

Abstract
microRNA (miRNA) is a class of small, noncoding, regulatory RNAs. The ∼ 21 nt mature miRNA is processed from larger precursor molecules following a coordinated series of events. In theory, miRNA processing may be regulated at any of these steps. A growing body of evidence has demonstrated various steps in the miRNA biogenesis process for which regulation occurs. RNA editing of miRNA precursors, SNPs or mutations in the miRNA precursors, regulation by RNA binding proteins, alterations in the levels of key processing proteins, as well as a number of unknown mechanisms contribute to the regulation of miRNA processing. This article reviews the available literature on the regulation of miRNA processing that occurs within normal cells, during development or in diseases such as cancer.
Keywords: microRNA biogenesis, microRNA processing
Introduction
microRNA processing and function
-
Regulation of microRNA processing during development, differentiation and normal cell function
Effects of RNA editing on miRNA processing
Effects of SNPs and mutations on miRNA processing
-
Examples of altered miRNA processing in cancer
Altered expression levels of Dicer in cancer
Evidence of post-transcriptional regulation of miRNA expression in cancer
Conclusion
Introduction
microRNAs (miRNAs) are a class of small, regulatory, noncoding RNAs. miRNA was discovered in C. elegans in 1993 by the Ambros and Ruvkun laboratories [1, 2]. The founding members of this class of RNAs include lin-4 and let-7. In 2001, interest in the field peaked when hundreds of miRNAs were discovered in Drosophila, C. elegans and mammals [3–5]. Over the past 7 years, numerous miRNAs have been discovered in many species including 678 in human, 472 in mouse and 154 in C. elegans[6]. miRNA represents a new class of gene regulation; it has been estimated that miRNA regulates up to one-third of human genes [7]. A number of excellent reviews on various aspects of miRNA have been published including overviews [8, 9], miRNA processing [10], post-transcriptional regulation of miRNA [11] and effect of RNA editing on miRNA [12]. This article reviews the literature on the regulation of miRNA processing that occurs within normal cells, during development or in diseases such as cancer.
microRNA processing and function
The genes encoding miRNAs are located within introns, intergenic regions and within exons [13]. The majority of human miRNA loci are encoded within intronic regions [14]. Although some exceptions exist [15], miRNA genes are transcribed by RNA polymerase II [16]. The primary transcript that results from the RNA polymerase II transcription is termed the primary miRNA (pri-miRNA) (Fig. 1). pri-miRNA contains a hairpin structure from which the mature miRNA is processed. Like mRNAs, the pri-miRNA is polyadenylated, contains a 5’ cap structure and may undergo splicing. The pri-miRNA is processed in the nucleus by a complex that includes the RNase III enzyme Drosha and the double stranded RNA binding domain protein DGCR8 to produce the pre-miRNA [17]. The complex contains a yardstick to measure the hairpin and to allow precise processing of the pri-miRNA [18]. Processing of the pri-miRNA appears to proceed prior to splicing [14]. The pre-miRNA contains the 2 nt 3’ overhang that is characteristic of Dicer processing of siRNA. The pre-miRNA is transported to the cytoplasm by Exportin 5, a Ran-GTP-dependent mechanism [19]. Once inside the cytoplasm, the pre-miRNA is subsequently loaded into a complex that includes the RNase III enzyme Dicer and TRBP/Loquacious. This complex cleaves the loop from the pre-miRNA to produce a double-stranded complex composed of the miRNA and miRNA*. The miRNA* strand is typically degraded and the mature miRNA strand, along with the Argonaute protein Ago 2, is further assembled into a ribonucleoprotein complex known as RISC (RNA-induced silencing complex).
Fig 1.
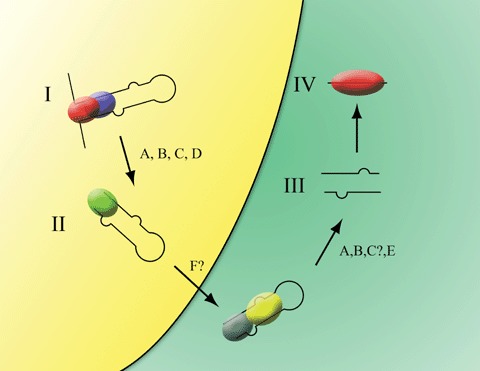
Biogenesis of miRNA and sites of regulation. miRNA is transcribed by RNA polymerase II to produce the primary precursor miRNA (I, pri-miRNA). The pri-miRNA is trimmed by Drosha/DGCR8 in the nucleus to produce the ∼ 70 nt precursor miRNA (II, pre-miRNA). The pre-miRNA is transported to the cytoplasm by Exportin 5 where it interacts with the RNAse III Dicer and TRBP/ Loquacious to produce the ∼ 21nt miRNA: miRNA* duplex (III). The miRNA strand (IV) is loaded into RNA-induced silencing complex (RISC) and the miRNA* strand is typically degraded. Possible sites of regulation include: A, A-I editing; B, SNPs or mutations; C, regulation of processing by miRNA-specific factors; D, altered levels of Drosha; E, altered levels of Dicer; F, sequestration into subcellular organelles.
