

Abstract
Information about polymorphism, population structure, and linkage disequilibrium (LD) is crucial for association studies of complex trait variation. However, most genomewide studies have focused on model systems, with very few analyses of undisturbed natural populations. Here, we sequenced 86 mapped nuclear loci for a sample of 46 genotypes of Boechera stricta and two individuals of B. holboellii, both wild relatives of Arabidopsis. Isolation by distance was significant across the species range of B. stricta, and three geographic groups were identified by structure analysis, principal coordinates analysis, and distance-based phylogeny analyses. The allele frequency spectrum indicated a genomewide deviation from an equilibrium neutral model, with silent nucleotide diversity averaging 0.004. LD decayed rapidly, declining to background levels in ∼10 kb or less. For tightly linked SNPs separated by <1 kb, LD was dependent on the reference population. LD was lower in the specieswide sample than within populations, suggesting that low levels of LD found in inbreeding species such as B. stricta, Arabidopsis thaliana, and barley may result from broad geographic sampling that spans heterogeneous genetic groups. Finally, analyses also showed that inbreeding B. stricta and A. thaliana have ∼45% higher recombination per kilobase than outcrossing A. lyrata.
ASSOCIATION studies are one of the fundamental tools for identification of the genes responsible for complex trait variation (Aranzana et al. 2005). The distance over which linkage disequilibrium (LD) persists will determine the number and density of markers and appropriate experimental design for association analysis (Flint-Garcia et al. 2003; Slatkin 2008). Inbreeding species are expected to have high levels of LD (Nordborg and Innan 2002). To date, genomic surveys of LD using SNP data are available for three highly selfing plant species, Arabidopsis thaliana, barley, and rice. In A. thaliana, LD decays within 10 kb at a genome level (Kim et al. 2007), which is more rapidly than expected considering its high levels of inbreeding. A recent study of LD in wild barley (which has a 98% selfing rate) showed very low levels of LD (Morrell et al. 2005). However, inbred rice populations show significant LD across >75 kb (Mather et al. 2007). Thus, some inbred plant species have surprisingly low levels of LD in specieswide samples. To test the generality of this pattern and examine possible evolutionary explanations for low levels of LD, we used genomewide single copy nuclear loci to investigate the extent and patterns of LD for Boechera stricta, a close relative of Arabidopsis, which has largely undisturbed natural populations and a high inbreeding rate.
Information on population structure is important in association studies (Goldstein and Weale 2001; Flint-Garcia et al. 2003). The primary obstacles to successful association studies in plants are population structure and levels of linkage disequilibrium which may be inappropriate for QTL identification. The presence of subpopulations can result in spurious associations due to confounding of unlinked markers with phenotypic variation (Buckler and Thornsberry 2002). Thus, population structure must be investigated to determine the potential for association analyses. Population structure is also crucial for understanding the evolutionary patterns of genes of interest. Most plant species studied to date do not fit the demographic assumptions of the standard equilibrium neutral model, and thus deviation from neutral expectation is relatively common (Wright and Gaut 2005; Mitchell-Olds and Schmitt 2006; Ross-Ibarra et al. 2008). One approach to understanding evolutionarily important polymorphisms is to identify loci that deviate from genomewide patterns of variation, which may indicate a locus-specific influence of natural selection. Thus, adaptation is best inferred by comparison to genomewide patterns of nucleotide variation and population structure (Mitchell-Olds and Schmitt 2006).
In the past decade most studies of molecular population genetics have used model organisms, while natural populations and organisms with complex life histories have received far less attention (Mitchell-Olds and Schmitt 2006). However, many key questions in ecology and evolutionary biology cannot be addressed solely using model systems (Gracey and Cossins 2003; Lee and Mitchell-Olds 2006). Recent progress in computational biology, comparative genomics, phylogenetics, and functional genomics makes it possible to apply these advances to diverse nonmodel systems (Schranz et al. 2007a). Here we focus on the genus Boechera, which is closely related to Arabidopsis, thus providing access to information and techniques from this model plant species. Boechera is a widespread North American group with great potential for studies of ecology and evolution, occurring in diverse habitats, with a range of mating systems, as well as highly variable phenotypes (Mitchell-Olds 2001). B. stricta (previously Arabis drummondii) is a morphologically and genetically well-defined, monophyletic, short-lived perennial crucifer species (Dobes et al. 2004). Genetic and molecular analyses indicate that B. stricta is predominantly diploid, sexual, and inbreeding (FIS = 0.9 and FIT = 0.95) (Schranz et al. 2005; Song et al. 2006). B. stricta populations are distributed in diverse habitats across much of western North America, providing opportunities to study evolutionary and ecological functional genomics in undisturbed natural environments.
