate binding domain (Hsp70386-640), which lacks the N-terminal ATP-binding regulatory domain (Supplementary Fig. 1B). These data together confirm and extend published studies that α-Syn fibril inhibition does not involve active ATP-mediated refolding by Hsp70.
Hsp70 inhibits assembly of terminally-truncated α-Syn
To identify the region(s) within α-Syn that interact with Hsp70 to mediate this inhibition, we examined the ability of Hsp70 to inhibit the fibrillization of a number of α-Syn mutants. Previous studies have shown that hydrophobic residues located within the central region of α-Syn are responsible for its propensity to assemble into fibrils (33). It is postulated that assembly of α-Syn monomers is suppressed by transient intramolecular interactions whereby the terminal domains conceal the aggregation-prone region. The acidic C-terminal domain plays a principal role in this process and α-Syn species wherein C-terminal regions are truncated exhibit accelerated fibril formation (33;35-37).
To determine whether Hsp70 suppresses α-Syn fibril formation by maintaining conformations favoring apposition of terminal regions with the hydrophobic domain, we examined the assembly of various α-Syn truncation mutants where this mechanism would be precluded (Fig. 2A). Consistent with previous reports, truncated forms lacking portions of the acidic C-terminal assembled more rapidly at equivalent molar concentrations compared to full-length α-Syn whereas loss of the extreme N-terminal (residues 1-20) did not alter the rate of assembly (33;36;37). Interestingly, addition of Hsp70 resulted in a 96-99% decrease in the ThT fluorescence of all the truncated α-Syn examined (Fig. 2A,B). Sedimentation analysis confirmed that soluble α-Syn predominated in the presence of chaperone (Fig. 2C,D), indicating that Hsp70 suppression of fibril assembly occurs through interactions within the central region (residues 21-110) of α-Syn. Furthermore, the similar efficiencies in inhibition observed at substoichiometric concentrations of chaperone (i.e. 10-fold lower than α–Syn) suggest a common mechanism underlying the inhibition of the various truncated α-Syn proteins.
FIG. 2. Hsp70 inhibits fibril assembly of full-length and truncated α-Syn proteins.
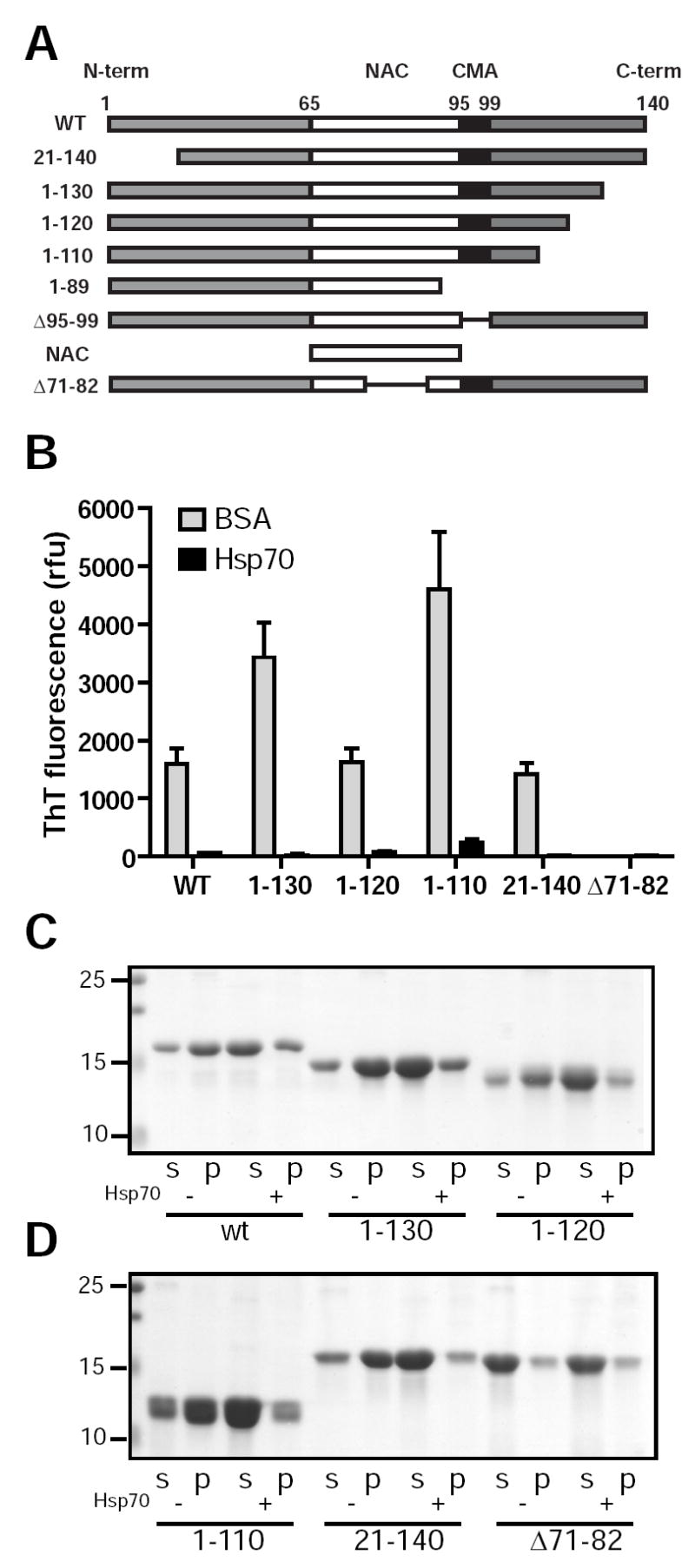
A, this shows the various α–Syn constructs used in this study. Full-length i.e. WT α–Syn possesses a central hydrophobic domain (NAC) adjacent to a putative recognition motif for chaperone-mediated autophagy (CMA). B, purified WT, C-terminal truncated (1-130, 1-120, 1-110), or N-terminal truncated (21-140) α-Syn (72 μM) was incubated in the presence of Hsp70 (7.2 μM) for 96h in 384-well plates. Control reactions contained 7.2 μM BSA in lieu of Hsp70. A mutant α-Syn lacking the NAC region required for fibril assembly (Δ71-82) was included as a non-assembling negative control. ThT readings show Hsp70 suppresses fibril formation of all species tested, suggesting that the extreme C- and N-termini of α-Syn are not required for chaperone-mediated inhibition (data expressed as means ± s.e.m. from two independent experiments, n = 4). C,D, fibril assembly reactions containing various α-Syn (72μM) were performed in microfuge tubes containing Hsp70 (+) or BSA (-). After 48-96h reactions were centrifuged at 100,000 × g and separated by 15% SDS-PAGE. Proteins were revealed by staining with Coomassie Blue (s: supernatant; p: pellet).
