

Abstract
Circulating endothelial progenitor cells (EPCs) are incorporated into foci of neovascularization where they undergo differentiation to mature endothelial cells (ECs). We show here that the enzyme sphingosine kinase-1 (SK-1) regulates the rate and direction of EPC differentiation without effect on the hematopoietic compartment. EPCs have high levels of SK-1 activity, which diminishes with differentiation and is, at least partially, responsible for maintaining their EPC phenotype. EPCs from SK-1 knockout mice form more adherent EC units and acquire a mature EC phenotype more rapidly. Conversely, EPCs from mice overexpressing SK-1 in the EC compartment are retarded in their differentiation. Exogenous regulation of SK-1 levels in normal EPCs, by genetic and pharmacologic means, including the immunomodulating drug FTY720, recapitulates these effects on EC differentiation. SK-1 knockout mice have higher levels of circulating EPCs, an exaggerated response to erythropoietin-induced EPC mobilization, and, in a mouse model of kidney ischemia reperfusion injury, exhibit a recovery similar to that of ischemic mice administered exogenous EPCs. Thus, SK-1 is a critical player in EPC differentiation into EC pointing to the potential utility of SK-1 modifying agents in the specific manipulation of endothelial development and repair.
Introduction
Endothelial progenitor cells (EPCs) functionally contribute to vasculogenesis during wound healing,1 recovery from limb ischemia,2,3 postmyocardial infarction,4,5 tumorigenesis,6 and endothelialization of vascular grafts.7,8 Circulating EPCs can be derived from several possible sources. They can be mobilized from their bone marrow (BM) niche and into the circulation in response to pathologic changes induced as a result of, for example, tissue hypoxia, vascular injury, and exercise.9 These insults result in the enhanced production of “mobilizing” cytokines into the circulation and include factors, such as vascular endothelial growth factor (VEGF), erythropoietin (EPO), as well as stromal-derived factor-1 (SDF-1). Their action results in enhanced numbers of EPCs in the circulation and their sequestration at sites of injury.9 EPCs can also be derived from mature endothelial cells (ECs), which are forced to dedifferentiate.10 More recently, studies suggest that EPC transplantation induces humoral effects, which are sustained by host tissues and play a critical role in repairing vascular injury.11 Despite these advances, key questions in this area of stem and progenitor cell research remain and pertain to the process, directionality, and rate of differentiation.
The isolation and definition of EPCs rely on the combination of properties rather than any single marker. For example, EPCs express high levels of CD34, CD133, and VEGFR2 and, as they differentiate, the expression of these markers is down-regulated and the mature EC markers, such as CD31 (PECAM-1), VE-cadherin, von Willebrand factor (VWF), endothelial nitric oxide synthase (eNOS), and E-selectin are up-regulated. EPCs have a weak ability to take up acetylated low-density lipoprotein (Ac-LDL) and are unable to form capillary tubes in Matrigel. However, on differentiation, Ac-LDL uptake and the capacity to generate capillary tubes are increased together with the ability to form mature cell-cell junctions.12–14 Circulating EPCs, in addition, are able to survive in the circulation, unattached to any underlying matrix and in the absence of locally elaborated growth factors.15 This is in sharp contrast to mature ECs, which are totally dependent on attachment to an extracellular matrix and to the survival effects of matrix bound growth factors.16,17 Although phenotypic and functional differences between EPCs and ECs are emerging, this system lags behind others, such as the hematopoietic system where the key regulatory molecules that control differentiation have been identified.
Sphingosine kinase (SK) is a highly conserved lipid kinase that catalyzes the phosphorylation of sphingosine to form sphingosine-1-phosphate (S1P), which has been implicated in regulating EC function in vitro and in vivo.18,19 Two isoforms of mammalian SK (SK-1 and SK-2) have been cloned and characterized,20 but SK-1 has been the major isoform implicated in ECs.18,21,22 SK-1 has 2 states: the basal state containing intrinsic catalytic activity, which has been proposed to fulfill a housekeeping role,23 and the activated state, which involves additional production of S1P via the phosphorylation at serine 225.24
There are 5 membrane-localized G protein–coupled receptors for S1P (S1P1-5), which display selective tissue distribution and after ligand binding result in the activation of different downstream signaling cascades.25 There is also increasing evidence to suggest that S1P functions not only as a ligand for its surface receptors, but also as an intracellular second messenger, through unidentified intracellular targets26,27 and may be regulated by changes in intracellular levels of sphingosine.28 In addition, SK-1 itself may be involved in signaling primarily through protein-protein interactions.29
A role for SK/S1P in progenitor cells is beginning to emerge30 with (1) S1P stimulating myogenic differentiation31 and (2) coincubation of S1P and platelet-derived growth factor in a serum-free culture medium successfully maintaining human embryonic stem cells in an undifferentiated state.32 Previous work from our laboratory has suggested that chronic overexpression of SK-1 in human umbilical vein endothelial cells (HUVECs) induces some characteristics of EPCs in that they show enhanced survival under growth factor and serum-free conditions and independent of extracellular matrix attachments.22 These observations suggested that SK-1 might be an important regulator of key aspects of the differentiation of EPCs to ECs. Based on these observations, we used genetically modified mice to investigate whether the regulation of SK-1 is a critical determinant in the EC differentiation pathway. Herein we demonstrate that when SK-1 levels are reduced, EPC mobilization of the bone marrow is increased, and the rate of EC differentiation is hastened.
Methods
Further details for all assays are provided in Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Mouse progenitor cell isolation, culture, and colony-forming assay
Whole BM cells were obtained by flushing femurs and tibiae as previously described.33 EPCs were identified as those from isolated BM cells that adhered within 24 hours after seeding,33,34 and colonies formed in both complete and S1P-depleted culture media were assessed periodically after seeding. Soft agar colonies and adherent EC units are defined as a cell mass composed of a central cord of round cells with elongated spindle-shaped cells sprouting at the periphery. Pure progenitor cells (Lin−/c-kit+) were isolated from murine BM using the Miltenyi Biotec (Bergisch Gladbach, Germany) magnetic cell sorting kit as previously described.33,35 Briefly, BM cells were collected as described in this paragraph and microbeads precoated with “Lineage” (CD5, CD45R, CD11b, anti-Gr-1, 7-4, and Ter119) or c-kit antibodies were used for cell purification.