In this study, we analyzed nucleotide polymorphism at 86 mapped loci in 46 individuals of B. stricta. We examined population structure across the species range, patterns of genomewide nucleotide polymorphism, and LD on a scale of nucleotides and centimorgans. Specifically, we ask the following questions: (a) Is the species distribution genetically structured? If so, what are the levels of population differentiation? (b) What are the levels of genomewide polymorphism? (c) What is the extent of LD at nucleotide and centimorgan scales, for both specieswide and population-level sampling?
MATERIALS AND METHODS
Sampling:
We analyzed 46 genotypes of B. stricta (one individual/site), as well as two B. holboellii plants from two locations (see supplemental Table 1; Figure 1A). The B. stricta individuals span most of the species range in the western United States. Although asexual hybrids between B. stricta and B. holboellii are found in some populations (Schranz et al. 2005; Kantama et al. 2007), these are readily identified by their divergent morphology and high levels of heterozygosity, and are not included in this study. Average FIS and FIT in B. stricta are ∼0.90 and 0.95, respectively (Song et al. 2006), and these studied genotypes had been propagated by self-pollination in the laboratory for one generation; hence, levels of residual heterozygosity were very low.
Figure 1.—

Geographic distribution and population structure. (A) Geographic distribution of the samples in this study. Blue, SOUTH; red, NORTH; yellow, WEST; and green, B. holboellii. (B) Population structure based on Bayesian clustering for the 46 B. stricta ecotypes and 2 B. holboellii individuals.
Loci, primer design, and sequencing:
The loci included in this study were chosen from the data set generated from end sequencing of lambda clones (Windsor et al. 2006) with the following filtering steps: exclude loci <600 bp; blast each locus against ∼43,000 B. stricta sequences in GenBank, and exclude those with E_values >1E-2 to any other sequences; and exclude loci containing microsatellites. We chose 69 loci located on linkage groups 2–7 of the B. stricta genetic map (Schranz et al. 2007b). Loci on linkage group 1 (LG1) were not selected because this chromosome showed low levels of recombination in the mapping population (Schranz et al. 2007b). Primers were designed using either PRIMER3 or PRIMACLADE software (Rozen and Skaletsky 2000; Gadberry et al. 2005), and the length of the PCR products ranged from 600 to 800 bp. In addition, 17 loci were chosen from previous studies (Schmid et al. 2005; Schranz et al. 2007b; Song and Mitchell-Olds 2007). Overall, a total of 86 mapped loci from linkage groups 2–7 (Schranz et al. 2007b) were examined. We used direct sequencing of PCR products as described (Song and Mitchell-Olds 2007). Base calling and sequence trimming followed Schmid et al. (2003). A few sites showing heterozygosity were considered as missing data.
Data analysis:
Polymorphism and diversity:
Sequences were aligned with ClustalW (Thompson et al. 1994) and diversity estimates (S, π, and θ) for the 86 loci that were obtained from DNASP 4.0 (Rozas et al. 2003). For the comparisons of polymorphisms at silent, synonymous, and nonsynonymous sites, 82 loci, with clear orthologs in A. thaliana, were analyzed with the MANVA program (http://www.ub.es/softevol/manva/).
Structure analyses:
Haplotype data were generated using original Python scripts for the 86 loci, with singleton and indel sites excluded from haplotype assignments. We used two Bayesian Markov Chain Monte Carlo programs, INSTRUCT and STRUCTURE, to infer historical lineages that show clusters of similar genotypes (Pritchard et al. 2000; Falush et al. 2003; Gao et al. 2007). STRUCTURE assumes Hardy–Weinberg equilibrium and linkage equilibrium within populations, although it can be applied to partially inbred genotypes by randomly choosing a single allele from each individual (J. Pritchard, personal communication). In contrast, INSTRUCT does not assume Hardy–Weinberg equilibrium and allows simultaneous inference of the selfing rate and the number and admixture of historical lineages (Gao et al. 2007). In these analyses, each locus was treated as a marker with multialleles, so that two individuals had a different type if they differed at any nonsingleton site.
Both INSTRUCT and STRUCTURE programs assume that the marker loci are statistically independent, hence, in linkage equilibrium and not closely linked with one another. To verify this assumption, LD was analyzed for these 86 loci with FSTAT version 2.9.3 (Goudet 2001). We ran INSTRUCT on the data for five independent chains for each K value (K = 2–9). Each chain was iterated 200,000 times after burn-in with 100,000 iterations, using the model for inferring population structure only with admixture. In addition, the STRUCTURE algorithm was run with a burn-in length of 100,000 MCMC iterations and then 50,000 iterations for estimating the parameters. This was repeated five times for each K, ranging from one to nine.