Hsp70 interacts with the central hydrophobic region of α-Syn
Apart from the N- and C-termini, two other domains of α-Syn could mediate Hsp70-dependent inhibition of fibrillization. First, the central portion of α-Syn (residues 61-95), also referred as non-amyloid component (NAC; see Fig. 2A) (4) was previously demonstrated to be required for α-Syn fibril formation. Indeed, more detailed analyses demonstrated that residues 71-82 represent a hydrophobic stretch that forms the β-sheet core of α-Syn fibrils and are essential for α-Syn assembly (29). Second, a pentapeptide sequence (95VKKDQ99) has been reported as a consensus motif for substrates of the chaperone-mediated autophagy (CMA) pathway (38). Given the well-described role of another member of the Hsp70 family (Hsc70) in CMA, we asked whether this latter sequence might be targeted by Hsp70 in our in vitro system by examining the ability of Hsp70 to inhibit assembly of α–Syn proteins lacking this motif.
Recombinant α-Syn lacking either the C-terminal 51 residues (i.e. α-Syn1-89) or the pentapeptide CMA-motif (α-SynΔ95-99) readily assembled into amyloid fibrils as confirmed by EM (data not shown). Interestingly, fibril formation of α-SynΔ95-99 was robustly inhibited by Hsp70 (Fig. 3A,B) indicating that this putative CMA-recognition sequence is not necessary for interaction with Hsp70 under these conditions.
FIG. 3. Hsp70 inhibits fibril assembly via the NAC domain of α-Syn.
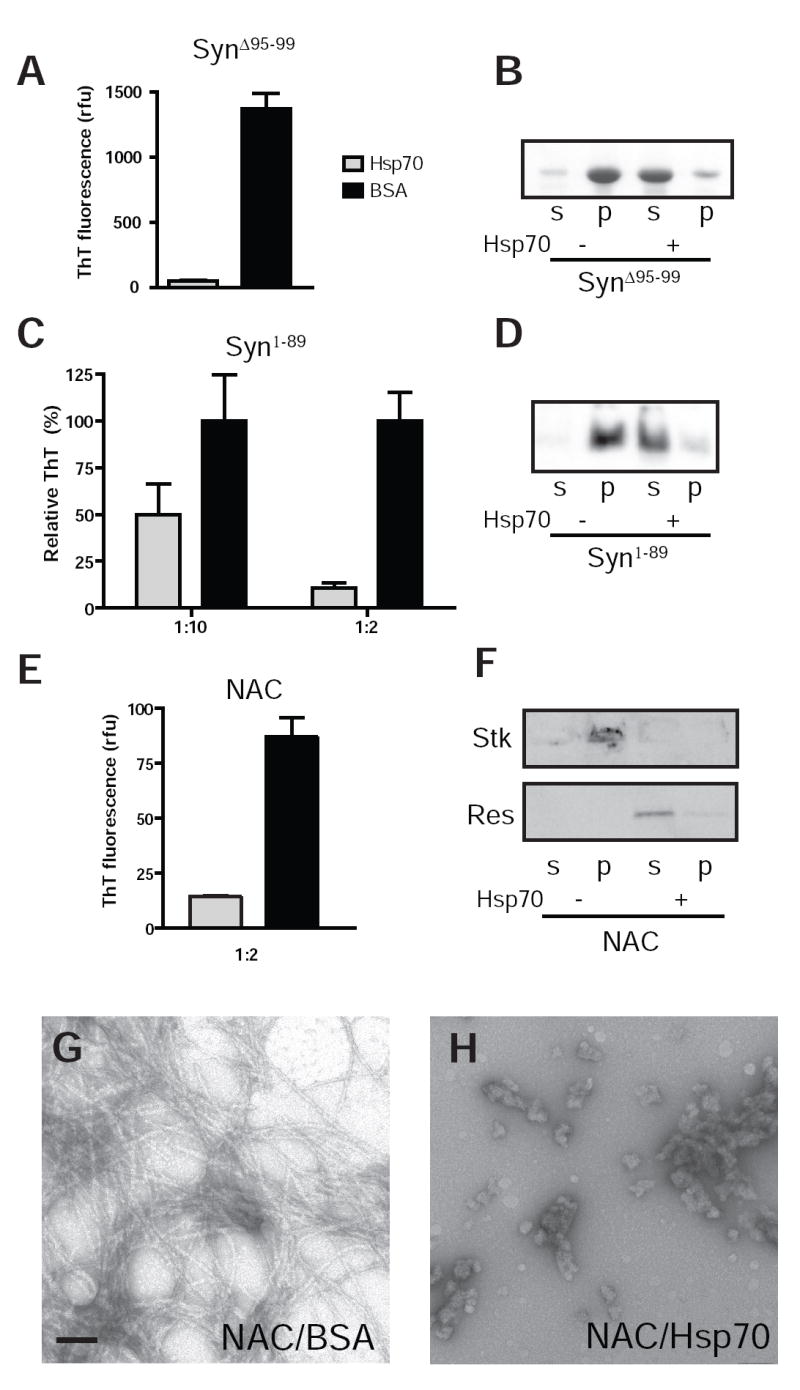
A, ThT measurement of α-SynΔ95-99 assembly reactions (72 μM) in the presence of BSA or Hsp70 after 96 h incubation. B, soluble (s) and pelletable (p) α–Syn visualized by Coomassie Blue following centrifugation showing inhibition of aggregation by Hsp70. C, ThT fluorescence of α-Syn1-89 (72 μM) reactions after 24 h incubation. Chaperone or BSA was added at an initial concentration of 7.2 μM (1:10) or 36 μM (1:2). D, sedimentation of reactions after 24h showing that the majority of α-Syn1-89 is aggregated within this period in the presence of BSA whereas solubility is maintained when Hsp70 is present at high concentrations (1:2). E, ThT measurement of NAC peptide assembly (80 μM) in the presence of either Hsp70 or BSA (1:2). Readings were taken 20 h after start of incubation. Similar to other α-Syn species, addition of Hsp70 resulted in markedly lower ThT fluorescence indicating reduced assembly of amyloid fibrils as confirmed by western blotting with NAC-1 following sedimentation (F) revealing aggregated NAC trapped in the stacker (Stk) in control samples while Hsp70 treated NAC appeared in the resolving gel (Res). EM showing negatively stained NAC fibrils formed in the presence of BSA (G) and amorphous aggregates when Hsp70 is added (H). Scale bars = 100nm.
Addition of Hsp70 at the 1:10 (chaperone: α-Syn) molar ratio used in previous experiments also inhibited assembly of α-Syn1-89 as detected by ThT fluorescence (Fig. 3C), further supporting that α-Syn residues 95-99 are dispensible for interaction with Hsp70. However, Hsp70 inhibition of α-Syn1-89 assembly was noticeably less efficient compared to other α-Syn species as measured by ThT fluorescence (40% inhibition vs. >95% for wildtype and other truncated forms). Increasing Hsp70 concentration to 1:2 relative to α-Syn1-89 restored a strong inhibitory effect corroborated by sedimentation analysis (Fig. 3D), likely reflecting the greater number of hydrophobic binding sites in the NAC domain exposed by α-Syn1-89 in the absence of the regulatory C-terminal domain.