Measurement of S1P
S1P levels were measured using fluorescence derivatization followed by high-performance liquid chromatography, as described previously.36
Matrigel tubule assay
As previously described,37 10 mg Matrigel (BD Biosciences, San Jose, CA) was added to a precooled 96-well plate (100 μL/well) and incubated at 37°C for 30 minutes. A cell suspension of 4 × 104 cells/well was seeded and monitored regularly using an inverted IX70 microscope 10×/0.3 NA objective, an S15 F view camera, and Analysis Life Sciences software (Olympus).
Measurement of cell proliferation
The number of living cells was measured [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Celltiter 96 Aqueous One solution cell proliferation assay; Promega, Madison, WI).
Attachment and migration assays
WT and SK-1-KO BM cells grown for 6 days were seeded at 104 cells/100 μL into a 96-well fibronectin-coated plate for 60 minutes (for adhesion) or onto Transwells (for migration).
Acetylated-LDL labeled with Dio-Ac-LDL uptake
3,3′-dioctadecyloxacarbocyanine perchlorate (Dio-Ac-LDL; Biomedical Technologies, Stoughton, MA) was incubated at 10 μg/mL with the cells at 37°C for 4 hours. Cells were then detached using 1% trypsin/1.1 mM ethylenediaminetetraacetic acid, and the fluorescence intensity was determined by flow cytometry.
RNA isolation and quantitative reverse transcriptase PCR analysis
Total RNA or mRNA was extracted using either RNeasy mini kit (QIAGEN, Valencia, CA) or MACS μ column with oligo(dT) microbeads, respectively (Miltenyi Biotec), according to the manufacturer's instructions.
Adenovirus infection
Generation of wild-type (WT) human SK-1, FLAG, and GFP tagged versions and mutants possessing an alanine mutation at serine 225 (SK-1S225A) or an aspartate at glycine 82 (SK1G82D) have been made as previously described.23,24,38
SK-1 activity assay
SK-1 activity was determined as previously described39 using D-erythro sphingosine (BIOMOL International, Plymouth Meeting, PA) solubilized by Triton X-100 and [γ32P]ATP (PerkinElmer Life and Analytical Sciences, Melbourne, Australia) as substrates.
EPC mobilization using erythropoietin
Mice were injected intraperitoneally with 1000 IU/kg recombinant human EPO (Eprex2000; Janssen-Cilag Australia, North Ryde, Australia) every day for 3 days before cardiac puncture40 and Lin−/c-kit+ cell isolation as detailed in “Mouse progenitor cell isolation, culture, and colony-forming assay.” All experimental procedures involving animals were approved by the Human and Animal Ethic Committee of the Institute of Medical and Veterinary Science and conform to the guidelines established by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Kidney ischemia reperfusion injury
Anesthetized mice (n = 6 per group) were incised (1.0-cm-long midline abdominal incision) and ischemia induced by applying a nontraumatic vascular clamp to the left renal pedicle for 30 minutes, during which time the wound was covered with phosphate-buffered saline (PBS)–soaked sterile cotton. After clamp removal, the left kidney was inspected for restoration of blood flow and the wound was closed in 2 layers with 0.05 mL of 1 mg/mL butorphanol tartrate (Intervet, Bendigo East, Australia) administered subcutaneously for postoperative pain management. The animals were killed 24 hours after reperfusion, at which time plasma was collected and the left and right kidneys harvested for morphologic, immunohistochemical, and mRNA analyses. Carboxyfluorescein succinimidyl ester (CFSE)–labeled EPCs (10 μM, 30 min; Invitrogen) were injected in the tail vein at 107/mouse immediately after ischemia optimal cutting temperature (OCT)–frozen kidney sections were stained for CD31 and cell nuclei using antibodies against CD31 (BD Biosciences) and 4,6-diamidino-2-phenylindole (DAPI; Roche, Basel, Switzerland) as per the manufacturer's instructions. Sections were analyzed using a Zeiss-Apotome Microscope and Axiovision, version 4.6.3 computer software (Carl Zeiss, Jena, Germany).
Statistical analysis
Data were expressed as mean plus or minus SEM from at least 4 experiments, unless stated otherwise. Statistical analysis was performed by Student t test or 2-way analysis of variance for multiple comparisons, and a P value less than .05 was considered significant.
Results
Phenotype, morphology, and function of BM-derived EC colonies
To determine whether SK-1 is involved in EC differentiation, we used SK-1 knockout mice and compared their capacity to produce mature ECs. BM cells were cultured with VEGF and fibroblast growth factor on fibronectin-coated plates, to promote the vascular endothelial lineage.33 Cells were cultured in charcoal-stripped serum (CS-FCS) where high-performance liquid chromatography confirmed a more than 90% reduction in S1P levels. Levels in undiluted FCS were 473 (± 38) nM and in CS-FCS 37 (± 5) nM. Because serum was used at a final concentration of 20%, the cells were being grown in approximately 7 nM S1P.
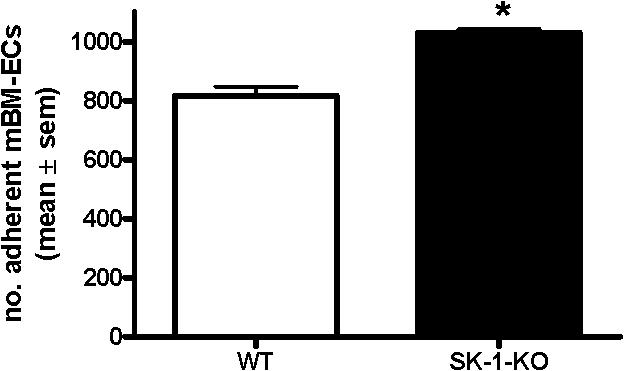
Figure 1 demonstrates that 10-day cultured BM cells from WT and SK-1-KO mice exhibit typical EC characteristics. They showed the typical structure of adherent EC units (ECU) with a cluster of round cells on top of the spindle-shaped adherent cells that emerge from the base (Figure 1A). When the adherent ECU were replated, they were capable of forming a compact monolayer, they expressed the endothelial specific marker VE-cadherin (Figure 1B and C, respectively), and they were able to form capillary-like tubes in a 3-dimensional Matrigel assay2 (Figure 1D). The cells are positive for CD34, c-kit, sca-1, Flk-1, CD133, CD146, CD31, eNOS, VWF, Tie2, CXCR2, CXCR4, α5-integrin, β1-integrin, CD45, and CD11b surface antigen, they were CD14 low and negative for CD105 and CD90 (data not shown). There was no difference in levels of expression of these antigens between the cells derived from WT and SK-1-KO BM. These cells were also negative for smooth muscle actin-alpha (data not shown), thereby negating the possibility of stromal cell and smooth muscle cell contamination. These results suggest that, when cultured under these conditions, both WT- and SK-1-KO–derived BM cells develop a mature EC-like phenotype. However, in contrast to these similarities, BM cells from SK-1-KO mice formed twice as many ECU within 7 days than WT cells with a time course revealing these differences to be significant within 2 days of culture (Figure 1E).