RISC will then scan cellular mRNA in an attempt to locate the miRNA's target. miRNAs function by binding through imperfect complimentarity to conserved sequences within the untranslated region (UTR) of mRNAs. Although most work has focused on miRNA binding to the 3’ UTR, miRNAs have also been shown to bind to the 5’ UTR [20]. Nucleotides 2 through 8 on the 5’ end of the mature miRNA, the so-called 5’ seed, interact with the reverse complement sequences on the mRNA. In situations of perfect complimentarity, the mRNA will be cleaved [21]. In animals, it is more likely that the miRNA will bind to the mRNA through imperfect complimentarity and produce translational repression. Translational repression proceeds without cleavage of the mRNA [22]. Translational repression is believed to occur by blocking translation, often by inhibiting translational initiation [23, 24]. miRNAs have been shown to be sequestered into processing-bodies (P-bodies) [25]. The role of P-bodies in target repression and miRNA biogenesis remain unclear. Knockdown of Drosha and DGCR8 resulted in a loss of the P-bodies suggesting that they are a consequence, rather than the cause, of miRNA biogenesis [26].
Regulation of microRNA processing during development, differentiation and normal cell function
Conceivably, production of the mature miRNA may be regulated at multiple steps in the biogenesis process (Fig. 1). Possibilities include Drosha processing, export from the nucleus, processing by Dicer or through altered stability of the mature miRNA. Changes in miRNA processing may be viewed experimentally by examining the levels of precursor and mature miRNAs on a Northern blot. An increased or decreased level of mature miRNA without noticeable changes in the levels of miRNA precursors is the hallmark of regulated miRNA processing. Such phenomenon was shown by Ambros et al. in studies of C. elegans development [27]. The precursor to miR-38 was evident during all stages of development, however the mature miR-38 was present only in the embryonic stage [27].
Various members of the let-7 family were regulated post-transcriptionally during neural differentiation of embryocarcinoma cells [28]. The levels of the precursor forms of the miRNAs were consistent during differentiation, whereas the mature form increased [28]. An increase in the let-7 precursor processing activity in vitro during neural differentiation of embryocarcinoma cells demonstrated that regulation occurred at the Dicer processing step [28]. Similar to [27], Thomson et al. showed that the levels of mature let-7g (but not precursor) miRNAs increased during development of the mouse [29]. A number of miRNAs (let-7g, let-7f-2, miR-24, miR-140, miR-103–1, etc.) were increased at the mature level at days 10.5 and 14.5 of gestation compared to an embryonic stem cell line [29]. However, the levels of pri-miRNAs were consistent throughout this period, demonstrating that regulation occurred at the miRNA processing steps and not transcription.
The answer to this puzzle came recently when it was discovered that the developmentally regulated RNA binding protein Lin-28 inhibits the processing of the primary precursor of the let-7 family members [30, 31]. Lin-28 protein was identified from cellular extracts of P19 cells using a biochemical approach [31]. Lin-28 protein levels decreased during the same time of differentiation as mature let-7 increased. As in the prior studies, there was no change in the precursor form of the let-7. Knockdown of Lin-28 using RNA interference increased the amount of mature let-7 but not miR-15a, miR-16–1, miR-122a or other miRNAs assayed by a micro-array demonstrating the specific nature of this interaction [31]. Lin-28 was shown to selectively bind to the terminal loop of the let-7 precursors and that the loop mediates miRNA processing inhibition [30, 32]. This is of particular importance as single nucleotide polymorphisms (SNPs) or mutations in the loop region of let-7 family members may cause Lin-28 to bind less effectively, resulting in increased levels of mature let-7.
