

Abstract
Escherichia coli muk mutants are temperature-sensitive and produce anucleate cells. A spontaneously occurring mutation was found in a ΔmukB∷kan mutant strain that suppressed the temperature-sensitive phenotype and mapped in or near topA, the gene that encodes topoisomerase I. Previously characterized topA mutations, topA10 and topA66, were found to be general suppressors of muk mutants: they suppressed temperature sensitivity and anucleate cell production of cells containing null or point mutations in mukB and null mutations in mukE or mukF. The suppression correlated with excess negative supercoiling by DNA gyrase, and the gyrase inhibitor, coumermycin, reversed it. Defects in topA allow 99% of cell division events in muk null mutants to proceed without chromosome loss or loss of cell viability. This observation imposes important limitations on models for Muk activity and is consistent with a role for MukBEF in chromosome folding and DNA condensation.
Accurate segregation of the Escherichia coli chromosome requires, among other factors, the action of the three muk gene products, MukB, -E, and -F (1, 2). Null muk mutants produce anucleate cells ≈100-fold more frequently than the wild type, and many cells have abnormal chromosome positioning and/or DNA content. All muk mutants are temperature sensitive (ts) for growth and form filaments before growth cessation (1–3). Suppressor mutations of mukB mutations (smb) have been isolated, but suppress only the temperature sensitivity, smbA (4), or act by overproducing the mutant mukB protein (5). A mutation in era, a gene encoding an essential GTPase involved in the E. coli cell cycle, was also found to suppress partially the temperature sensitivity of the mukB null mutation (6). Multicopy suppressors, cspC and cspE, suppress the ts and anucleate cell formation phenotypes of mukB106 (3). The cspE effect requires overexpression of the wild-type cspE, crcA, and crcB genes, which have been implicated in chromosome condensation (7).
MukB resembles eukaryotic force-generating proteins, with an extended coiled-coil domain joining N-terminal ATP/GTP binding and C-terminal DNA binding domains (8). MukB was therefore suggested as a candidate for a segregation motor (1, 8). The structure (but not the sequence) of MukB is also similar to SMC proteins (7, 9), which are involved in DNA condensation and chromosome segregation in eukaryotes (10–12). Smc proteins have recently been discovered in Bacillus subtilis and Caulobacter crescentus, and smc mutants of these species have phenotypes similar to mukB mutants of E. coli (13–15). E. coli MukB and B. subtilis Smc share a feature not found in motor proteins such as the kinesins and myosins: the dimers are in an antiparallel configuration (9).
The 13S condensin of Xenopus is a five-protein complex containing two proteins in the SMC family. It introduces a global (+) writhe into DNA (12, 16) and may act by nonplanar bending of the DNA into coils that remodel the chromosome into a compact structure (16). The E. coli chromosome is organized into ≈50 negatively supercoiled domains (17), and its compact structure depends on bound protein and RNA (18). We present evidence suggesting that MukB, in conjunction with the MukE and MukF proteins, may play a somewhat similar role to that proposed for 13S condensin in remodeling the nucleiod structure into a more compact form as a requirement for accurate segregation.
Materials and Methods
Bacterial Strains, Phage, and Plasmids.
Strains GC7528, ΔmukB∷kan dadR trpE61 trpA62 tna-5 immφ80 (1); SH9019, mukB33 zcb∷Tn10 (3); AZ5381, mukF∷kan, AZ5450, mukE∷kan (2); CAG5053, KL208: PO43 relA1 zbc-280∷Tn10, CAG18455, trpB83∷Tn10, CAG12094, zcb-3059∷Tn10 (19); DPB923, topA10, DPB924, topA+, and DPB636, topA66 zch-2250∷mini-kan (20) were as described.
Strain CC3955 is recA56 lacZam (λ cI857 recA+). CC3974 is a rec+ recombinant derived from CC3955 that was transduced to ΔmukB∷kan by a P1 lysate grown on GC7528, cured of λ by heat pulse, and lysogenized by λ xis6 ind−. It was subsequently found to have acquired an immφ80 prophage from GC7528 and a spontaneous mutation linked to topA. This strain was transduced (21, 22) to muk+ zcb-3059∷Tn10 by selecting a tetracycline-resistant (Tcr), kanamycin-sensitive (Kms) colony to give CC3976, lacZam zcb-3059∷Tn10 (λ xis6 ind−). CC3977, lacZam rpsL (λ xis6 ind−), is a Tcs derivative made as described (23). Both CC3976 and CC3977 have the immφ80 prophage and putative topA mutation. DPB923 and DPB924 gave rise to CC4207 and CC4208, respectively, by transduction to ΔmukB∷kan using a P1 lysate grown on CC3979 (MC4100 ΔmukB∷kan), kindly supplied by Nancy Trun (National Cancer Institute). CC4209, CC4210, CC4213, and CC4214 are pAX804 transformants of DPB923, DPB924, CC4207, and CC4208, respectively. CC4209 and CC4210 were transduced with P1 lysates grown on AZ5381 or AZ5450 to produce CC4211, mukF∷kan topA10 [pAX804]; CC4212, mukF∷kan [pAX804]; CC4217, mukE∷kan topA10 [pAX804]; and CC4218, mukE∷kan [pAX804], respectively. CC4221, mukB33 topA10 zcb∷Tn10; and CC4222, mukB33 zcb∷Tn10 were made by transducing DPB923 and DPB924 with P1 grown on SH9019. CC4220, mukB33 zcb∷Tn10, was derived from CC4208 by transduction with P1 grown on SH9019, and three independent topA66 transductants, CC4223–4225, were made by transduction of CC4220 using a P1 lysate grown on DPB636. CC4226 is a mukB33 zch-2250∷mini-kan zcb∷Tn10 (topA+) transductant.