Figure 1.

Cultured mBM cells exhibit EC-like morphology and function. Cultured BM cells were seeded onto fibronectin-coated plated plates in CS-FCS–containing media. (A) The ECU formed from WT and SK-1-KO cells within 24 hours of plating. (B) Cells harvested at day 7 after seeding were replated and within 24 hours formed compact EC-like monolayers. (C) Cells were harvested after 10 days and stained for expression of the endothelial-specific marker, VE-cadherin. Ctl indicates an isotype-matched control. Expression was analyzed by flow cytometry. (D) Ten-day cultured BM cells were seeded onto Matrigel at 4 × 104 cells/well. Tube formation was monitored periodically with images depicting tube formation 30 hours after seeding. Classic branching patterns protruding from large tube-like structures (arrows) can be seen. Results are representatives of at least 3 separate experiments. (E) ECU generated by WT and SK-1-KO BM cells on fibronectin were counted at times specified as “days in culture.” Results show the mean plus or minus SEM of 5 separate mice. *P < .05 compared with WT, 1-way analysis of variance.
Reduced SK-1 does not affect hematopoietic colony formation
Because BM cells also contain hematopoietic progenitor cells, we investigated whether SK-1 influences their differentiation. BM cells from WT and SK-1-KO mice were cultured in soft agar medium containing CS-FCS and a cocktail of growth factors and cytokines suitable for the expansion of myeloid, hematopoietic, and erythroid lineages.41 After 7 and 14 days of culture, the number, size, and type of colonies were counted. As shown in Table 1, similar numbers of colonies were obtained from the WT and SK-1 KO BM cells irrespective of the cocktail used. This correlated with similarities in the proportions of small, medium, and large colony sizes as well as similar numbers of neutrophils, macrophage, and mixed colonies as determined by tri-staining (data not shown). Thus, SK-1 expression appears to selectively regulate the EC lineage and not the hematopoietic compartment.
Table 1.
Colony-forming units from WT and SK-1-KO BM cells cultured in soft agar for 14 days with growth factors and cytokines known to promote myeloid, hematopoietic, and erythroid colony growth
| Cytokine/GF cocktail | WT | SK-1-KO |
|---|---|---|
| Nil | 0 ± 0 | 0 ± 0 |
| GM-CSF + G-CSF | 22 ± 6 | 28 ± 10 |
| IL-3 + SCF | 144 ± 26 | 135 ± 26 |
| IL-6 + SCF | 117 ± 30 | 100 ± 26 |
| IL-6 + SCF + G-CSF + EPO | 123 ± 30 | 99 ± 23 |
| IL-6 + SCF + IL-3 + EPO | 83 ± 5 | 85 ± 3 |
Data are mean CFU plus or minus SEM from 9 mice per group.
WT indicates wild-type; and BM, bone marrow.
Cell proliferation is regulated by SK-1
There is generally an inverse relationship between differentiation and cell proliferation. We therefore examined WT and SK-1-KO BM cell accumulation on fibronectin-coated plates using the MTS cell viability assay. As shown in Figure 2, a significant reduction in SK-1-KO BM cells was observed within 48 hours of culture.
Figure 2.

Proliferative rate of EPCs in vitro. Accumulation of WT and SK-1-KO BM cells cultured for 5 days on fibronectin-coated plates in CS-FCS–containing media as assessed by the MTS assay. Results show the mean plus or minus SEM of a representative of 4 separate mice, where each group was performed in quadruplicate. *P < .05, compared with WT, 1-way analysis of variance.
Role for intracellular SK-1/S1P during EC maturation
The increase in the number of ECU derived from SK-1 knockout BM cells is probably not the result of exogenous S1P based on several different experimental approaches. First, in “add-back” experiments, when SK-1-KO BM cells were grown in “normal” FCS (ie, not charcoal stripped), they still showed significantly higher ECU compared with WT (Figure 3A). CS-FCS slightly, but not significantly, enhanced ECU formation in all cell types (Figure 3A). SK-1-KO cells cultured with 1 μM S1P, a concentration known to result in receptor-mediated signaling events,18 exhibited no change in ECU (Figure 3B). Chemical inhibition of the 3 S1P receptors identified on ECs (ie, S1P1, S1P2, and S1P3)42 using pertussis toxin, JTE-013, and VPC23019, inhibits S1P-induced SK activity on HUVECs (Figure S1) but failed to alter the number of WT ECU (Figure 3B). Finally, analysis of the S1P receptors by quantitative RT-PCR showed that S1P1 and S1P2 mRNA, although readily detectable in the mixed population of freshly isolated BM cells (Figure 3C), is low to undetected in day 3 cultured early EPCs and are up-regulated on mature 10-day cultured ECs (Figure 3C,D). S1P3 mRNA was below our level of detection at all time points investigated for both groups (data not shown). Although the current lack of antibodies to murine S1P receptors prevents investigation of their surface expression, our results recapitulate previously published data of receptor expression on progenitor cells.43
Figure 3.

ECU formation is extracellular S1P and receptor independent. (A) BM cells derived from WT and SK-1-KO mice were cultured in CS-FCS or complete FCS and ECU numbers determined 7 days after culture. Results show the mean plus or minus SEM for 5 mice per group. *P < .05, compared with WT, 2-tailed paired t test. (B) ECU were counted 4 days after culture in CS-FCS–containing media with administration of S1P (1 μM), pertussis toxin (50 ng/mL), JTE-013 (1 μM), or VPC23019 (10 μM) at initial seeding and every 48 hours thereafter. Results are the mean plus or minus SEM of 3 mice. (C) Semiquantitative RT-PCR was determined for S1P1 and S1P2 mRNA expression in BM cells cultured under EPC differentiating conditions for 0 (BM), 3, and 10 days and represents 1 of 5 separate experiments; vertical line(s) have been inserted to indicate repositioned gel lanes. (D) Quantitative RT-PCR was used to determine expression of S1P1 and S1P2 in BM cells cultured for 3 and 10 days. Results are the mean plus or minus SEM of receptor expression normalized to the PPIA gene and shown as a ratio over day 3 WT, for 5 separate mice.