To confirm our hypothesis that direct interactions between Hsp70 and the hydrophobic domain are responsible for inhibition, assembly of a synthetic peptide corresponding to the NAC sequence was examined in the presence of either Hsp70 or BSA. Fibril formation of NAC occurred spontaneously and rapidly in vitro (29;39), reaching completion within 10 h. Significant ThT fluorescence was detectable at 20 h in reactions containing BSA, but was 84% lower in the presence of Hsp70 at a 1:2 relative concentration (Fig. 3E). Sedimentation analysis and EM of 20 h samples showed the majority of Hsp70 treated NAC peptide remained soluble, in contrast to control samples (Fig. 3F-H), demonstrating that Hsp70 inhibition of α-Syn fibril formation indeed occurs at the level of the NAC region.
Visualization of α-Syn assembly inhibition by EM
Previous studies have suggested that Hsp70 recognizes non-fibrillar multimeric α–Syn intermediates, thus accounting for the substoichiometric concentrations required to inhibit α–Syn assembly (19;20). Hsp70 is also reported to recognize and bind fibrillar α–Syn, preventing α–Syn fibrils from acting as elongation nuclei for fibril extension (19). Indeed these scenarios do not appear to be mutually exclusive as Hsp70 can readily inhibit assembly reactions containing α–Syn monomer that are seeded by pre-formed fibrils (Ref. 19 and Supplementary Fig. 2).
In order to further investigate the mechanism of assembly inhibition by Hsp70, we used EM to examine chaperone: α–Syn complexes in fibril assembly reactions. As expected, negatively-stained samples from α–Syn reactions containing BSA revealed an abundance of highly ordered fibrils ranging from 10-14 nm in diameter (Fig. 4A) characteristic of that described for fibrils formed by amyloidgenic proteins in this system (24). In contrast, addition of Hsp70 resulted in numerous structures similar to those previously described as amorphous aggregates (19;20). Larger aggregates appeared to be comprised of smaller, mainly elliptoid units 20-50 nm in diameter, or approximately 3-4 times the axial thickness of fibrils formed in the absence of chaperone (Fig. 4A–D). Surprisingly, structures resembling whole or fragmented fibrils were not detectable when chaperone was present, suggesting that Hsp70 was not associating with and sequestrating small fibrils in these reactions (Fig. 4B,E).
FIG. 4. Characterization of α-Syn in the presence of Hsp70 by EM.

Fibril assembly reactions containing full-length α-Syn (72 μM) were incubated for 96h in the presence of either 7.2 μM BSA (A) or Hsp70 (B). Reactions containing Hsp70 alone were also included (C). Three μL of each reaction was adsorbed onto carbon/formvar-coated grids and visualized by transmission EM following negative stain with 1% uranyl acetate. In contrast to uniform fibrils 10-14 nm in caliber, addition of Hsp70 resulted in amorphous objects ranging from 20-50 nm and larger aggregates. Samples from α-Syn assembly reactions containing Hsp70 were immunostained using antibodies against Hsp70 (3a3, black arrowheads) and α-Syn (SNL-1, white arrowheads). Single labeling with anti-Hsp70 antibody revealed strong staining of amorphous aggregates (D). In contrast, double labeling did not show colocalization of anti-α-Syn with either aggregates or Hsp70 (E,F,G). Scale bars: 500nm (A,B,C); 100nm (D,E); 50nm (F,G). H, Hsp70, GST-Hsp70386-640, or BSA were incubated in the absence of α-Syn for 72 h with agitation and separated by SDS-PAGE following centrifugation (100,000 × g). Coomassie Blue staining of proteins shows that Hsp70 and its substrate binding alone are capable of forming pelletable aggregates.