λ cI857 recA+ contains the wild-type recA gene and promoter and was kindly provided by Frank Stahl (University of Oregon). The P1 miniplasmids λ-P1:5R and λ-P1:5RparB34 (24) were as described, as was λ-P1:5RΔ1005∷pALA1952 (25). λ-P1:5RΔ1005∷pALA1991 is similar to the latter but lacks the parS site. Plasmid pALA1413 produces the proper levels of ParA and ParB for accurate P1 partition (25) and pAX804 carries the wild-type mukB gene (1). The mukB alleles in wild-type or topA mutant strains were confirmed by using standard PCR techniques, screening for the increased length of the ΔmukB∷kan gene or the SspI site introduced by mukB33.
Media.
LB-broth and plates were as described (26) but with NaCl at 5 g/liter. Ampicillin (100 μg/ml), tetracycline (12.5 μg/ml), chloramphenicol (2.5 μg/ml or 10 μg/ml), streptomycin (100 μg/ml), kanamycin (50 μg/ml), or tetracycline (6 μg/ml) plus sodium citrate (5 mM) were used as needed. Coumermycin A1 (coumermycin, Sigma) at 5 mg/ml in DMSO was added as needed.
Determining the topA Status of Cells.
Alleles of topA were confirmed by their ability to suppress par mutant plasmids (27, 28). Additionally, pBR322 plasmid DNA introduced into the cells that were grown to an optical density (OD600) of ≈1.0 in LB broth at 23°C. The DNA was extracted from the cells and was analyzed on 1% agarose gels containing chloroquine at 5 or 15 μg/ml. Gels were run at ≈1.6 V/cm with recirculated TA buffer (29) containing chloroquine for ≈40 hr at 4°C.
Mapping the Suppressor Mutation.
Initial crosses were carried out in the presence of 5 mM sodium citrate at 42°C to prevent reinfection by the immφ80 phage (30) and were designed to remove the prophage at the φ80 attachment site. CC3977 was mated with the Hfr strain, CAG5053 (KL208: PO43 zbc-280∷Tn10), selecting for Tcr and against the donor with streptomycin resistance (19). Forty-seven of one hundred exconjugants had lost immφ80. Eight of these retained the lacZam and immλ markers needed for the P1 partition assay (25). None of the eight retained the suppressor of P1 Par−, suggesting that the suppressor locus was linked to the φ80 attachment site. P1 transduction (21, 22) of Tcr from CAG18455 (trpB83∷Tn10: a Tcr insertion linked to attφ80) to CC3977 to give Tcr at 42°C resulted in 11 of 19 transductants losing the P1par suppressor mutation. Five of the eleven retained immφ80. Thus, the suppressor mutation is ≈58% linked to the trpB83∷Tn10 on the side opposite to attφ80, in the vicinity of the topA gene (≈51% linked to the trpB83∷Tn10 marker).
Mini-P1 Partition Assays.
Mini-P1 partition assays were carried out by measuring retention of the plasmid during 25 generations of unselected growth. Tests used λ-P1:5R (Par+) or λ-P1:5RparB34 as described (31), or the colony color test that measures the maintenance of λ-P1:5RΔ1005∷pALA1952 (parS+) or λ-P1:5RΔ1005∷pALA1991 (no parS) when P1 ParA and ParB are supplied in trans from pALA1413 (25).
Growth of Cells in Coumermycin.
Fresh single colonies were inoculated into LB broth at 23°C, were grown to an optical density (OD600) of 0.1, were diluted 25-fold, and were grown for 1 hour. Coumermycin or the equivalent amount of DMSO was then added at the indicated concentrations, and growth (OD600) was monitored at 30-min intervals for 6–8 hours. Doubling times were determined from the growth occurring after the 4-hour time point because there appeared to be a lag period before the full effect of the drug was seen (data not shown). Cells for microscopy were grown from a single colony at 23°C with coumermycin at a concentration of 1 μg/ml. Samples were taken after 6 hours and were 4′,6-diamidino-2-phenylindole-stained as outlined below.
Microscopy.