The expression profiles of 142 xenopus tropicalis miRNAs were studied by Northern blotting [33]. The tissue-specific expression profile of the pri-miRNA was different from that of the mature miRNA. For example, xtr-miR-98 and xtr-let-7f are clustered on a 427 nt DNA fragment and can be assumed to be cotranscribed as a common pri-miRNA. The expression of the primary precursors of xtr-miR-98 and xtr-let-7f was consistent among seven different tissues studied, whereas the expression of the mature xtr-miR-98 and xtr-let-7f differed. Mature xtr-let-7f was present in the heart, whereas xtr-miR-98 was not, demonstrating differential processing of the precursor forms of these two miRNAs. Sometimes the pri-miRNA was present but not the pre- or mature forms (e.g. xtr-miR-215), other times the pri- and pre-miRNAs were present but not the mature miRNA (e.g. xtr-miR-200b), suggesting differential precursor processing in both the nucleus and cytoplasm [33].
Lugli et al. studied the expression of mature and precursor miRNAs in synaptic fractions of the adult mouse forebrain [34]. Both the precursor and mature forms of several miRNAs expressed within synaptic fractions of the adult mouse forebrain were present at levels that were similar to or greater than total forebrain homogenate. Mature miRNAs were predominantly associated with soluble components of the synaptic fractions, whereas miRNA precursors were predominantly associated with post-synaptic densities [34]. Dicer that is present in the post-synaptic densities is enzymatically inactive until conditions arise that cause activation of calpain. These authors propose that mature miRNAs are formed locally within synaptic fractions following synaptic stimulation via processing of miRNA precursors [34].
Obernosterer et al. studied the post-transcriptional regulation of miRNA in the developing mouse and in HeLa cells [35]. They found that many miRNAs are transcribed at the precursor level, however upon differentiation, the ubiquitously expressed precursor miRNAs are processed to mature miRNA in a tissue-specific manner. In situ hybridization revealed that pre-miR-138–2 is present in nearly all of the tissues in the E17 mouse embryos, whereas mature miR-138 was expressed only in neuronal tissues (brain, CNS) and in the foetal liver [35]. HeLa cells are another example of a cell type that expressed the miR-138–2 precursor but not the mature miR-138. Conceivably, inhibition of Exportin 5 transport would prevent the pre-miR-138 from reaching the cytoplasm and be processed to mature. However, pre-miR-138–2 was present in the cytoplasm of HeLa cells effectively ruling out this possibility. HeLa cytoplasmic extracts inhibited pre-miR-138–2 processing by recombinant Dicer but not pre-miR-19a (a miRNA that is normally expressed in HeLa cells) [35]. Therefore, the presence of some unknown inhibitory factor appears to be preventing processing of pre-miR-138–2 in the cytoplasm of HeLa cells. These authors have developed a protocol to assist in the identification of cytoplasmic, inhibitory factors of miRNA processing [36].
The miR-17–92 polycistron is composed of six miRNA precursors (i.e. miR-17, -18a, -19a, -19b-1, -20a and -92–1). Forced expression of the polycistron along with c-myc was tumourigenic, suggesting that this group of miRNAs may function as oncogenes [37]. The splicing regulatory factor heterogeneous nuclear ribonucleoprotein (hnRNP) A1 is required for Drosha processing of miR-18a from the primary precursor [38]. hnRNP A1 was shown to interact specifically with the miR-18a hairpin and this interaction occurred prior to the processing by Drosha [7]. Using RNA interference knockdown of hnRNP A1, it was shown that miR-18a and not the other five miRNAs on the pri-miRNA required hnRNP A1 for processing. This is the first example of an hnRNP regulating the processing of miRNA. There are at least 28 known human hnRNPs and mRNA–protein complex proteins (mRNPs) [39]. Conceivably, some of these hnRNPs or mRNPs may be involved in regulating the processing of other miRNAs.
Effects of RNA editing on miRNA processing
RNA editing is a process in which adenosine deaminases (ADARs) convert adenosine to inosine on double-stranded RNA (reviewed in [40]). This process will have profound differences when protein-coding mRNA is edited as inosine is translated as guanosine. On double-stranded noncoding RNA, guanosine will base pair with cytidine, thus altering the base pairing specificity and ultimately the three-dimensional structure of the RNA. Therefore, processes such as miRNA processing that relies heavily upon three-dimensional RNA structure would be expected to be affected by RNA editing. This in fact has been shown to be the case. Pri-miR-22 undergoes A-to-I editing at both the -1 and +1 positions that are near the cleavages site [41]. A-to-I editing of pri-miR-142 resulted in suppression of its processing by Drosha [42]. The edited pri-miR-142 was degraded by Tudor-SN, a ribonuclease specific to inosine-containing dsRNAs and a component of RISC.