Enhanced EC differentiation in SK-1 null mice
Increased ECU numbers generated by SK-1-KO BM cells could be the result of either a higher number of progenitor cells within the BM or a greater tendency of these cells to differentiate. As shown in Figure 4A, the number of purified Lin−/c-kit+ progenitors isolated from 6.5 × 107 BM cells was equivalent between WT and SK-1 KO populations. The Lin−/c-kit+/flk+ endothelial progenitors were also equivalent between the 2 groups, with an average of 1.0 × 104 and 0.9 × 104 cells isolated from the WT and SK-1-KO BM cells, respectively.
Figure 4.

Depletion of SK-1 promotes EC maturation. (A) BM was harvested from WT and SK-1-KO mice, the Lin−/c-kit+ cells isolated by magnetic sorting. Results show the mean plus or minus SEM isolated from 6.5 × 107 cells per group from 3 separate experiments. (B) BM cells from WT and SK-1-KO mice (solid and broken lines, respectively) were examined for their ability to take up Ac-LDL at days 3 and 9 after seeding. Results show the mean plus or minus SEM of the percentage uptake. *P < .05, compared with WT day 3. #P < .05, compared with WT day 10, 1-way analysis of variance. (C) Flow cytometric analysis of Ac-LDL uptake by WT (thin line) and SK-1-KO (thick line) BM cells after 3 days of culture from a representative of 3 experiments performed. (D) WT and SK-1-KO mBM cells (□ and ■, respectively) were harvested for Sca-1 surface expression and VE-cadherin mRNA expression analysis by flow cytometry and quantitative RT-PCR, respectively. Messenger RNA results are normalized to their PPIA gene. Data are expressed as mean plus or minus SEM from 3 separate experiments for Sca-1 surface expression (at day 7 after seeding) and VE-cadherin mRNA levels (at day 10 after seeding). *P < .05, compared with WT levels on day of harvest. (E) Six-day cultured BM cells were seeded into Matrigel. Images depict tube formation after 5 and 30 hours and are representative of 5 separate experiments. Inset numbers reflect mean plus or minus SEM of total tubes formed at 30 hours from 5 experiments.
The increase in ECU without differences in the number of Lin−/c-kit+/flk+ progenitors suggests that the differentiation step may be hastened in SK-1-KO BM cells. When EPCs differentiate into mature ECs, their ability to take up Ac-LDL increases.12,44 BM cells were assessed for Ac-LDL uptake on days 3 and 9 of culture. As shown in Figure 4B, the percentage of SK-1-KO BM cells taking up Ac-LDL at day 3 of culture was significantly greater than that of WT BM cells and was maintained over time. As Ac-LDL can also be taken up by myeloid cells,44 we double-stained with the EC marker VE-cadherin to demonstrate a more than 95% population of Ac-LDL+/VE-cadherin+ cells (data not shown).
Further evidence for an enhanced rate of differentiation was identified in Sca-1 surface protein levels, which were significantly reduced in SK-1-KO cells and VE-cadherin mRNA levels, which were significantly greater in the SK-1-KO cells (Figure 4D). EPC differentiation depends on adhesion. There was a 25% (± 5%) increase in SK-1-KO cell adhesion to fibronectin compared with WT cell adhesion (Figure S2), but the cells exhibit no differences in migration to VEGF (data not shown). On Matrigel, both WT and SK-1-KO BM cells (previously cultured for 6 days on fibronectin) exhibited the cellular alignment essential for tubule development. However, after 30 hours, the SK-1-KO formed more tubes than the WT cells, and these tended to break down very early (Figure 4E). Cells from both WT and SK-1 KO mice cultured for 3 days were unable to form tubes (suggesting EC immaturity), and 10-day cultured cells from the WT and SK-1 KO mice formed similar numbers of tubes (suggesting an equivalent stage of EC maturation, Figure 2). Together, these results support the possibility that SK-1 may regulate the rate of EPC differentiation.
SK-1 activity decreases with EC development
The data suggest that low SK-1 levels are associated with EC differentiation and that SK-1 activity may decrease as EPCs differentiate into mature ECs. The SK-1 activity in these cells was determined in the presence of Triton X-100, which largely inhibits SK-2 activity.45 When cultured for 9 days, WT BM cells exhibit a 78% (± 8%) reduction in SK activity (Figure S3) with differentiation. In mature ECs, this level, although low, is always significantly above background. The SK-1 activity in SK-1-KO BM cells was small, although above control levels, and probably the result of SK-2 activity.
Manipulation of SK-1 in mBM cells alters EC development
If SK-1 regulates EC differentiation, then we would expect exogenous manipulation of SK-1 levels to impact on ECU formation and differentiation. Using ECU as the readout assay for EC differentiation, we first determined whether the SK-1-KO BM cell phenotype could be reversed by the reintroduction of SK-1 using adenovirus. As shown in Figure 5A, within 48 hours of the WT SK-1 cDNA being reintroduced into the SK-1 KO cells (SK-1-KOSK), SK activity returned to normal levels (1.3 ± 0.4) and was correlated with a 50% reduction in ECU formation compared with empty vector control (SK-1-KOEV). Second, we investigated transgenic mice (SK-1-Tg), which overexpress SK-1 in the EC compartment via the Tie2 promoter/enhancer and exhibited an increase in SK activity (1.8 ± 3 vs 1.0 ± 0, SK-1-Tg vs WT, respectively). SK-1-Tg showed a decrease in ECU numbers compared with WT (Figure 5B). Third, blocking SK-1 activity in WT BM cells using the inhibitor SKi46 resulted in a 60% (± 10%) reduction in SK activity as well as a significant increase in ECU formation (Figure 5C). Finally, FTY720, initially thought to be solely an S1P analog but recently shown to be an inhibitor of SK-1,43 induced a 30% (± 10%) reduction in human EC SK-1 protein (data not shown) and increased ECU formation of WT BM cells (Figure 5D). Importantly, FTY720 did not change ECU in SK-1-KO cultures (data not shown). The ability of FTY720 to enhance EPC differentiation was further supported with the tube formation assay wherein a significant increase in tube numbers from WT BM cells cultured with FTY720 for 6 days was observed (Figure 5E). Taken together, these results suggest that manipulation of SK-1 results in the regulation of EC differentiation.
Figure 5.