Rather, the observed amorphous aggregates resembled Hsp70 oligomers described in a previous study (40) and formed under conditions similar to those used here. Similar species could also be readily detected by EM in reactions containing only Hsp70 or its substrate binding domain (Fig. 4C and data not shown), leading us to hypothesize that the aggregates observed by negatively-stained EM are comprised primarily of Hsp70 rather than α-Syn as previously postulated by others (20). In agreement with this, the Hsp70 species in the inhibited assembly reactions were distributed equally between the soluble and insoluble fractions (Fig. 4H) whereas α-Syn remained mostly in the supernatant (Fig. 2B).
Immunogold EM labeling using an antibody specific for Hsp70 robustly decorated the amorphous aggregates (Fig. 4D) confirming that they are composed of Hsp70. In non-sedimented samples, α-Syn-specific antibodies labeled presumably soluble species adsorbed onto grids, but only rarely the Hsp70-positive aggregates (Fig. 4E). Double immunogold EM labeling corroborated these findings, further supporting our hypothesis that these aggregates are comprised of Hsp70 rather than α-Syn (Fig. 4E-G). Taken together, these data are consistent with the reversible oligomerization observed with a number of Hsp70 family members that is mediated via interactions between substrate binding domains of individual chaperone molecules in the absence of either alternative substrates or nucleotide (41).
Reversing the size of immunogold particles used for EM labeling Hsp70 and α–Syn gave similar results, indicating that our observations did not arise from steric hindrance created by the conjugated secondary antibodies. Although it is still possible that sequestration within Hsp70 complexes rendered α–Syn epitopes undetectable by the antibody used, the partition of Hsp70 and α-Syn into largely separate compartments suggests that a minimal amount of α-Syn is interacting with Hsp70 and that interaction with a minor population of α-Syn may be sufficient to inhibit fibril assembly altogether. Considering the already substoichiometric initial quantities of Hsp70 required for inhibition, the actual quantity of α-Syn recognized as substrate by Hsp70 likely represents a small fraction of the total α-Syn present.
Monitoring assembly inhibition by fluorescence polarization (FP)
Our EM observations also suggest that α–Syn likely does not form large oligomeric complexes following exposure to Hsp70. Therefore, we further examined the molecular mass of the predominant soluble α–Syn species using a sensitive FP method capable of detecting both oligomeric and fibrillar forms of α–Syn in a non-destructive manner (30). Assembly reactions containing full-length α-Syn spiked with small quantities of fluorescently labeled α–Syn (α–Syn488) were monitored over the extended periods using FP in the presence of either Hsp70 or BSA (Fig. 5). FP, which correlates with the molecular mass of α–Syn488-containing species, reached 50% of its maximal value within 30 h in the absence of Hsp70 and remained elevated for the duration of the reaction, corresponding to the formation of oligomers, and eventually insoluble fibrils. Addition of Hsp70 prevented this increase, although a much smaller increase (~15 mP) could be detected at all timepoints after 20 h. Similar results were observed when α-Syn1-110 and labeled α-Syn1-110 were used instead of full-length α–Syn (data not shown), in agreement with our observations made using ThT and K114.
FIG. 5. Monitoring Hsp70 inhibition of α-Syn488 assembly by FP.
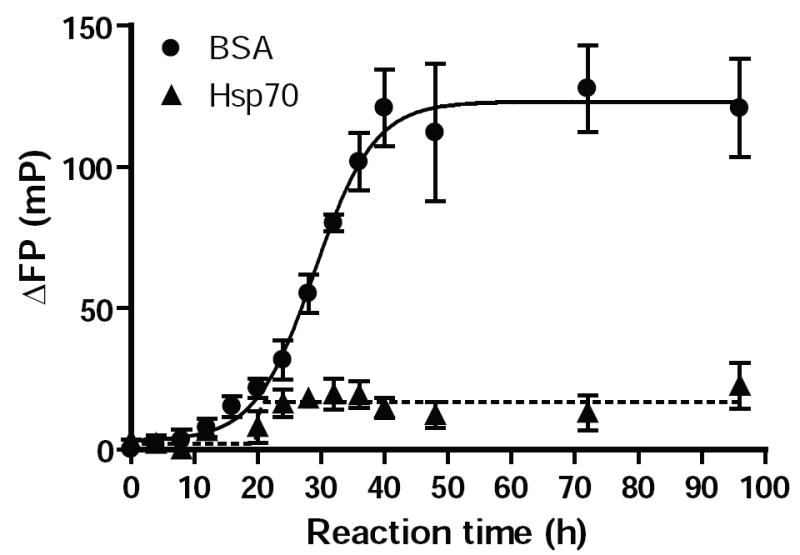
Reactions containing labeled full-length (72μM) were monitored by measuring FP in samples at various timepoints after the addition of Hsp70 or BSA (7.2μM). Control reactions containing BSA showed large increases in FP corresponding to α–Syn fibrils. However, addition of Hsp70 at the start of the reaction resulted in only a minor increase in FP at 20-24h which remained constant thereafter, likely reflecting formation of a small quantity of α–Syn:Hsp70 complexes.