Cells for microscopy were grown for a minimum of 10 generations of balanced growth and were stained as described (7). Cells were viewed with a 100/1.25× plan objective on a Nikon Eclipse E600 microscope using a combination of UV and visible (phase contrast) light. Photographs were taken with a Nikon FDX-35 camera with Kodak 1000 color print film.
Results
A mutation that Suppresses a P1 Plasmid Partition Defect also Suppresses mukB Temperature Sensitivity.
Defects in the P1 plasmid par genes increase the plasmid loss rate some 100-fold (24). However, a ΔmukB∷kan mutant strain, CC3974, maintained a par mutant mini-P1 plasmid considerably better than its Muk+ progenitor, CC3955 (Table 1). The partial suppression was not due to the ΔmukB∷kan allele itself because other ΔmukB∷kan mutant strains did not show this effect (GC7528, Table 1). Moreover, the suppressor effect persisted on replacing the ΔmukB∷kan allele of CC3974 by the wild-type mukB+ allele (CC3977, Table 1). The suppressor mutation mapped in or near topA, the gene for topoisomerase I (Materials and Methods). Mutations in topA are known to promote the stability of partition-defective P1 and F mini-plasmids (27, 28).
Table 1.
Plasmid stability of mini-P1 in mukB mutant cells
| Relevant genotype (strain number) | Percent retention of P1 miniplasmid, 25 generations
|
|
|---|---|---|
| Mini-P1Par+ | Mini-P1Par− | |
| ΔmukB∷kan (GC7528) | 87 | 1 |
| mukB+ (CC3955) | 86 | 2 |
| ΔmukB∷kan topA? (CC3974) | 93 | 32 |
| ΔmukB∷kan topA? (CC3974)* | 78 | 35 |
| mukB+topA? (CC3977)* | 90 | 34 |
*Colony color partition tests. See Materials and Methods.
As the putative topA mutation arose spontaneously in strain CC3974, we reasoned that it might confer an advantage on the strain and may have been inadvertently selected. Consistent with this, CC3974 grew well at 37°C on LB plates whereas other ΔmukB∷kan mutant strains such as GC7528 failed to grow at 30°C or above. It seemed probable that a topA mutation could suppress both the P1 partition defect and mukB temperature sensitivity.
Suppression of muk Mutant Temperature-Sensitivity by Known topA Alleles.
The topoisomerase I activity of topA10 is ≈1% of wild-type (32). Isogenic topA10 ΔmukB∷kan and ΔmukB∷kan mutant cells were constructed at 23°C. The ΔmukB∷kan mutant failed to grow at 30°C and above unless plasmid pAX804 (1), containing the wild-type mukB gene, was introduced. However, the ΔmukB∷kan topA10 double mutant cells grew at all temperatures between 23°C and 42°C (Fig. 1). Temperature sensitivity of a point mutation in mukB, mukB33, was also substantially suppressed by topA10 (data not shown), as were null mutants in mukE (data not shown) and mukF (Fig. 1). The suppressed cells were fully viable at the higher temperatures but grew somewhat more slowly than wild-type cells (data not shown). The topA66 mutation also suppressed muk mutant temperature-sensitivity (data not shown).
Figure 1.

Suppression of muk mutant temperature sensitivity. Colonies grown at 23°C were streaked onto LB-agar plates at the indicated temperatures and were grown for 24 hours. Strain numbers starting with “WT” and proceeding clockwise are DPB924, CC4211, CC4213, CC4207, DPB923, CC4212, CC4214, and CC4208. CC4211, CC4212, CC4213, and CC4214 contain the plasmid pAX804 that produces wild-type MukB protein. At 30°C, the wild-type and topA10 and topA10 ΔmukB∷kan mutant strains had generation times of 35, 34, and 43 min, respectively, whereas the ΔmukB∷kan strain failed to grow.
Anucleate Cell Formation of muk Mutants Is Suppressed by topA10.
The ΔmukB∷kan mutant cultures contained ≈12% anucleate cells at 23°C (Table 2 and Fig. 2C; refs. 33 and 34). Only 1% of ΔmukB∷kan topA10 double mutant cells were scored as anucleate (Table 2, Fig. 2D), representing only a small increase over the proportion of anucleate cells seen in wild-type cultures (≈0.3%, Table 2). The topA10 mutation also suppressed anucleate cell formation of mukB33 15-fold (Table 2). The mukB null and point mutants produce a number of filamentous cells at 23°C (4–10%), and the topA10 mutation also suppressed this phenotype (Fig. 2).
Table 2.
Microscopic observation of nucleoids of muk mutant cells
| Strain: genotypePercent anucleate cells | |
| DPB924: wild-type | 0.3 |
| DPB923: topA10 | <0.3 |
| CC4208: ΔmukB∷kan | 12.4 |
| CC4207: topA10 ΔmukB∷kan | 1.0 |
| CC4220: mukB33 | 14.5 |
| CC4221: topA10 mukB33 | <0.2 |
| CC4223: topA66 mukB33* | 1.0 |
| CC4218: mukE∷kan | 19.0 |
| CC4217: topA10 mukE∷kan | 1.0 |
| CC4212: mukF∷kan | 19.0 |
| CC4211: topA10 mukF∷kan | 2.1 |
Cells were grown in LB broth at 23°C. From 375–2,500 cells were scored for each strain, and those with little or no 4′, 6-diamidino-2-phenylindole staining were considered “anucleate.”