Manipulation of SK-1 levels alters ECU formation. ECU numbers were determined from: (A) BM cells from SK-1-KO mice infected with adenovirus containing either control EV (SK-1-KOEV) or SK-1 (SK-1-KOSK); (B) WT and SK-1-Tg BM cells; (C) WT BM cells cultured with the SK inhibitor SKi (5 μM) administered at initial seeding and every 48 hours thereafter; and (D) WT and SK-1-KO BM cells cultured without (□ and with FTY720 (100 nM, ■) administered at initial seeding and every 48 hours thereafter. All experiments show the mean plus or minus SEM of 4-day cultured ECU from at least 3 separate experiments. SK-1 levels shown numerically leading the figure (activity for panels A-C and protein for panel D) identified successful SK-1 manipulation. Results are expressed as U/mg protein normalized to WT BM cells at day 4 of culture plus or minus SEM. *P < .05, compared with controls using a 2-tailed, paired t test. (E) Six-day cultured WT BM cells plus or minus FTY720 (100 nM) administered at initial seeding and every 48 hours thereafter, seeded into Matrigel. Images show tube formation; numbers reflect mean plus or minus SEM tubes per experiment;  identify tubes formed at 30 hours. Results are representatives of 3 separate experiments.
identify tubes formed at 30 hours. Results are representatives of 3 separate experiments.
Intrinsic SK-1 activity may regulate EC differentiation
We have previously generated 2 mutants of SK-1 to differentiate the intrinsic activity from the activated state of this enzyme.47 One mutant is a catalytically inactive version of SK-1 (SK-1G82D).38 The other is a nonphosphorylatable mutant (SK-1S225A) that, although having full intrinsic catalytic activity, cannot be activated.24 As shown in Figure 6, overexpression of WT SK-1 in WT BM cells results in a 10-fold (± 4-fold) increase in SK activity and the expected reduction in ECU formation. Cells expressing the SK-1G82D mutant show neither a detectable change in cellular SK activity (1 ± 0 normalized U/mg protein) nor ECU formation compared with the empty vector controls. Cells expressing the SKS225A mutant increased cellular SK activity (12 ± 2 normalized U/mg protein) and also decreased the formation of ECU to levels similar to that seen when WT SK-1 was overexpressed. Western blotting confirmed that transfection efficiency and protein expression were equivalent between WT and the mutants of SK-1 (data not shown). Together, these data suggest that the modulation of EC maturation is regulated through the intrinsic activity of SK and is independent of SK-1 activation via serine 225 phosphorylation.
Figure 6.

Intrinsic activity of SK-1 regulates EC differentiation. WT BM cells containing the empty vector (EV) control, WT SK-1 (SK-1), catalytically inactive mutant (G82D), or the phosphorylation-dead mutant (S225A). ECU numbers determined at day 4 of culture with results showing the mean plus or minus SEM from 3 separate experiments. SK-1 activity was determined at completion of the experiments to confirm the successful manipulation (shown numerically as U/mg protein normalized to WTEV). *P < .05, compared with EV and G82D.
Increased circulating EPC in SK-1–deficient mice and restitution of vascular injury
EPCs are present in the circulation and can be further mobilized from the BM on stimulation with agents, such as granulocyte colony-stimulating factor and EPO.40,48 Isolation of circulating Lin−/c-kit+ progenitor cells showed a 30% increase in the number of circulating progenitors in the SK-1-KO animals compared with WT (Figure 7A). Circulating Lin−/c-kit+/flk-1+ cells in SK-1-KO mice was 2.1 × 104 per 107 WBC vs 0.3 × 104 per 107 WT WBC. EPO administration for 3 consecutive days resulted in an increase in the number of circulating EPCs in WT mice. However, a further increase in circulating progenitors was mobilized from the SK-1 KO mice (Figure 7A). Thus, although similar numbers of progenitors exist in the BM of WT and SK-1 KO animals, the lack of SK-1 potentiates higher circulating numbers under both basal conditions and after treatment with EPO.
Figure 7.

EPC mobilization is enhanced in SK-deficient mice and protects against ischemic injury. (A) WT and SK-1-KO mice, no treatment (−) or administered with PBS or EPO (1000 IU/kg) for 3 consecutive days. Circulating blood was collected and Lin−/c-kit+ cells isolated by magnetic sorting. Results show the mean plus or minus SEM of cell numbers from at least 3 mice in each group. *P < .05, compared with WT PBS. #P < .05, compared with SK-1-KO PBS. (B) CFSE-labeled EPCs injected intravenously into WT mice after kidney ischemia and visualized after 24 hours of reperfusion. Frozen tissue sections stained for CD31 (red) and DAPI (blue) allowed visualization of kidney vasculature as well as recruited CFSE-EPCs (green) by fluorescence microscopy. (C) mRNA levels of hsp70 and TLR4 were investigated in ischemic kidneys 24 hours after IRI by quantitative RT-PCR. Expression is normalized to their GAPDH gene, and results show the mean plus or minus SEM for 5 mice in each group. *P < .05, 1-way analysis of variance, compared with sham controls.
As EPCs contribute to the restitution of vascular injury,49 we next investigated our BM-derived EPCs in a mouse model of kidney ischemia reperfusion injury (IRI) and compared WT to SK-1-KO vascular repair 24 hours after injury. As shown in Figure 7B, when labeled with CFSE and injected intravenously into mice immediately after ischemic insult, our WT BM-derived EPCs were detected adjacent to and within the vasculature of the ischemic kidney. This is shown by the green (injected CFSE-EPCs) localized adjacent to the red (CD31)–stained vasculature and blue (DAPI) nuclear counterstain in the left panel of Figure 7B. The orange glomerulus in the right panel suggests a merged area of green and red (ie, CFSE-EPCs into the local vasculature). No CFSE-EPCs were detectable in the right contralateral control kidney (data not shown). The restitution of IRI by exogenously added EPCs was examined 24 hours after IRI using quantitative RT-PCR for 2 key markers of kidney injury, hsp70 and TLR4.50 These markers were significantly lower in mice that received EPCs than in control WT IRI mice (Figure 7C). As we would predict, IRI in SK-1-KO mice, with their inherently greater numbers of circulating EPCs and propensity to release BM-derived EPCs, showed similar reductions in hsp70 and TLR4 to EPC-injected mice (Figure 7C).
Discussion
In the present study, we demonstrate a new mechanism for regulation of EC differentiation. The cellular level of SK, which declines with EC maturity, dictates the rate and direction of EPC/EC differentiation. Our results also suggest that it is the intracellular component of SK-1/S1P that regulates this differentiation process and that the intrinsic but not the activation state of the enzyme is critical.