The minor increases in FP observed for both full-length and truncated α–Syn indicate that reduced molecular mobility occurs in only a small fraction of α–Syn, and that oligomeric α–Syn is not the prevalent species in the presence of Hsp70. Although we can not exclude the possibility that small fibrils or oligomers may contribute to this signal, our EM observations indicate this would be limited. Instead, α–Syn interactions with a much larger protein such as Hsp70 likely account for the changes in FP. Taken together, the majority of α–Syn remains in the monomeric state when Hsp70 is added to α–Syn assembly reactions. These observations further suggest that interactions between Hsp70 and α-Syn are transient in nature, which likely accounts for the inability to isolate complexes containing both of these proteins (see below).
Hsp70 interactions with α–Syn are transient
Because Hsp70 appears to interact with only a limited fraction of the total α–Syn present in reactions, we attempted to purify α–Syn:Hsp70 complexes. We were unable to detect Hsp70 following immunoprecipitation of α–Syn in inhibited assembly reactions (Fig. 6A). Similarly, pull-down experiments using a GST-fusion protein containing the Hsp70 substrate binding domain (GST-Hsp70386-640) did not yield detectable quantities of α–Syn (data not shown), suggesting that interactions between α–Syn and Hsp70 are transient or unstable in pull down conditions.
FIG. 6. Reversible interactions between Hsp70 and α–Syn.

A, immunoprecipitation of α–Syn from inhibition reactions. Five μg of α–Syn was immunoprecipitated from Hsp70-containing reactions at 0, 24, and 72h after incubation using Syn211 antibody. Total input (T), flow through (F), and immunoprecipitate (IP) representing 20%, 10% and 40% of each fraction were separated by SDS-PAGE and immunoblotted for Hsp70 (3a3) or α–Syn (Syn303). B, schematic of α–Syn594 displacement assay. Assembly reactions containing labeled α–Syn (α–Syn594) were inhibited using substoichiometric quantities (1:10) of chaperone or GST as a control. After 72 h incubation, an equal amount of fresh monomeric α–Syn488 was added to each reaction. Aggregation was assessed by measuring the relative fluorescence of α–Syn594 and α–Syn488 in the soluble fractions following centrifugation at 100,000 × g. Reactions containing GST (C) or GST-Hsp70386-640 (D) were monitored immediately, 24, or 48 h after α–Syn488 was added. Black and grey lines denote soluble α–Syn594 and α–Syn488, respectively. Similar proportions of both α–Syn594 and α–Syn488 are found in the soluble fraction following extended incubation, indicating that previously bound α–Syn594 is readily displaceable. Data represent means from 4 independent reactions ± s.e.m.
To further establish the dynamic nature of the interaction between α–Syn and Hsp70, we examined whether labeled α–Syn inhibited by chaperone could be displaced by a fresh pool of α–Syn tagged with a different fluorophore (Fig. 6B). Assembly reactions containing Alexa fluor-594-labeled monomeric α–Syn (α–Syn594) were incubated in the presence of either GST or GST-Hsp70386-640. As expected, the majority of the 590/620 nm fluorescence signal was detected in the pellet of reactions containing GST alone after the initial 72 h incubation period (Fig. 6C). In contrast, the soluble fraction accounted for the majority of fluorescence in reactions with Hsp70, indicating inhibition of α–Syn aggregation (Fig. 6D). Following the addition of an equal quantity of monomeric α–Syn488, reactions were resumed and aliquots retrieved for sedimentation analysis at various time points. Within 24 h post-addition, the majority of both α–Syn594 and α–Syn488 were aggregated together in GST-containing reactions, reflecting continued assembly and a robust seeding effect from fibrils formed during the initial incubation. An increase in the amount of insoluble α–Syn was also detected in reactions containing Hsp70, indicating the progression of aggregation as the ratio of Hsp70 to α–Syn was reduced. Interestingly, while ~ 60% of total α–Syn was soluble at equilibrium, the proportion of pelletable α–Syn594 closely mirrored that of α–Syn488 suggesting that Hsp70 did not discriminate between the two pools of α–Syn and that interactions of α–Syn with Hsp70 are readily displaced by fresh α–Syn. Collectively, these data point to a mechanism of assembly inhibition by Hsp70 that is mediated by transient interactions between α–Syn and Hsp70 that also are highly dynamic.
Hsp70 does not alter the assembly competency of α–Syn
We next asked whether such brief interactions with Hsp70 might be sufficient to alter the capacity for α–Syn to aggregate, thereby preventing assembly even when Hsp70 was no longer present. Assembly rates were measured by ThT fluorescence in reactions from which GST-Hsp70386-640 was depleted by incubation with glutathione agarose (Fig. 7). While ThT levels remained at baseline levels in reactions containing GST-Hsp70386-640 (non-depleted), assembly was restored by depletion of chaperone, as indicated by a sharp increase in ThT signal within 48 h. Moreover, the time course for assembly was similar to reactions without Hsp70 (Fig.1), suggesting that Hsp70 does not significantly alter the misfolding capacity of α–Syn.
FIG. 7. α–Syn treated with Hsp70 maintains its assembly capacity.