Of three topA66 mutant transductants examined, all behaved identically. One topA+ transductant from the same cross, CC4226, gave similar results to CC4220.
Figure 2.

Anucleate cell formation in muk mutant cells. Cells were grown in LB broth at 23°C. (A) DBP924 wild-type. (B) DPB923, topA10. (C) CC4208, ΔmukB∷kan. (D) CC4207, topA10 ΔmukB∷kan. (E) CC4218, mukE∷kan. (F) CC4217, topA10 mukE∷kan mutant cells. White arrows highlight some anucleate cells. Black arrows highlight filamentous cells with “DNA-free bays.”
Anucleate cells and filaments are also produced by mukF∷kan and mukE∷kan mutants and are a direct effect of the disruption of the respective gene rather than of polarity on downstream genes (2). On transferring these mutations into topA10 mutant cells, both anucleate (Table 2) and filamentous cell formation was suppressed. Thus, topA10 can suppress the temperature sensitivity, anucleate cell production, and filamentation of all muk mutations tested.
Suppression of mukB in topA Mutant Strains Correlates with Increased Negative Supercoiling.
Defects in topA increase negative DNA supercoiling (32) because of the unrestrained activity of DNA gyrase. As this can limit growth, mutants such as topA10 can acquire compensating DNA gyrase mutations upon propagation (35). Thus, the resulting strains may not have excess negative supercoils. Plasmid pBR322 was introduced into the relevant strains and was reisolated from cells grown at 23°C, and the degree of supercoiling was estimated by electrophoresis in agarose gels containing chloroquine (Materials and Methods). The wild-type strain and its ΔmukB∷kan derivative showed indistinguishable levels of plasmid supercoiling (Fig. 3). However, plasmid DNA from the suppressed ΔmukB∷kan topA10 strain, CC4207, showed evidence of considerably increased negative supercoiling and was indistinguishable from that of the topA10 parent, DPB923 (Fig. 3). Virtually identical results were obtained on analysis of the mukE∷kan mutant strain and its suppressed derivative (Fig. 3) and of mukB33 strains suppressed by topA66 (data not shown). Thus, suppression of the muk mutants correlates with increased negative supercoiling. Presumably, suppression is a direct effect of the increase in negative supercoiling induced by the defects in topA.
Figure 3.

Supercoiling of plasmid pBR322 isolated from wild-type and topA10 mutant cells. A 1% agarose gel containing 5 μg/ml chloroquine was used to separate topoisomers, as outlined in Materials and Methods. More highly supercoiled species migrate further than less supercoiled species. The change in the topoisomer ladders seen when 15 μg/ml chloroquine was used (data not shown) indicates that all of the topoisomers produced from the topA and topA muk mutant cells are negatively supercoiled species in this gel. Likewise, most if not all of the bands produced by the top+ cells are negatively supercoiled. Thus, the topA suppressed cells produce plasmid DNA that contains on average nine or more additional negative supercoils relative to their top+ parents. DNA was isolated from pBR322 transformants of DPB924, DPB923, CC4207, CC4208, CC4215, and CC4216, from left to right, respectively.
The topA66 allele does not readily acquire secondary mutations (20). When this topA allele was introduced into mukB33 mutant cells by generalized transduction, it suppressed the ts phenotype (data not shown) and anucleate cell production (Table 2) of the mukB33 strain equally well in three separate isolates. This result is also consistent with suppression being a direct effect of the topA mutation rather than the acquisition of secondary mutations.
A DNA Gyrase Inhibitor Reverses the Suppression of ΔmukB∷kan by topA10.
If increased negative supercoiling is responsible for muk mutant suppression, inhibition of DNA gyrase should reverse it. The effect of low levels of the DNA gyrase inhibitor, coumermycin (36), on the ΔmukB∷kan suppressed cells was tested. With no coumermycin, the ΔmukB∷kan mutant grew slowly and only at 23°C (110-min doubling time) whereas the ΔmukB∷kan topA10 double mutant had a similar growth rate to wild-type cells (89- vs. 82-min doubling time at 23°C) and was not temperature-sensitive (Fig. 4). Coumermycin at low concentrations (e.g., 5 μg/ml) had no significant effect on the growth or temperature resistance of wild-type or topA10 mutant cells, and they grew at all temperatures from 23°C and 42°C (data not shown). However, the drug strongly inhibited the growth of topA10 ΔmukB∷kan double mutant cells (>170-min doubling time at 23°C), and the cells failed to grow above 23°C even at coumermycin concentrations as low as 1 μg/ml (Fig. 4). Thus, when DNA gyrase is partially inhibited, the suppression by topA is reversed, and the cells show the same low growth rate and temperature sensitivity as the unsuppressed ΔmukB∷kan mutant cells. Note also that ΔmukB∷kan mutant cells are hypersensitive to coumermycin (Fig. 4), such that they fail to grow at 23°C with only 1 μg/ml coumermycin. This suggests that these cells are critically dependent on supercoiling levels for survival.