The evidence for SK-1 involvement in EPC differentiation relies on SK-1 mutant mice and on genetic and pharmacologic regulation of SK-1. Important in our proposal of SK-1 activity regulating EC differentiation is that: (1) immature EPCs have high SK-1 activity levels, which decrease on differentiation; and (2) exogenous manipulation of SK-1 activity levels influences EPC maturation. Knockdown of SK-1 in WT EPCs by a functional inhibitor of SK (SKi)46 and also by FTY720, a drug that has recently been shown to inhibit SK-1, but not SK-2, function,43 recapitulates the phenotype seen with cells from the SK-1 knockout animals. Moreover, reinsertion of SK-1 cDNA in cells from the knockout animals rescues their differentiation to levels similar to that seen in normal cells. In addition, we observed a reversal of phenotype, where retarded EPC differentiation occurred from transgenic animals overexpressing SK-1 in the EC compartment.
The regulation in the rate of differentiation by SK-1 was reflected in steady state and stimulated circulating endothelial progenitor cell (Lin−/ckit+/flk-1+) number. The progenitor cells were significantly greater in the circulation of the SK-1 KO mice and were further amplified after mobilization with EPO. The use of EPO is of therapeutic interest for ischemic injury as it is associated with tissue protective effects through activation of EPO receptor-related antiapoptotic pathways, for example, PI3K/Akt51 as well as neovascularization of the ischemic tissue.52 This neovascularization occurs primarily via the CXCR4/SDF-1 axis, which is the key cellular anchor within the BM microenvironment and a pivotal element of EPC mobilization and homing.52,53 Interestingly, the WT and SK-1-KO groups showed no difference in either the number of Lin−/ckit+/flk-1+ cells in the BM or their capabilities to migrate toward VEGF. These results highlight the possibility that SK-1 acts as a rheostat for determining numbers of EPCs in the circulation and their differentiation to ECs. This directive of differentiation is supported by SK-1 regulation of cell cycle, as we and others observe that reduced SK-1 retards cell proliferation, probably via an increase in the G0/G1 population and concomitant decrease in S-phase population.54 This system of regulation would seem especially suited to the bidirectional differentiation in the endothelial system, which also allows dedifferentiation from ECs back into EPCs.10 Indeed, preliminary work by us in HUVECs suggests that overexpression of SK-1 can also influence the human EPC/EC pathway of differentiation with a significant increase in CD34 and VEGFR-2 expression (Figure S4). With the possible exception of monocyte-macrophage, this bidirectionality is absent from the hematopoietic system; correspondingly, SK-1 does not appear to play a role. Thus, modulation of SK-1 activity could be therapeutically beneficial in the attempt to specifically alter the EC compartment.
The mechanism whereby SK-1 works was also examined. SK-1 has 2 functional states24,55: one is the intrinsic or basal catalytic activity, which is independent of posttranslational modifications; and the other an extrinsic or agonist-induced activation, which involves the phosphorylation of serine 225 and translocation to the plasma membrane for its oncogenic function.55 The regulation of the rate of mEPC to EC differentiation is reliant on the intrinsic or basal activity of SK-1, demonstrated in 2 ways. First, whereas overexpression of WT SK-1 in WT and the SK-1-Tg BM-derived EPCs inhibited ECU formation, the catalytically inactive G82D SK-1 mutant failed to alter ECU number. Overexpression of the S225A SK-1 mutant, which has full basal activity but is incapable of being activated, inhibited ECU formation. Second, it is probable that SK (or its bioactive metabolite) acts intracellularly as we show that the S1P1-3 receptors are not involved in the rate of EPC differentiation. Notably, S1P1-5 appear to be involved in the earliest stages of vascular development56 and have a range of effects in embryonic stem cell and myogenic differentiation that appear quite cell type–specific.31,57 Our results here demonstrate that, together with regulation of EC permeability,58 the basal/intrinsic activity of this enzyme acts intracellularly to control EPC differentiation. At present, the mechanism underlying this regulation of differentiation is unknown.
Our results with FTY720 are particularly interesting because this molecule is in clinical trials (with success in phase 3 for treatment of multiple sclerosis59). Although the mechanism of action of FTY720 was initially thought to center on its phosphorylated form acting on S1P receptors, it is now evident that its unphosphorylated form also activates PP2A and inhibits SK-1.43 High concentrations (> 1 μM) promote the apoptosis of cells (including leukemic cells60 and fibroblasts61) and lower concentrations (< 1 μM) promote pluripotent hematopoietic stem and progenitor cell survival and proliferation62 and fibroblast differentiation.61 Because phosphorylation of FTY720 is not needed, at least for some of these effects, the mechanism is not through S1P receptors. Here, administration of FTY720 at 100 nM significantly enhanced EPC differentiation without compromising the survival of EPCs or ECs (C.S.B., unpublished observations, June 2008). Because our effects also do not involve S1P receptors and the effects take place at lower doses, the probable mechanism of FTY720 is the inhibition of SK-1. Together, these observations raise the possibility that some of the clinical benefits of FTY720 are mediated by its effects on EPC or other non–S1P receptor-mediated mechanisms.
Our mouse model of kidney ischemia reperfusion injury supports our in vitro data. The EPCs generated in our system are able to influence IRI. Intravenous injection of EPCs home to the ischemic kidney and integrate into the vasculature within 24 hours after insult. In these same mice, a significant reduction in 2 key markers of kidney IRI was observed, hsp70 and TLR4.50 Thus, although there are building concepts of multiple EPC subpopulations that promote different aspects of neovascularization,63 the population we derive from the BM under our conditions is functionally significant. Furthermore, our SK-1-KO mice, which have an inherently greater number of circulating EPCs with increased adhesion capabilities to fibronectin, also exhibited a reduction in hsp70 and TLR4 mRNA levels 24 hours after IRI. As circulating endothelial cells are thought to represent mature nondividing ECs of the vascular intima that are sloughed off after a pathologic process, one must take into consideration that the cells contributing to the recovery from kidney ischemia/reperfusion injury in SK-1-KO mice may not be solely BM derived. Alternatively, and indistinguishable in this study as our BM-derived ECs are CD146 positive, it is possible that modulation of SK-1 regulates both the angiogenic pathway (via EC survival, proliferation, and migration) and vasculogenic pathway (via EPC mobilization from the BM) for vascular repair. Nevertheless, these results support the increasing evidence for EPC transplantation to induce autocrine, paracrine, and humoral effects to abrogate IRI.11 The use of whole-body SK-1-KO mice also begs the question of whether the IRI resolution includes additional effects (eg, IRI-induced adhesion molecule expression and leukocyte recruitment).