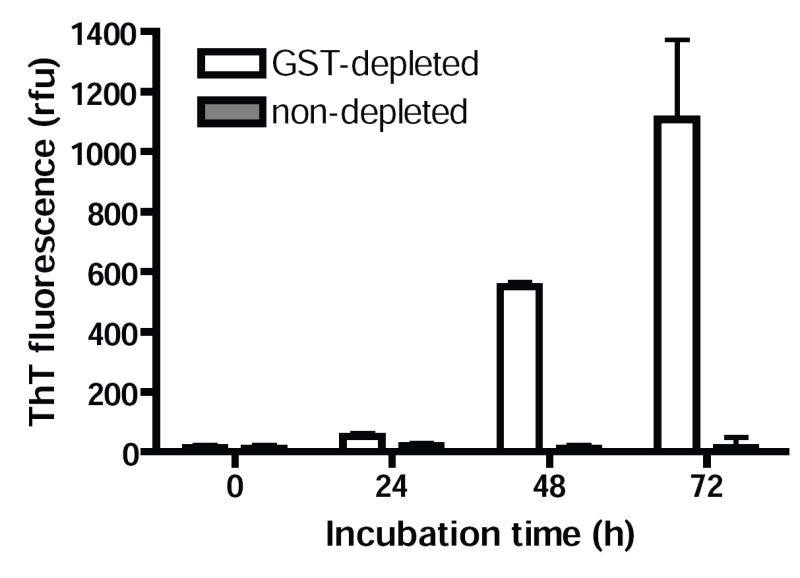
ThT fluorescence was measured in assembly reactions containing GST-Hsp70386-640 and those in which chaperone had been depleted. Glutathione resin was used to deplete reactions 72 h after initiation. Unlike reactions with Hsp70, where ThT fluorescence remained unchanged, ThT increased in Hsp70-depleted reactions after a lag phase similar to that seen in naïve reactions. Data represent means of two independent experiments ± s.e.m.
DISCUSSION
Insoluble cytoplasmic inclusions comprised predominantly of misfolded α–Syn are a prominent feature of PD, DLB, and MSA. Commonly present in such inclusions, and those found in multiple other neurodegenerative diseases is the stress-induced chaperone Hsp70 (8;16). While Hsp70 upregulation is postulated to reflect a cellular response to aggregated proteins, its exact function remains unclear in the context of synucleinopathies. Using an in vitro model of α–Syn aggregation, we show that Hsp70 efficiently inhibits fibril formation at substoichiometric concentrations relative to α-Syn monomer, confirming two recent studies (19;20).
Unlike in vitro models of Hsp70-mediated inhibition of amyloid-beta (42) or mutant polyQ-huntingtin assembly (43), inhibition of α-Syn occurred in the absence of nucleotide or the co-chaperones Hsp40, suggesting that an active refolding process mediated by Hsp70 is unlikely. Moreover, truncated Hsp70 lacking the N-terminal nucleotide-binding domain (Hsp70386-640) retained similar inhibition capacity to that of full-length Hsp70. This indicates that capture of α-Syn by the substrate-binding region alone is sufficient for preventing assembly, and that the N-terminal domain, which regulates the accessibility and affinity of the substrate binding pocket (25;44;45), does not influence this process. In agreement with this, addition of Bag-1 or Hsp40, which regulate Hsp70 ATPase activity, did not diminish the effectiveness of Hsp70 in preventing α-Syn aggregation (data not shown). Similarly, addition of ATP, which accelerates substrate exchange, did not significantly influence inhibition (Supplementary Fig. 1A).
In addition to these and previous findings, our assembly experiments using various α–Syn deletion mutants identified the core hydrophobic region as the main component interacting with Hsp70, since aggregation was effectively prevented even in the absence of essentially all α–Syn N- or C-terminal regions not critical for fibril formation (Figs. 2 and 3). Thus, Hsp70 appears to recognize and interact directly with the NAC region of α–Syn, which contains a 12-residue stretch critical for fibril formation (29) and whose high hydrophobic content is typical of classical Hsp70 substrates (46;47). These findings rule out a mechanism whereby Hsp70 stabilizes soluble α-Syn by preserving (or restoring) intramolecular interactions which conceal the hydrophobic core region as postulated by models based on NMR and FRET studies (35;48). Interestingly, we also found that a previously reported consensus motif for chaperone-mediated autophagy (residues 95-99; ref. 38) was dispensable for Hsp70-mediated assembly inhibition. Therefore, we argue that Hsp70 interactions at this site are unlikely to influence α–Syn fibril assembly although its importance for other functions cannot be ruled out.
The interaction between Hsp70 and the α-Syn core region also distinguishes Hsp70 from other inhibitors of α-Syn assembly, as most small-molecule inhibitors reported to date act upon the C-terminal region of α-Syn (32;49-52). The activities of these compounds, which include porphyrins (52), polyphenols (32;51), and the antibiotic rifampicin (53), also depend on the formation of adducts, which is not evident in Hsp70-mediated inhibition. In addition, many of these molecules, most notably the catecholamines, result in the formation of stable α-Syn oligomers (32;49;54), suggesting that their mechanism of inhibition differs significantly from that of Hsp70.
Consistent with previous reports (19;20), we observed that substoichiometric concentrations of Hsp70 efficiently inhibited assembly. As the core hydrophobic region was present on all full-length α–Syn molecules, this relationship suggested that individual Hsp70 molecules can recognize α–Syn multimers. Accordingly, Hsp70 suppressed elongation in fibril-seeded assembly reactions, likely by preventing the addition of monomer at fibril ends (55;56). However, although previous size exclusion chromatography experiments showed that Hsp70 co-elutes with prefibrillar species following co-incubation with monomeric α–Syn (19;20), our ultrastructural examination of WT α–Syn monomer following incubation with Hsp70 revealed a conspicuous absence of fibrils, indicating that inhibition is not mediated primarily via “capping” of small, early fibrils. In contrast, amorphous species similar to those described by others (19;20) were abundant (Fig. 5). To our surprise however, immunolabeling of these aggregates established them to be composed mainly of Hsp70 and not α–Syn.