Figure 4.

Temperature-resistance of topA10 ΔmukB∷kan is reversed by coumermycin. Cells were streaked out on prewarmed plates containing DMSO (controls) or coumermycin at 1 μg/ml at the indicated temperatures. 30°C plates were incubated for 24 hours, and 23°C plates were incubated for 48 hours. Strains were DPB924, wild type (a), DPB923, topA10 (b), CC4207, topA10 ΔmukB∷kan (c), and CC4208, ΔmukB∷kan (d).
As shown above, ≈1% of topA10 ΔmukB∷kan cells are anucleate in the absence of the drug. However, in the presence of 1 μg/ml coumermycin, many of the cells were anucleate, and the cells showed the frequent filamentation and nucleoid disorganization typical of the parent ΔmukB∷kan strain (Fig. 5B). The topA10 cells were unaffected by coumermycin at this concentration (Fig. 5A), as were wild-type cells (data not shown). We conclude that partial inhibition of DNA gyrase reverses topA-mediated suppression of mukB mutant cells for all three of the measured MukB phenotypes. This strongly suggests that the excess negative supercoiling in the topA mutants is responsible, directly or indirectly, for the suppression.
Figure 5.
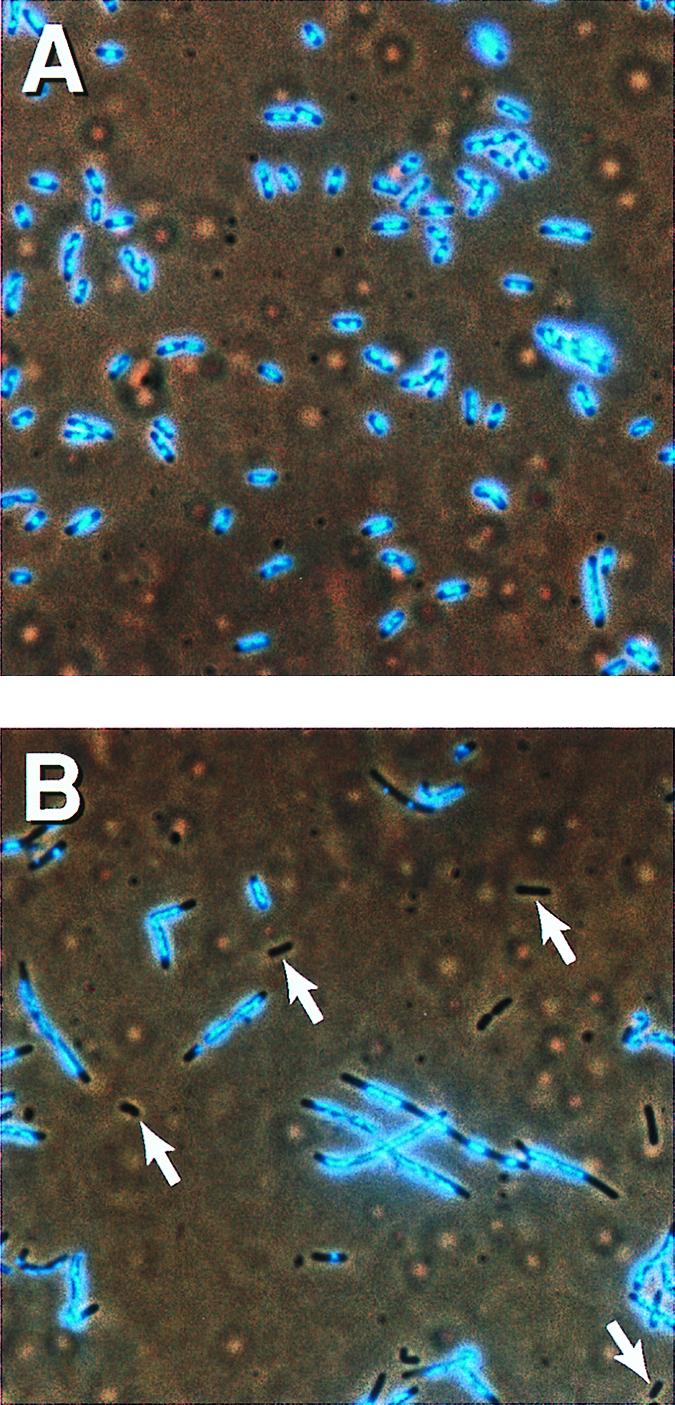
Coumermycin reverses suppression of ΔmukB∷kan by topA10. Cells were grown with 1 μg/ml coumermycin for 6 hours and were 4′,6-diamidino-2-phenylindole-stained for microscopy. (A) DPB923, topA10. (B) CC4207, topA10 ΔmukB∷kan. White arrows highlight some examples of anucleate cells.