Pri- and pre-miR-151 undergoes A-to-I editing at two specific positions, −1 and +3, with the +3 site being edited at a greater frequency than the −1 site [43]. RNA editing inhibited the pre- to mature miRNA step and not the pri- to pre-miRNA step. This is the first example of inhibition of Dicer-TRBP cleavage due to RNA editing. Interestingly, evidence was provided to suggest that A-to-I editing at the +3 site may occur in the cytoplasm by cytoplasmic ADAR even after the processing of the pri- to pre-miR-151 [43]. The frequency of A-to-I editing in human amygdala and mouse cerebral cortex was 100%, suggesting that in vivo cleavage of unedited pre-miR-151 by Dicer-TRBP is highly efficient [43].
Effects of SNPs and mutations on miRNA processing
Conceivably, SNPs or mutations within the primary miRNA precursor gene can affect miRNA processing. A number of artificial mutations were created in both the stem and loop portion of miRNA precursors [44]. These mutations caused both reduction of mature miRNA and reduced miRNA activity. Data on SNPs and/or mutations that affect miRNA processing in cells, tissues or cell lines are sparse. A germ-line mutation in the miR-16–1-miR-15a primary precursor was identified in two patients with chronic lymphocytic leukaemia [45]. Northern blotting and miRNA microchip analysis showed a reduction in the mature miR-16 and -15a in patients harbouring these mutations.
Ten SNPs were identified among 173 human pre-miRNA genes from 96 subjects [46]. Each of these SNPs was located in the stem or loop region of the pre-miRNA. With the exception of miR-30c (C-A polymorphism), the effects of these SNPS on miRNA processing were not evaluated. Pre-miRNA expression vectors harbouring either the wild-type or A SNP were processed to the mature form in CHO or HEK293T cells, demonstrating that this particular SNP had no effect on miRNA processing [46]. A total of 323 SNPs were identified among 227 human miRNA genes; 12 were located within miRNA precursors [47]. A G-U polymorphism was located at the +8 position of the mature form of miR-125a. Transfection of HEK393T cells with a vector expressing the G or U forms of the miR-125a precursor demonstrated that only the G form was processed into mature miR-125a [47]. Using quantitative RT-PCR and primers specific to the pri- and pri-/pre-forms, it was shown that miRNA biogenesis was blocked at the level of pri-to pre-miR-125a processing [47]. This G-U SNP introduced base-pairing mismatches, altered free energy values and created an enlarged RNA bulge in the predicted secondary structure of the miRNA precursor.
Examples of altered miRNA processing in cancer
Numerous studies have identified widespread alterations in the expression of miRNAs in human cancer (reviewed in [48, 49]). In fact, differential miRNA expression has been identified in all malignancies studied to date. There is no clear trend in the direction of the differentially expressed miRNA in human cancer as the expression is increased or decreased compared to normal tissue. Whether the expression is increased or decreased depends upon the individual miRNA. For example, some miRNAs, e.g. miR-15, -16 [50], miR-143, -145 [51–53], let-7 [54], miR-199a, -199a*[55–57] and 122a [56, 58] are reduced in cancer. Other miRNAs, e.g. miR-21 [59–62], miR-221, -222 [63–65], miR-100 [63] and miR-155 [66] are increased in cancer. At least three different mechanisms have been proposed to explain the alterations in miRNA expression in human cancer. These include (i) miRNAs that are located at cancer associated genomic regions [67, 68], (ii) epigenetic regulation of miRNA expression [69, 70] and (iii) abnormalities in miRNA processing. This review will discuss only the later mechanism.
The initial study demonstrating the relationship between alterations in miRNA processing in cancer was that of Michael et al. [51]. Two miRNAs, miR-143 and miR-145, had reduced expression of the mature form in colorectal adenocarcinoma compared to the matched normal mucosa. Interestingly, the Northern blots showed that the levels of pre-miR-143 and -145 were identical between the tumour and normal tissues. These authors went on to examine the miR-143 and -145 expression in several cancer cell lines and showed that like the tumours, the cell lines had reduced levels of mature miRNA, whereas equivalent levels of the precursors were detectable. Thus, the reduction in mature miR-143 and -145 in colorectal cancer cell lines and tissues is a result of reduced processing of the miRNA precursors.