Monitoring α–Syn assembly by FP also confirmed that α–Syn oligomers were not a major product resulting from Hsp70 mediated assembly inhibition (Fig. 6). Rather, the low FP values following incubation with Hsp70 were more consistent with small, soluble α–Syn species, most likely monomeric α–Syn, although contribution from a small population of oligomers cannot be excluded. Interestingly, Hsp70-inhibited α-Syn reactions readily resumed assembly after being depleted of chaperone, suggesting that such soluble α–Syn species remained on the assembly pathway as their capacity for assembly was maintained. In addition, the rates at which these depleted reactions progressed were more similar to initial monomer-containing reactions than the rapid rate expected if nucleation had already been achieved and pre-fibrillar species were present (Fig. 7 and Supplementary Fig. 2). Moreover, we found no evidence of SDS-insoluble α–Syn oligomers typical of stable off-pathway intermediates in Hsp70-inhibited assembly reactions containing either WT α–Syn or any of the mutants used. Together with our EM observations, these data point to a mechanism whereby Hsp70 recognizes the hydrophobic regions in soluble α–Syn species (monomer or low-order oligomers) exposed during the early stages of the assembly process and preventing their progression along this pathway. The extended lag phase observed for α–Syn assembly relative to other amyloidgenic proteins such as Aβ and tau also suggests that conversion of unfolded monomer occurs at an infrequent rate, which may explain how weak transient interactions with a chaperone protein may be sufficient for halting assembly.
One obstacle to identifying the precise α-Syn species bound to Hsp70 is the difficulty in isolating complexes of the two proteins. The transient nature of this interaction was evident in dual-labeling experiments in which α-Syn at various stages of assembly was readily displaced by fresh α-Syn, suggesting that binding of α–Syn intermediates is a dynamic equilibrium. Although additional studies are required to elucidate the rate of association and the exact mechanism of assembly inhibition, we propose that a much larger Hsp70 molecule transiently or reversibly binds the core α-Syn region in misfolded species, thereby preventing further progression of aggregation and fibrillization.
In summary, our data here suggest that Hsp70 recognizes α-Syn early in the fibril assembly process and is highly efficient in preventing fibril formation. While an intimate relationship between molecular chaperones and neurodegenerative diseases clearly exists, it remains unclear whether Hsp70 participates in normal physiology of α-Syn. Future studies on the nature of the interactions presented here should help elucidate the in vivo interplay between these two molecules and set the stage for the use of chaperones as disease modifying therapies in synucleinopathies.
Supplementary Material
Supplementary Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
VMYL is the John H. Ware, 3rd Professor of Alzheimer’s disease research. JQT is the William Maul Measey-Truman G. Schnabel, Jr. Professor of Geriatric Medicine and Gerontology.
This work was supported by NIH grants AG09215, NS044233, NS053488 and the Picower Foundation.
ABBREVIATIONS
- α-Syn
Alpha-synuclein
- ATP
Adenosine triphosphate
- BSA
Bovine serum albumin
- CMA
Chaperone-mediated autophagy
- DLB
Dementia with Lewy bodies
- EM
Electron microscopy
- FP
Fluorescence polarization
- FRET
Fluorescence resonance energy transfer
- GST
Glutathione S-transferase
- Hsp70
Heat-shock protein 70
- K114
(trans,trans)-1-bromo-2,5-bis-(4-hydroxy)styrylbenzene
- Mab
Monoclonal antibody
- NAC
Non-amyloid component
- NMR
Nuclear magnetic resonance
- PD
Parkinson’s disease
- ThT
Thioflavin T
- WT
Wild-type
References
- 1.Forman MS, Lee VMY, Trojanowski JQ. Nosology of Parkinson’s disease: Looking for the way out of a quackmire. Neuron. 2005;47:479–482. doi: 10.1016/j.neuron.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG. Parkinson’s disease, dementia with Lewy bodies and multiple system atrophy are alpha-synucleinopathies. Parkinsonism & Related Disorders. 1999;5:157–162. doi: 10.1016/s1353-8020(99)00031-0. [DOI] [PubMed] [Google Scholar]
- 4.Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, Takahashi H. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathologica. 1998;96:445–452. doi: 10.1007/s004010050918. [DOI] [PubMed] [Google Scholar]
- 5.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 6.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Tortosa EG, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 7.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, DiIorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 8.Auluck PK, Chan HYE, Trojanowski JQ, Lee VMY, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 9.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 10.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VMY. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 11.Hartl FU, Hayer-Hartl M. Protein folding - Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 12.Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. Journal of Neuroscience. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warrick JM, Chan HYE, Gray-Board, Chai YH, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nature Genetics. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 15.Klucken J, Shin Y, Masliah E, Hyman BT, Mclean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. Journal of Biological Chemistry. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- 16.Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, Pham CT, Yazawa I, Hilburger K, Micsenyi M, Giasson BI, Bonini NM, Lee VMY, Trojanowski JQ. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. American Journal of Pathology. 2006;168:947–961. doi: 10.2353/ajpath.2006.050770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mclean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. Journal of Neurochemistry. 2002;83:846–854. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- 18.Yaglom JA, Gabai VL, Meriin AB, Mosser DD, Sherman MY. The function of HSP72 in suppression of c-Jun N-terminal kinase activation can be dissociated from its role in prevention of protein damage. Journal of Biological Chemistry. 1999;274:20223–20228. doi: 10.1074/jbc.274.29.20223. [DOI] [PubMed] [Google Scholar]
- 19.Huang CJ, Cheng H, Hao SF, Zhou H, Zhang XJ, Gao JN, Sun QH, Hu HY, Wang CC. Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. Journal of Molecular Biology. 2006;364:323–336. doi: 10.1016/j.jmb.2006.08.062. [DOI] [PubMed] [Google Scholar]
- 20.Dedmon MM, Christodoulou J, Wilson MR, Dobson CM. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. Journal of Biological Chemistry. 2005;280:14733–14740. doi: 10.1074/jbc.M413024200. [DOI] [PubMed] [Google Scholar]