Discussion
When topoisomerase I activity was limited by mutation, the chromosome segregation defect of all muk mutants tested was suppressed. This suppression appears to be the result of the excess negative supercoils in the DNA because it is completely reversed by partial inhibition of DNA gyrase. In suppressing conditions, chromosome segregation proceeded successfully in 99% of cells without Muk function. This imposes important limitations on the possible roles for the Muk proteins. The resemblance of MukB to eukaryotic motor proteins and the phenotype of mutant cells suggested that the MukB protein might actively segregate E. coli chromosomes (8). However, it is difficult to imagine how an increase in negative supercoiling could so effectively compensate for loss of such a function. We favor an alternative idea: that the Muk system is required for condensation and organization of the chromosome into a compact structure that is important for proper segregation, and that this activity can be substituted for, in large part, by increased negative supercoiling. Consistent with this, we found that Muk mutants are hypersensitive to coumermycin. This suggests that, in the absence of the Muk system, the supercoiling density of the chromosome is in a critical range such that an increase is beneficial for segregation but a decrease is lethal.
Recent views of the mechanism for bacterial chromosome segregation are largely based on microscopic observations (37–43). Studies on the localization of origins and their associated proteins suggested a polar attachment and condensation mechanism for chromosome segregation (42). The probable location of the chromosome replication forks at the cell center further suggested a modified model in which segregation is concurrent with replication, and origins are attached to new cell division sites rather than at the cell pole (Fig. 6; ref. 43). Attachment of the replication forks to the cell center and transport of the newly replicated origins to the 1/4 and 3/4 positions in the cell should be specific and fundamental elements of the segregation process (Fig. 6). Without these events, chromosome segregation should fail, and restoration of segregation by increased negative supercoiling seems unlikely. Therefore, we do not regard the Muk proteins as good candidates for the essential effectors of these processes. The Muk system does, however, seem to be a good candidate for the condensation and folding of the nucleoids that provide the motive force for moving the bulk of the DNA during segregation in these models. The action of DNA gyrase in forming independently supercoiled loops in the newly synthesized DNA (17) is presumably critical for this step. Certain mutants defective in DNA gyrase activity are blocked for chromosome segregation (44), likely as a result of reduced negative supercoiling (45). We suggest that the MukBEF proteins are also involved in DNA condensation and folding. Perhaps they aid in the higher order organization of DNA into the compact nucleoid structure, somewhat along the lines proposed for the 13S condensin of Xenopus (16). Coiling or some other form of condensation could be necessary to provide sufficient space between the daughter nucleoids to permit cell division (Fig. 6). An increase in negative supercoiling is expected to make the DNA considerably more compact (46). Suppression of muk mutations by topA mutations could be explained if the excess negative supercoiling found in topA mutant cells results in sufficient compaction of the nucleoid to make further organization by the Muk system largely unnecessary (Fig. 6F). However, we have not ruled out that the change in supercoiling in topA mutant cells increases the expression of other genes involved in nucleoid condensation, thus achieving the same effect.
Figure 6.

Directed condensation model for E. coli chromosome segregation. The model is based on that of Lemon and Grossman (43). (A) The nucleoid is supercoiled and further condensed to some unspecified compact form (represented as stacked loops). During initiation, the origin (circle) is attached to a site (triangles) at the cell center. (B) The new origins (circles) attach to new sites at the 1/4 and 3/4 positions. The DNA replication forks remain attached to the central site. (C) Newly replicated DNA (black lines) is supercoiled by DNA gyrase and remodeled into the compact form by the action of the Muk proteins. As the origins are tethered in opposite halves of the cell, the nascent nucleoids form as separate masses. (D) On completion of replication, the daughter nucleoids are sufficiently separated to allow cell division. (E) In the absence of the Muk system, the supercoiled loops of the completed nucleoids are disorganized and the resulting masses fail to separate. (F) The additional negative supercoiling caused by unrestrained DNA gyrase activity in topA mutant cells increases superhelical density and allows for a more reliable separation.
Apart from the resemblance of MukB to SMC proteins, additional observations suggest a chromosome folding or DNA condensation role for Muk proteins. First, muk mutant chromosomes appear decondensed by microscopy (refs. 1 and 8; Fig. 2), and the presence of the suppressing topA allele appears to favor a more compact form (Fig. 2). Second, simultaneous overexpression of crcA, crcB, and cspE suppresses at least one mukB point mutation, and these small proteins are implicated in chromosome condensation (7).