miR-143 was subsequently shown to target ERK5 [71]. ERK5 is a growth-related mitogen-activated protein kinase. Transfection of a miR-143 and miR-145 precursor oligonucleotides into colorectal carcinoma cell lines reduced cell viability and reduced the levels of ERK5 protein [52]. Furthermore, the genomic loci of the majority of the cancer cell lines were unchanged and alterations in DNA methylation among the cell lines did not occur [52]. miR-143 and -145 was found to be reduced in most of the B-cell malignancies examined, including chronic lymphocytic leukaemias, B-cell lymphomas, Epstein–Barr virus-transformed B-cell lines and Burkitt lymphoma cell lines; these authors did not examine the levels of miR-143 and miR-145 precursors [53]. The results of these studies strongly suggest that miR-143 and -145 function as tumour suppressors and that deregulation in colorectal cancer and B-cell malignancies contributes to the tumourigenesis. This is of particular interest because it would exemplify the first tumour suppressive miRNAs whose expression is regulated by processing of the precursor.
miR-7 inhibits the epidermal growth factor receptor and the Akt pathway [72]. Mature miR-7 was predominately reduced in glioblastoma tumours and in the glioblastoma cell line U87MG [72]. The mature miR-7 is processed from three different primary precursors, miR-7–1, miR-7–2 and miR-7–3. Using a quantitative PCR approach, levels of all three pri-miR-7 isoforms were similar between the glioblastomas and the normal tissues, however the levels of the pre-miR-7 isoforms were reduced in the glioblastomas [72]. The reduction in the miR-7 processing was shown to occur at the pri- to pre-miRNA level and not at the pre-miRNA to mature level and was specific to the U87MG cell line and not to HeLa cells [72]. Deletions or mutations were not responsible for the reduction in miR-7 processing. These results indicate that there is a specific defect in the processing of the pri-miR-7 in glioblastomas.
Altered expression levels of Dicer in cancer
One possibility to explain changes in the expression levels of miRNA in neoplasia is if the expression of the processing enzymes (Dicer and/or Drosha) were altered in cancer. Increased or reduced expression of these enzymes would result in a global increase or decrease in the expression levels of the miRNA, respectively. Data on the potential role of altered Drosha or Dicer expression in cancer are inconsistent. Some studies report an increase in Dicer levels in cancer, some report reduced Dicer expression and some studies report no change. There is likely no one size fits all explanation for altered processing in cancer and each malignancy must be examined on an individual bases with regards to Dicer or Drosha processing.
In their study of reduced mature miR-143 and -145 expression in colorectal carcinoma, Michael et al. did not examine the Dicer expression levels in the neoplastic and normal tissues, so a conclusion to describe their data on the basis of Dicer processing cannot be made at this time [51]. Lu et al. reported predominately reduced expression of multiple miRNAs in several different solid tumours [73]. They did not, though, detect a reduction in Drosha or Dicer mRNA in these tumour specimens and conclude that other mechanisms are responsible for the reduction in miRNA expression [73].
In a study of non-small-cell-lung cancer (NSCLC), it was determined that the expression levels of Dicer but not Drosha were reduced in a fraction of lung tumour tissues and that those patients with reduced Dicer expression had a poorer prognosis [74]. The expression of Drosha as well as other genes on chromosome 5p was increased in cervical cancer [75]. In a tissue micro-array consisting of 232 prostate specimens, Dicer protein was found to be up-regulated in prostatic intraepithelial neoplasia and in 81% of the prostate adenocarcinomas [76]. It was proposed that increased Dicer expression in prostate carcinoma was partially responsible to explain the global increased miRNA expression in prostate cancer that was identified by this group [76]. However, others have concluded that there is a predominate reduced expression of miRNAs in prostate cancer [73, 77, 78], so a conclusion on the contribution of Dicer expression in prostate cancer awaits further study.
Precursor lesions of peripheral adenocarcinoma of the lung (i.e. atypical adenomatous hyperplasia and bronchoalveolar carcinoma) showed increased Dicer protein by immunohistochemistry [79]. The opposite was true for invasive and advanced adenocarcinoma, where Dicer protein was reduced [79]. The later phenomena may be partially explained by deletions in the Dicer locus [79]. This same group of researchers studied the Dicer protein expression in specimens of mucoepidermoid carcinoma (MEC) and correlated the Dicer expression with survival [80]. They determined that Dicer protein expression could either be increased or decreased in MEC. The high-grade, stage-III/IV, margin positive MECs showed predominate increased Dicer expression, whereas the tumour specimens from patients over 55 years of age were predominately reduced [80].