We have defined a novel system that not only regulates the rate and extent of EPC differentiation but also influences the function of EPCs. The dominant role of SK-1 is evidence by both genetic and pharmacologic means and provides a road map for the manipulation of EPCs in vivo.
Supplementary Material
Acknowledgments
The authors thank Jen Drew and Anna Sapa for excellent technical assistance, Paul Moretti for molecular expertise, the Animal Care Facility at the Institute of Medical and Veterinary Science, the mothers and staff at the Women's & Children's Hospital, and Burnside War Memorial Hospital for collection of umbilical cords.
This work was supported in part by grants from National Health & Medical Research Council of Australia with a Peter Doherty Fellowship (C.S.B.), Project Grant 430907 (C.S.B.), Program Grant 349332 (J.R.G., M.A.V., A.F.L.), a Peter Nelson Leukemia Research Fund Senior Research Fellowship (H.S.R.), and by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (R.L.P.). J.R.G. is a Medical Foundation Fellow at the University of Sydney (Sydney, Australia).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.S.B. designed and performed research, contributed vital new reagents or analytical tools, analyzed data, and wrote the paper; W.Y.S., T.M., and C.C. performed research and analyzed data; X.L. and H.S.R. performed research; S.M.P., P.T.C., and R.L.P. contributed vital new reagents or analytical tools; A.F.L. designed research; and M.A.V. and J.R.G. designed research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Claudine S. Bonder, Vascular Biology and Cell Trafficking Laboratory, Division of Human Immunology, Hanson Institute, Institute of Medical and Veterinary Science, PO Box 14, Rundle Mall, Adelaide, South Australia, Australia 5000; e-mail: claudine.bonder@imvs.sa.gov.au; or Jennifer R. Gamble, Vascular Biology Program, Centenary Institute, University of Sydney, Locked Bag No. 6, Newtown, New South Wales, Australia, 2042; e-mail: j.gamble@centenary.org.au.
References
- 1.Gill M, Dias S, Hattori K, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 4.Edelberg JM, Tang L, Hattori K, Lyden D, Rafii S. Young adult bone marrow-derived endothelial precursor cells restore aging-impaired cardiac angiogenic function. Circ Res. 2002;90:E89–E93. doi: 10.1161/01.res.0000020861.20064.7e. [DOI] [PubMed] [Google Scholar]
- 5.Shintani S, Murohara T, Ikeda H, et al. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 2001;103:2776–2779. doi: 10.1161/hc2301.092122. [DOI] [PubMed] [Google Scholar]
- 6.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2:826–835. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 7.Shi Q, Rafii S, Wu MH, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92:362–367. [PubMed] [Google Scholar]
- 8.Bhattacharya V, McSweeney PA, Shi Q, et al. Enhanced endothelialization and microvessel formation in polyester grafts seeded with CD34(+) bone marrow cells. Blood. 2000;95:581–585. [PubMed] [Google Scholar]
- 9.Aicher A, Zeiher AM, Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi: 10.1161/01.HYP.0000154789.28695.ea. [DOI] [PubMed] [Google Scholar]
- 10.Thum T, Haverich A, Borlak J. Cellular dedifferentiation of endothelium is linked to activation and silencing of certain nuclear transcription factors: implications for endothelial dysfunction and vascular biology. FASEB J. 2000;14:740–751. doi: 10.1096/fasebj.14.5.740. [DOI] [PubMed] [Google Scholar]
- 11.Cho HJ, Lee N, Lee JY, et al. Role of host tissues for sustained humoral effects after endothelial progenitor cell transplantation into the ischemic heart. J Exp Med. 2007;204:3257–3269. doi: 10.1084/jem.20070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 13.Vittet D, Prandini MH, Berthier R, et al. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- 14.Bellik L, Ledda F, Parenti A. Morphological and phenotypical characterization of human endothelial progenitor cells in an early stage of differentiation. FEBS Lett. 2005;579:2731–2736. doi: 10.1016/j.febslet.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Chavakis E, Dimmeler S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:887–893. doi: 10.1161/01.atv.0000017728.55907.a9. [DOI] [PubMed] [Google Scholar]
- 16.Meredith JE, Jr, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res. 1999;76:1–20. doi: 10.1016/s0065-230x(08)60772-1. [DOI] [PubMed] [Google Scholar]
- 18.Xia P, Gamble JR, Rye KA, et al. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A. 1998;95:14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chae SS, Paik JH, Allende ML, Proia RL, Hla T. Regulation of limb development by the sphingosine 1-phosphate receptor S1p1/EDG-1 occurs via the hypoxia/VEGF axis. Dev Biol. 2004;268:441–447. doi: 10.1016/j.ydbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Alemany R, van Koppen CJ, Danneberg K, Ter BM, Meyer Zu HD. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–428. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- 21.Xia P, Wang L, Gamble JR, Vadas MA. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J Biol Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 22.Limaye V, Li X, Hahn C, et al. Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. Blood. 2005;105:3169–3177. doi: 10.1182/blood-2004-02-0452. [DOI] [PubMed] [Google Scholar]
- 23.Pitson SM, D'Andrea RJ, Vandeleur L, et al. Human sphingosine kinase: purification, molecular cloning and characterization of the native and recombinant enzymes. Biochem J. 2000;350:429–441. [PMC free article] [PubMed] [Google Scholar]
- 24.Pitson SM, Moretti PA, Zebol JR, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22:5491–5500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 26.Kluk MJ, Hla T. Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochim Biophys Acta. 2002;1582:72–80. doi: 10.1016/s1388-1981(02)00139-7. [DOI] [PubMed] [Google Scholar]
- 27.Spiegel S, Milstien S. Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem Soc Trans. 2003;31:1216–1219. doi: 10.1042/bst0311216. [DOI] [PubMed] [Google Scholar]
- 28.Van B, Jr, Lee MJ, Menzeleev R, et al. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol. 1998;142:229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukuda Y, Aoyama Y, Wada A, Igarashi Y. Identification of PECAM-1 association with sphingosine kinase 1 and its regulation by agonist-induced phosphorylation. Biochim Biophys Acta. 2004;1636:12–21. doi: 10.1016/j.bbalip.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Pebay A, Bonder CS, Pitson SM. Stem cell regulation by lysophospholipids. Prostaglandins Other Lipid Mediat. 2007;84:83–97. doi: 10.1016/j.prostaglandins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Donati C, Meacci E, Nuti F, et al. Sphingosine 1-phosphate regulates myogenic differentiation: a major role for S1P2 receptor. FASEB J. 2005;19:449–451. doi: 10.1096/fj.04-1780fje. [DOI] [PubMed] [Google Scholar]