If muk mutant cells lack a DNA condensation activity, how can the anucleate cell production of these mutants be explained by the model in Fig. 6? Tangling of the nascent daughter nucleoids might occasionally hinder the attachment of origin regions to their appropriate division sites, thus causing sporadic missegregation. Additionally, lack of proper DNA condensation might leave some of the daughter nucleoids overlapping and blocking the plane of cell division (Fig. 6E). Anucleate cells might then arise by breakage of one or both chromosomes during septation (“guillotining”), followed by exonucleolytic digestion of the broken chromosome. Some cells in muk mutant cultures do appear to have partial nucleoids (refs. 1 and 2; data not shown). In other cases, the failure of chromosomes to separate properly might inhibit normal cell septation, leading to the production of the filamentous cells that are seen in cultures of all muk mutants (Fig. 2; Table 2). These often contain DNA-free bays, usually at one end (Fig. 2). Abnormal divisions near these ends would produce anucleate cells. Recent observations of the location of FtsZ cell division rings in mukB mutant cells suggest that at least some anucleate cells may be produced by divisions near the DNA-free ends of filamentous cells (33).
In conclusion, our results are consistent with a chromosome condensation or folding role for the Muk proteins. Increased negative supercoiling of the DNA appears to provide a Muk-independent route to adequate chromosome condensation, either as a direct effect on DNA topology or via some change in gene expression that aids condensation.
Acknowledgments
We thank Don Biek, Sota Hiraga, Mitchell Singer, Frank Stahl, and Nancy Trun for providing strains, phage, and plasmids. We thank Rob Britton and Don Court for critical reading of the manuscript. This research was sponsored by the National Cancer Institute, Department of Health and Human Servies, under contract with Advanced BioScience Laboratories. The contents of this publication do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. J.A.S. was also supported by National Institutes of Health National Research Service Award Fellowship No. 5 F32 GM16971.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030528397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030528397
References
- 1.Niki H, Jaffé A, Imamura R, Ogura T, Hiraga S. EMBO J. 1991;10:183–193. doi: 10.1002/j.1460-2075.1991.tb07935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamanaka K, Ogura T, Niki H, Hiraga S. Mol Gen Genet. 1996;250:241–251. doi: 10.1007/BF02174381. [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka K, Mitani T, Feng J, Ogura T, Niki H, Hiraga S. FEMS Microbiol Lett. 1994;123:27–32. doi: 10.1111/j.1574-6968.1994.tb07196.x. [DOI] [PubMed] [Google Scholar]
- 4.Yamanaka K, Ogura T, Niki H, Hiraga S. J Bacteriol. 1992;174:7517–7526. doi: 10.1128/jb.174.23.7517-7526.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kido M, Yamanaka K, Mitani T, Niki H, Ogura T, Hiraga S. J Bacteriol. 1996;178:3917–3925. doi: 10.1128/jb.178.13.3917-3925.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Britton R A, Powell B S, Dasgupta S, Sun Q, Margolin W, Lupski J R, Court D L. Mol Microbiol. 1998;27:739–750. doi: 10.1046/j.1365-2958.1998.00719.x. [DOI] [PubMed] [Google Scholar]
- 7.Hu K H, Liu E, Dean K, Gingras M, DeGraff W, Trun N J. Genetics. 1996;143:1521–1532. doi: 10.1093/genetics/143.4.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niki H, Imamura R, Kitaoka M, Yamanaka K, Ogura T, Hiraga S. EMBO J. 1992;11:5101–5109. doi: 10.1002/j.1460-2075.1992.tb05617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melby T E, Ciampaglio C N, Briscoe G, Erickson H P. J Cell Biol. 1998;142:1595–1604. doi: 10.1083/jcb.142.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koshland D, Strunnikov A. Annu Rev Cell Dev Biol. 1996;12:305–333. doi: 10.1146/annurev.cellbio.12.1.305. [DOI] [PubMed] [Google Scholar]
- 11.Hirano T, Mitchison T J, Swedlow J R. Curr Opin Cell Biol. 1995;7:329–336. doi: 10.1016/0955-0674(95)80087-5. [DOI] [PubMed] [Google Scholar]
- 12.Kimura K, Hirano T. Cell. 1997;90:625–634. doi: 10.1016/s0092-8674(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 13.Britton R, Lin D, Grossman A. Genes Dev. 1998;12:1254–1259. doi: 10.1101/gad.12.9.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moriya S, Tsujikawa E, Hassan A K M, Asai K, Kodama T, Ogasawara N. Mol Microbiol. 1998;29:179–187. doi: 10.1046/j.1365-2958.1998.00919.x. [DOI] [PubMed] [Google Scholar]