- 21.Wu B, Hunt C, Morimoto R. Structure and Expression of the Human-Gene Encoding Major Heat-Shock Protein Hsp70. Molecular and Cellular Biology. 1985;5:330–341. doi: 10.1128/mcb.5.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown IR. Heat shock proteins and protection of the nervous system. Stress Responses in Biology and Medicine. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- 23.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giasson BI, Uryu K, Trojanowski JQ, Lee VMY. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. Journal of Biological Chemistry. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 25.Freeman BC, Myers MP, Schumacher R, Morimoto RI. Identification of A Regulatory Motif in Hsp70 That Affects Atpase Activity, Substrate-Binding and Interaction with Hdj-1. Embo Journal. 1995;14:2281–2292. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crystal AS, Giasson BI, Crowe A, Kung MP, Zhuang ZP, Trojanowski JQ, Lee VMY. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. Journal of Neurochemistry. 2003;86:1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x. [DOI] [PubMed] [Google Scholar]
- 27.Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson’s disease. Journal of Neuroscience Research. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Jakes R, Crowther RA, Lee VMY, Trojanowski JQ, Iwatsubo T, Goedert M. Epitope mapping of LB509, a monoclonal antibody directed against human alpha-synuclein. Neuroscience Letters. 1999;269:13–16. doi: 10.1016/s0304-3940(99)00411-5. [DOI] [PubMed] [Google Scholar]
- 29.Giasson BI, Murray IVJ, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. Journal of Biological Chemistry. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 30.Luk KC, Hyde EG, Trojanowski JQ, Lee VMY. Sensitive fluorescence polarization technique for rapid screening of (X-Synuclein Oligomerization/Fibrillization inhibitors. Biochemistry. 2007;46:12522–12529. doi: 10.1021/bi701128c. [DOI] [PubMed] [Google Scholar]
- 31.Hashimoto M, Hsu LJ, Sisk A, Xia Y, Takeda A, Sundsmo M, Masliah E. Human recombinant NACP/alpha-synuclein is aggregated and fibrillated in vitro: Relevance for Lewy body disease. Brain Research. 1998;799:301–306. doi: 10.1016/s0006-8993(98)00514-9. [DOI] [PubMed] [Google Scholar]
- 32.Zhu M, Rajamani S, Kaylor J, Han S, Zhou FM, Fink AL. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. Journal of Biological Chemistry. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 33.Murray IVJ, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VMY. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 34.Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong GZ, Gee AD, Brown J. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. Journal of Biological Chemistry. 2005;280:7677–7684. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 35.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. Febs Letters. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 38.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 39.Han HY, Weinreb PH, Lansbury PT. The Core Alzheimers Peptide Nac Forms Amyloid Fibrils Which Seed and Are Seeded by Beta-Amyloid - Is Nac A Common Trigger Or Target in Neurodegenerative Disease. Chemistry & Biology. 1995;2:163–169. doi: 10.1016/1074-5521(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 40.Benaroudj N, Batelier G, Triniolles F, Ladjimi MM. Self-Association of the Molecular Chaperone Hsc70. Biochemistry. 1995;34:15282–15290. doi: 10.1021/bi00046a037. [DOI] [PubMed] [Google Scholar]
- 41.Benaroudj N, Triniolles F, Ladjimi MM. Effect of nucleotides, peptides, and unfolded proteins on the self-association of the molecular chaperone HSC70. Journal of Biological Chemistry. 1996;271:18471–18476. doi: 10.1074/jbc.271.31.18471. [DOI] [PubMed] [Google Scholar]
- 42.Evans CG, Wisen S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. Journal of Biological Chemistry. 2006;281:33182–33191. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 43.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nature Structural & Molecular Biology. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 44.Buchberger A, Theyssen H, Schroder H, Mccarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-Induced Conformational-Changes in the Atpase and Substrate-Binding Domains of the Dnak Chaperone Provide Evidence for Interdomain Communication. Journal of Biological Chemistry. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 45.Greene LE, Zinner R, Naficy S, Eisenberg E. Effect of Nucleotide on the Binding of Peptides to 70-Kda Heat-Shock Protein. Journal of Biological Chemistry. 1995;270:2967–2973. doi: 10.1074/jbc.270.7.2967. [DOI] [PubMed] [Google Scholar]
- 46.Rudiger S, Germeroth L, SchneiderMergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. Embo Journal. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fourie AM, Sambrook JF, Gething MJH. Common and Divergent Peptide Binding Specificities of Hsp70 Molecular Chaperones. Journal of Biological Chemistry. 1994;269:30470–30478. [PubMed] [Google Scholar]
- 48.Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. Journal of Molecular Biology. 2005;353:357–372. doi: 10.1016/j.jmb.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 49.Norris EH, Giasson BI, Hodara R, Xu SH, Trojanowski JQ, Ischiropoulos H, Lee VMY. Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. Journal of Biological Chemistry. 2005;280:21212–21219. doi: 10.1074/jbc.M412621200. [DOI] [PubMed] [Google Scholar]
- 50.Rao JN, Dua V, Ulmer TS. Characterization of alpha-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry. 2008;47:4651–4656. doi: 10.1021/bi8002378. [DOI] [PubMed] [Google Scholar]
- 51.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nature Structural & Molecular Biology. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 52.Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Zhu M, Rajamani S, Uversky VN, Fink AL. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chemistry & Biology. 2004;11:1513–1521. doi: 10.1016/j.chembiol.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 54.Conway KA, Rochet JC, Bieganski RM, Lansbury PT. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 55.Yagi H, Kusaka E, Hongo K, Mizobata T, Kawata Y. Amyloid fibril formation of alpha-synuclein is accelerated by preformed amyloid seeds of other proteins - Implications for the mechanism of transmissible conformational diseases. Journal of Biological Chemistry. 2005;280:38609–38616. doi: 10.1074/jbc.M508623200. [DOI] [PubMed] [Google Scholar]
- 56.Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent - Implications for the pathogenesis of Parkinson’s disease. Journal of Biological Chemistry. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org