- 32.Pebay A, Wong RC, Pitson SM, et al. Essential roles of sphingosine-1-phosphate and platelet-derived growth factor in the maintenance of human embryonic stem cells. Stem Cells. 2005;23:1541–1548. doi: 10.1634/stemcells.2004-0338. [DOI] [PubMed] [Google Scholar]
- 33.De Falco E, Porcelli D, Torella AR, et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 2004;104:3472–3482. doi: 10.1182/blood-2003-12-4423. [DOI] [PubMed] [Google Scholar]
- 34.Tarzami ST, Wang G, Li W, Green L, Singh JP. Thrombin and PAR-1 stimulate differentiation of bone marrow-derived endothelial progenitor cells. J Thromb Haemost. 2006;4:656–663. doi: 10.1111/j.1538-7836.2006.01788.x. [DOI] [PubMed] [Google Scholar]
- 35.Jin H, Aiyer A, Su J, et al. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J Clin Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leclercq TM, Moretti PA, Vadas MA, Pitson SM. Eukaryotic elongation factor 1A interacts with sphingosine kinase and directly enhances its catalytic activity. J Biol Chem. 2008;283:9606–9614. doi: 10.1074/jbc.M708782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamble JR, Matthias LJ, Meyer G, et al. Regulation of in vitro capillary tube formation by anti-integrin antibodies. J Cell Biol. 1993;121:931–943. doi: 10.1083/jcb.121.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitson SM, Moretti PA, Zebol JR, et al. Expression of a catalytically inactive sphingosine kinase mutant blocks agonist-induced sphingosine kinase activation: a dominant-negative sphingosine kinase. J Biol Chem. 2000;275:33945–33950. doi: 10.1074/jbc.M006176200. [DOI] [PubMed] [Google Scholar]
- 39.Roberts JL, Moretti PA, Darrow AL, et al. An assay for sphingosine kinase activity using biotinylated sphingosine and streptavidin-coated membranes. Anal Biochem. 2004;331:122–129. doi: 10.1016/j.ab.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 40.Heeschen C, Aicher A, Lehmann R, et al. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003;102:1340–1346. doi: 10.1182/blood-2003-01-0223. [DOI] [PubMed] [Google Scholar]
- 41.Bradley TR, Metcalf D. The growth of mouse bone marrow cells in vitro. Aust J Exp Biol Med Sci. 1966;44:287–299. doi: 10.1038/icb.1966.28. [DOI] [PubMed] [Google Scholar]
- 42.Whetton AD, Lu Y, Pierce A, Carney L, Spooncer E. Lysophospholipids synergistically promote primitive hematopoietic cell chemotaxis via a mechanism involving Vav 1. Blood. 2003;102:2798–2802. doi: 10.1182/blood-2002-12-3635. [DOI] [PubMed] [Google Scholar]
- 43.Vessey DA, Kelley M, Zhang J, et al. Dimethylsphingosine and FTY720 inhibit the SK1 form but activate the SK2 form of sphingosine kinase from rat heart. J Biochem Mol Toxicol. 2007;21:273–279. doi: 10.1002/jbt.20193. [DOI] [PubMed] [Google Scholar]
- 44.Loomans CJ, Wan H, de Crom R, et al. Angiogenic murine endothelial progenitor cells are derived from a myeloid bone marrow fraction and can be identified by endothelial NO synthase expression. Arterioscler Thromb Vasc Biol. 2006;26:1760–1767. doi: 10.1161/01.ATV.0000229243.49320.c9. [DOI] [PubMed] [Google Scholar]
- 45.Allende ML, Sasaki T, Kawai H, et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 46.French KJ, Schrecengost RS, Lee BD, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 47.Wattenberg BW, Pitson SM, Raben DM. The sphingosine and diacylglycerol kinase superfamily of signaling kinases: localization as a key to signaling function. J Lipid Res. 2006;47:1128–1139. doi: 10.1194/jlr.R600003-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Powell TM, Paul JD, Hill JM, et al. Granulocyte colony-stimulating factor mobilizes functional endothelial progenitor cells in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2005;25:296–301. doi: 10.1161/01.ATV.0000151690.43777.e4. [DOI] [PubMed] [Google Scholar]
- 49.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 50.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–2053. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- 52.Brunner S, Winogradow J, Huber BC, et al. Erythropoietin administration after myocardial infarction in mice attenuates ischemic cardiomyopathy associated with enhanced homing of bone marrow-derived progenitor cells via the CXCR-4/SDF-1 axis. FASEB J. 2009;23:351–361. doi: 10.1096/fj.08-109462. [DOI] [PubMed] [Google Scholar]
- 53.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 54.Kohno M, Momoi M, Oo ML, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26:7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pitson SM, Xia P, Leclercq TM, et al. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med. 2005;201:49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kono M, Mi Y, Liu Y, et al. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 57.Walter DH, Rochwalsky U, Reinhold J, et al. Sphingosine-1-phosphate stimulates the functional capacity of progenitor cells by activation of the CXCR4-dependent signaling pathway via the S1P3 receptor. Arterioscler Thromb Vasc Biol. 2007;27:275–282. doi: 10.1161/01.ATV.0000254669.12675.70. [DOI] [PubMed] [Google Scholar]
- 58.Li X, Stankovic M, Bonder CS, et al. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008;111:3489–3497. doi: 10.1182/blood-2007-05-092148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martini S, Peters H, Bohler T, Budde K. Current perspectives on FTY720. Expert Opin Investig Drugs. 2007;16:505–518. doi: 10.1517/13543784.16.4.505. [DOI] [PubMed] [Google Scholar]
- 60.Liu Q, Zhao X, Frissora F, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111:275–284. doi: 10.1182/blood-2006-10-053884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keller CD, Rivera GP, Tolle M, et al. Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. Am J Pathol. 2007;170:281–292. doi: 10.2353/ajpath.2007.060485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimura T, Boehmler AM, Seitz G, et al. The sphingosine 1-phosphate receptor agonist FTY720 supports CXCR4-dependent migration and bone marrow homing of human CD34+ progenitor cells. Blood. 2004;103:4478–4486. doi: 10.1182/blood-2003-03-0875. [DOI] [PubMed] [Google Scholar]
- 63.Sieveking DP, Buckle A, Celermajer DS, Ng MK. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol. 2008;51:660–668. doi: 10.1016/j.jacc.2007.09.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}