- 15.Jensen R B, Shapiro L. Proc Natl Acad Sci USA. 1999;96:10661–10666. doi: 10.1073/pnas.96.19.10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura K, Rybenkov V V, Crisona N J, Hirano T, Cozzarelli N R. Cell. 1999;98:239–248. doi: 10.1016/s0092-8674(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 17.Sinden R R, Pettijohn D E. Proc Natl Acad Sci USA. 1981;78:224–228. doi: 10.1073/pnas.78.1.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pettijohn D E. In: Escherichia coli and Salmonella cellular and Molecular Biology. Neidhardt F C, Curtiss R III, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 158–166. [Google Scholar]
- 19.Singer M, Baker T A, Schnitzler G, Deischel S M, Goel M, Dove W, Jaacks K J, Grossman A D, Erickson J W, Gross C A. Microbiol Rev. 1989;53:1–24. doi: 10.1128/mr.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biek D P, Cohen S N. J Bacteriol. 1989;171:2066–2074. doi: 10.1128/jb.171.4.2066-2074.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 22.Willetts N S, Clark A J, Low B. J Bacteriol. 1969;97:244–249. doi: 10.1128/jb.97.1.244-249.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maloy S R, Nunn W D. J Bacteriol. 1981;145:1110–1112. doi: 10.1128/jb.145.2.1110-1111.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman S A, Austin S J. Plasmid. 1988;19:103–112. doi: 10.1016/0147-619x(88)90049-2. [DOI] [PubMed] [Google Scholar]
- 25.Radnedge L, Davis M A, Austin S J. EMBO J. 1996;15:1155–1162. [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Austin S J, Eichorn B G. J Bacteriol. 1992;174:5190–5195. doi: 10.1128/jb.174.16.5190-5195.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller C A, Beaucage S L, Cohen S N. Cell. 1990;62:127–133. doi: 10.1016/0092-8674(90)90246-b. [DOI] [PubMed] [Google Scholar]
- 29.Haniford D B, Pulleyblank D E. Nature (London) 1983;302:632–634. doi: 10.1038/302632a0. [DOI] [PubMed] [Google Scholar]
- 30.Signer E R, Beckwith J R. J Mol Biol. 1966;22:33–51. doi: 10.1016/s0022-2836(66)80003-7. [DOI] [PubMed] [Google Scholar]
- 31.Martin K A, Friedman S A, Austin S J. Proc Natl Acad Sci USA. 1987;84:8544–8547. doi: 10.1073/pnas.84.23.8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sternglanz R, DiNardo S, Voelkel K A, Nishimura Y, Hirota Y, Becherer K, Zumstein L, Wang J C. Proc Natl Acad Sci USA. 1981;78:2747–2751. doi: 10.1073/pnas.78.5.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q, Yu X-C, Margolin W. Mol Microbiol. 1998;29:491–503. doi: 10.1046/j.1365-2958.1998.00942.x. [DOI] [PubMed] [Google Scholar]
- 34.Hiraga S, Ichinose C, Hironori N, Yamazoe M. Mol Cell. 1998;1:381–387. doi: 10.1016/s1097-2765(00)80038-6. [DOI] [PubMed] [Google Scholar]
- 35.DiNardo S, Voekel K A, Sternglanz R, Reynolds A E, Wright A. Cell. 1982;31:43–51. doi: 10.1016/0092-8674(82)90403-2. [DOI] [PubMed] [Google Scholar]
- 36.Gellert M, O'Dea M H, Itoh T, Tomizawa J. Proc Natl Acad Sci USA. 1976;73:4474–4478. doi: 10.1073/pnas.73.12.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon G S, Sitnikov D, Webb C D, Teleman A, Straight A, Losick R, Murray A W, Wright A. Cell. 1997;90:1113–1121. doi: 10.1016/s0092-8674(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 38.Niki H, Hiraga S. Genes Dev. 1998;12:1036–1045. doi: 10.1101/gad.12.7.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glaser P, Sharp M E, Raether B, Perego M, Ohlsen K, Errington J. Genes Dev. 1997;11:1160–1168. doi: 10.1101/gad.11.9.1160. [DOI] [PubMed] [Google Scholar]
- 40.Lin D C H, Levin P A, Grossman A D. Proc Natl Acad Sci USA. 1997;94:4721–4726. doi: 10.1073/pnas.94.9.4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohl D A, Gober J W. Cell. 1997;88:675–684. doi: 10.1016/s0092-8674(00)81910-8. [DOI] [PubMed] [Google Scholar]
- 42.Webb C D, Teleman A, Gordon S, Straight A, Belmont A, Lin D C-H, Grossman A D, Wright A, Losick R. Cell. 1997;88:667–674. doi: 10.1016/s0092-8674(00)81909-1. [DOI] [PubMed] [Google Scholar]
- 43.Lemon K P, Grossman A D. Science. 1998;282:1516–1519. doi: 10.1126/science.282.5393.1516. [DOI] [PubMed] [Google Scholar]
- 44.Steck T R, Drlica K. Cell. 1984;36:1081–1088. doi: 10.1016/0092-8674(84)90058-8. [DOI] [PubMed] [Google Scholar]
- 45.Zechiedrich E L, Khodursky A B, Cozzarelli N R. Genes Dev. 1997;11:2580–2592. doi: 10.1101/gad.11.19.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rybenkov V V, Vologodskii A V, Cozzarelli N R. J Mol Biol. 1997;267:312–323. doi: 10.1006/jmbi.1996.0877. [DOI] [PubMed] [Google Scholar]