Evidence of post-transcriptional regulation of miRNA expression in cancer
Thomson et al. used published messenger RNA data to approximate the pri-miRNA xpression of those miRNAs that are encoded within introns [29]. Thus, the expression levels of a large number of pri-miRNAs were determined in several tumours and normal tissues without additional profiling. Because the mature miRNA was profiled in this same data set [73], it was possible to establish the correlation between the levels of pri-miRNA and mature miRNA in a number of normal tissues and tumours. Consistent with their data in the developing mouse, the expression of the pri-miRNA was more or less consistent between tumour and normal, whereas the levels of mature miRNA were considerably reduced in the malignancy compared to the normal tissue [29]. These authors conclude that the widespread down-regulation of miRNA in cancer is due to a failure at the Drosha processing step. This failure is not due to reduction in Drosha mRNA expression as the levels of Drosha mRNA in this set of tissues did not change [73] and therefore alternative explanations are necessary.
Perhaps the strongest case for a relationship between reduced mature miRNA expression and cancer comes from the study by Kumar et al. [81]. shRNAs to three regulators of miRNA processing (Drosha, DGCR8 and Dicer) were used to dramatically reduce the levels of these genes and thus impair miRNA processing. Transformed cells transfected with the shRNAs showed enhanced transformation properties in vitro and in vivo. These include enhanced colony formation and growth in soft agar. miRNA processing impaired cells injected into nude mice formed tumours that grew both faster and larger than control cells. Protein levels of the oncogenes K-Ras and c-Myc were elevated in the cells with impaired processing compared to control [81]. Since the let-7 family of miRNAs regulate K-Ras [82], this is but one example of the effects of reduced mature miRNA on an oncogene. Unlike the transformed cells, primary cells with experimentally impaired processing did not show the transformed properties and grew slower than wild-type cells. This suggests that reduction in mature miRNA levels is not an initiating event but perhaps an event that maintains the transformation properties.
Lee et al. used real-time PCR assays specific to the miRNA precursors [83, 84] and mature miRNA [85] to expression profile the precursor and mature miRNAs in a number of cancer cell lines, tumours and normal tissues [86]. Quite often, the miRNA precursor was present but not the mature miRNA. For example, the brain-specific miR-128a precursor was present in most of the 22 normal tissues at levels of several hundreds of copies per cell, however the mature miR-128a was expressed only in brain and skeletal muscle [86]. As expected, plenty of examples of pre-miRNAs being processed to mature miRNA exist. This, in effect, rules out the possibility of widespread reduction in the expression of Drosha or Dicer being responsible for reduced mature miRNA as one would expect the reduction in processing to be consistent across all tumours, tissues and cell lines.
The processing of miR-31 was of particular interest [86]. Equivalent miR-31 precursor levels were detectable by real-time PCR and Northern blotting in a number of cancer cell lines. The mature miRNA was detectable by real-time PCR and Northern blotting in some of the cell lines (e.g. HS766T cells) but not others (e.g. MCF-7). In situ hybridization of the mature miR-31 showed the presence of mature miR-31 in the cytoplasm of HS766T cells but was undetectable in the MCF-7 cells. Detection of the pri- and pre-miR-31 with RT in situ PCR demonstrated that the miR-31 precursors were present in both the nucleus and cytoplasm of cells that expressed mature miR-31 (e.g. HS766T) but in cells that lacked mature miR-31 (e.g. MCF-7); the miRNA precursors were confined to the nucleolus [86]. Thus, miR-31 is an example of a miRNA that is processed to the mature form in some cell lines, but in other cell lines it is processed to the pre-miRNA and is not processed to the mature miRNA because it is retained in the nucleolus.
A large-scale comparison was made to determine the Pearson coefficients for correlations between the precursor and mature miRNA expression. This comparison allowed an approximation of the degree to which the miRNA precursors are being processed to mature miRNA. The data demonstrated that the miRNA precursors were processed to a greater extent in pancreas tumours, whereas in hepatocellular cancer a greater percentage of the precursor miRNA were not processed to mature miRNA [86]. In pancreas cancer, there was a predominate increase in the expression of a number of miRNAs [63, 64], whereas in hepatocellular carcinoma, there was a predominate decrease in the miRNA expression [55, 56]. These findings suggest that in pancreatic cancer, the increased expression of miRNA is regulated at the level of transcription or perhaps post-transcriptionally with no apparent reduction in processing, but in hepatocellular carcinoma the reduced expression of miRNA in the tumours may result from a reduction in miRNA processing.
Recently, it was shown that the SMAD proteins (SMAD1, SMAD2 and SMAD3) control processing by Drosha of miR-21 and miR-199a [87]. Human primary pulmonary artery smooth muscle cells exposed to TGF-β or bone morphogenic factor (BMP) displayed increased expression of the precursor and mature forms of miR-21 and miR-199a but not the primary precursor forms of these miRNAs, demonstrating that the increased expression of these mature miRNAs was due to post-transcriptional regulation. The post-transcriptional regulation was due to a specific interaction between the pri-miRNAs, SMAD proteins, p68 and the Drosha complex [87]. The post-transcriptional regulation of pri-miR-21 and pri-miR-199a is independent of SMAD4, the SMAD required for most transcriptional responses to TBF-β and BMP. TGF-β signalling was inhibited in MDA-MB-468 breast cancer cells using a mutated form of the TGF-β type I receptor. MDA-MB-468 cells with impaired TGF-β signalling produced reduced levels of both basal and TGF-β-induced pre- and mature miR-21 but did not alter the pri-miR-21 levels. This study identifies a critic link between the TGF-β family signalling pathways and miRNA biogenesis and suggests that one explanation for increased miR-21 expression present in a number of solid tumours results from increased TGF-β signalling.
Conclusion
This review highlighted the available data that describe situations where alterations in the levels of mature miRNA may be explained by alterations in miRNA processing. This phenomenon occurs in terminally differentiated cells, during development and in at least one human disease, cancer. As reviewed here, several known and unknown mechanisms are responsible for causing reduced miRNA processing (Fig. 1). Known mechanisms include binding of hnRNPs to the pri-miRNA [38], A-to-I editing of the pri-/precursor miRNA [41–43], mutations on the miRNA precursors that likely affect processing [45], the presence of inhibitory proteins that regulate processing [30, 31], altered levels of Dicer and/or Drosha [74, 76], regulation by the TGF-β family signalling pathways [87] and sequestration in different cells types [34]. Because processing proteins (e.g. Drosha, Dicer, DCGR8, etc.) produce global effects on miRNA processing, it is unlikely that situations of altered processing result from altered levels of these factors. More than likely, regulation of processing results from miRNA-specific factors that aid or inhibit processing of the precursors [30–32, 38, 87]. In this way, individual miRNAs may be regulated without affecting global miRNA expression.
A number of important questions await further study. Do inhibitory factors besides Lin-28 exist that regulate the processing of the miRNA precursors? What is the prevalence of SNPs and/or mutations in the miRNA precursors and how do they affect processing? Do hnRNPs modulate processing of miRNAs other than miR-18? Are specific factors present that assist in the intracellular trafficking of miRNA precursors and could alterations in these factors account for altered trafficking within the cell (e.g. reduced cytoplasmic transport). How many miRNAs are regulated post-transcriptionally by the TBF-β/SMAD signalling pathway and what other signalling pathways regulate miRNA processing? How prevalent is the reduction or induction of miRNA processing in human cancers? And finally, do other human diseases exist in which alterations in miRNA processing is a major contributor? Clearly, understanding the mechanisms responsible for alterations in miRNA processing will be an active area of research for years to come.
Acknowledgments
Dr. Schmittgen is supported by grant CA114304–01 from the National Cancer Institute.
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the hete-rochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 3.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 4.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–4. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 5.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–62. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–11. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 8.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 11.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 12.Ohman M. A-to-I editing challenger or ally to the microRNA process. Biochimie. 2007;89:1171–6. doi: 10.1016/j.biochi.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Kim VN, Nam JW. Genomics of microRNA. Trends Genet. 2006;22:165–73. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–83. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–60. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–9. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 18.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 19.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–8. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 20.Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5’ UTR as in the 3’ UTR. Proc Natl Acad Sci USA. 2007;104:9667–72. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–6. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- 22.Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–60. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 23.Humphreys DT, Westman BJ, Martin DI, Preiss T. MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proc Natl Acad Sci USA. 2005;102:16961–6. doi: 10.1073/pnas.0506482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–6. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol. 2005;7:719–23. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauley KM, Eystathioy T, Jakymiw A, Hamel JC, Fritzler MJ, Chan EK. Formation of GW bodies is a consequence of microRNA genesis. EMBO Rep. 2006;7:904–10. doi: 10.1038/sj.embor.7400783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol. 2003;13:807–18. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 28.Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, Strehle M, Seiler A, Schumacher S, Nitsch R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–26. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 29.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–7. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–49. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piskounova E, Viswanathan SR, Janas M, Lapierre RJ, Daley GQ, Sliz P, Gregory RI. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J Biol Chem. 2008;283:21310–4. doi: 10.1074/jbc.C800108200. [DOI] [PubMed] [Google Scholar]
- 33.Tang GQ, Maxwell ES. Xenopus microRNA genes are predominantly located within introns and are differentially expressed in adult frog tissues via post-transcriptional regulation. Genome Res. 2008;18:104–12. doi: 10.1101/gr.6539108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lugli G, Torvik VI, Larson J, Smalheiser NR. Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J Neurochem. 2008;106:650–61. doi: 10.1111/j.1471-4159.2008.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006;12:1161–7. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leuschner PJ, Martinez J. In vitro analysis of microRNA processing using recombinant Dicer and cytoplasmic extracts of HeLa cells. Methods. 2007;43:105–9. doi: 10.1016/j.ymeth.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 37.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol. 2007;14:591–6. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 39.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 40.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–7. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8:763–9. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9:112–23. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 46.Iwai N, Naraba H. Polymorphisms in human pre-miRNAs. Biochem Biophys Res Commun. 2005;331:1439–44. doi: 10.1016/j.bbrc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 47.Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16:1124–31. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 48.Barbarotto E, Schmittgen TD, Calin GA. MicroRNAs and cancer: profile, profile, profile. Int J Cancer. 2008;122:969–77. doi: 10.1002/ijc.23343. [DOI] [PubMed] [Google Scholar]
- 49.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 50.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michael MZ, SM OC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–91. [PubMed] [Google Scholar]
- 52.Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol Rep. 2006;16:845–50. [PubMed] [Google Scholar]
- 53.Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007;98:1914–20. doi: 10.1111/j.1349-7006.2007.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 55.Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, Roberts LR, Schmittgen TD. Association of microRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res. 2008;14:419–27. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L, Negrini M. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–9. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 57.Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Comprehensive analysis of microRNA expression patterns in hepato-cellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–45. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 58.Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, Jacob ST, Ghoshal K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99:671–8. doi: 10.1002/jcb.20982. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–36. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 61.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–58. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 63.Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD. Expression profiling identifies microRNA signature in pancreatic cancer. Int. J. Cancer. 2007;120:1046–54. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C, Croce CM. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–8. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 65.He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, Kloos RT, Croce CM, de la Chapelle A. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA. 2005;102:19075–80. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makunin IV, Pheasant M, Simons C, Mattick JS. Orthologous microRNA genes are located in cancer-associated genomic regions in human and mouse. PLoS ONE. 2007;2:e1133. doi: 10.1371/journal.pone.0001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–9R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 70.Yang N, Coukos G, Zhang L. MicroRNA epigenetic alterations in human cancer: one step forward in diagnosis and treatment. Int J Cancer. 2008;122:963–8. doi: 10.1002/ijc.23325. [DOI] [PubMed] [Google Scholar]
- 71.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–5. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 72.Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, Lee J, Fine H, Chiocca EA, Lawler S, Purow B. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–72. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 73.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 74.Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, Yatabe Y, Takamizawa J, Miyoshi S, Mitsudomi T, Takahashi T. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–5. doi: 10.1111/j.1349-7006.2005.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Schneider A, Arias-Pulido H, Murty VV. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008;7:58. doi: 10.1186/1476-4598-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, Dhir R. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–20. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–5. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 78.Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27:1788–93. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 79.Chiosea S, Jelezcova E, Chandran U, Luo J, Mantha G, Sobol RW, Dacic S. Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res. 2007;67:2345–50. doi: 10.1158/0008-5472.CAN-06-3533. [DOI] [PubMed] [Google Scholar]
- 80.Chiosea SI, Barnes EL, Lai SY, Egloff AM, Sargent RL, Hunt JL, Seethala RR. Mucoepidermoid carcinoma of upper aerodigestive tract: clinicopathologic study of 78 cases with immunohistochemical analysis of dicer expression. Virchows Arch. 2008;452:629–35. doi: 10.1007/s00428-007-0574-5. [DOI] [PubMed] [Google Scholar]
- 81.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–7. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 82.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–47. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 83.Schmittgen TD, Jiang J, Liu Q, Yang L. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 2004;32:E43. doi: 10.1093/nar/gnh040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang J, Lee EJ, Gusev Y, Schmittgen TD. Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 2005;33:5394–403. doi: 10.1093/nar/gki863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA. 2007;14:35–42. doi: 10.1261/rna.804508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]