Both INSTRUCT and STRUCTURE analyses consistently identified four clusters for the 48 individuals, with three clusters for the 46 B. stricta individuals (see results). We also analyzed population subdivision employing principal coordinates analysis (PCA) implemented in GENALEX (Peakall and Smouse 2006) on pairwise genetic distances among all 46 individuals of B. stricta and two individuals of B. holboellii. Finally, a neighbor-joining tree on the basis of genetic distance was constructed in MEGA (Tamura et al. 2007). We obtained the same grouping patterns from all these methods. On the basis of the three inferred groups in B. stricta, we analyzed species-level and within-group diversity based on S and θ using MANVA (http://www.ub.es/softevol/manva/). The statistical comparisons employed paired t-tests in SYSTAT (version 11). Genetic differentiation between pairs of inferred populations was calculated with Snn (Hudson 2000) and FST (Hudson et al. 1992). Snn (nearest-neighbor statistic) is a powerful statistic for detecting genetic differentiation using sequence-based analysis over a wide range of sample size and levels of variation (Hudson 2000). If two populations are highly differentiated, Snn is expected to be near one. For FST, we employed Hudson et al.'s (1992) Equation 3: FST = 1 − Hw/Hb (Hw is mean number of differences between sequences from the same population, and Hb is mean number of differences between sequences from the different populations). The hierarchical analysis of population differentiation was conducted using AMOVA implemented in GENALEX (Peakall and Smouse 2006).
Intralocus and interlocus LD:
The level of LD between pairs of sites within each locus was estimated using the software package TASSEL (http://www2.maizegenetics.net/index.php?page=home/index.html). Only biallelic SNPs with at least 10% frequency were considered. SNPs typed in <75% of the individuals in each group were excluded from LD analyses. We evaluate LD using the correlation coefficient r2 between each pair of SNPs (Hartl and Clark 2007). The intralocus LD was estimated for the entire B. stricta sample, as well as for each structure group (NORTH, SOUTH, and WEST, see results). Decay of intralocus LD with distance in base pairs (bp) was evaluated by nonlinear regression using SigmaPlot (Windows version 10.0).
To examine the population recombination parameter (ρ = 4Nr) for the whole data set, as well as each population, we obtained the estimates for each locus using a composite-likelihood approach (Hudson 2001) as implemented in the LDhat software package (McVean et al. 2002).
We evaluated interlocus LD by using the correlation coefficient r2 between all pairs of SNPs along each linkage group using HAPLOVIEW (version 4.0) (Barrett et al. 2005). Interlocus r2 was calculated for the total B. stricta data set, as well as for each group (NORTH, SOUTH, and WEST). Decay of interlocus LD with distance (cM) for all samples and the three groups was plotted using SYSTAT, on the basis of the estimate that 1 cM = 100 kb (Schranz et al. 2007b). A Mantel permutation test implemented in the ZT program (Bonnet and Van de Peer 2002) was used to test for significant correlation between recombination distance and LD for each linkage group.
To better understand the observed patterns of linkage disequilibrium in our species, we calculated variance components of linkage disequilibrium for each linkage group on the basis of Ohta (1982) with the program LINKDOS (Black and Krafsur 1985). By analogy with the partitioning of the inbreeding coefficient, Ohta (1982) defined several variance components of disequilibrium to account for within- (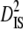 and
and 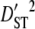 ) and between- (
) and between- (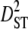 and
and 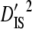 ) subpopulation effects, to discriminate among possible evolutionary forces shaping patterns of LD (Black and Krafsur 1985; Kremer and Zanetto 1997; Vitalis et al. 2002). When variances of LD among subpopulations (
) subpopulation effects, to discriminate among possible evolutionary forces shaping patterns of LD (Black and Krafsur 1985; Kremer and Zanetto 1997; Vitalis et al. 2002). When variances of LD among subpopulations (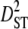 and
and 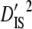 ) are greater than those within populations (
) are greater than those within populations (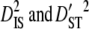 ), this suggests that genetic drift plays an important role in shaping observed patterns of LD (Ohta 1982; Black and Krafsur 1985; Vitalis et al. 2002). Alternatively, epistatic selection among loci also might be reflected by these components of LD.
), this suggests that genetic drift plays an important role in shaping observed patterns of LD (Ohta 1982; Black and Krafsur 1985; Vitalis et al. 2002). Alternatively, epistatic selection among loci also might be reflected by these components of LD.
RESULTS
Polymorphism and population structure in B. stricta:
Genome-level polymorphism:
We surveyed 86 single copy nuclear loci with average length (including indels) of 591 bp. The total length of all analyzed amplicons was ∼51 kb, and the number of nucleotides sequenced reached 2.2 million. Overall, a total of 687 polymorphic sites were detected, averaging S = 8 per locus and 0.013 per nucleotide. Pairwise sequence diversity averages π = 0.0030 (range from 0.0001 to 0.028) and mean θ = 0.0035 (range from 0.0005 to 0.018) (Table 1). Polymorphism patterns along each linkage group showed little evidence for local windows of high or low diversity. Instead, we found apparently haphazard differences among adjacent loci (see supplemental Figure 1). The 82 loci, with clear orthologs in A. thaliana, show 13.5% divergence between B. stricta and A. thaliana at silent sites, and average levels of nucleotide diversity of 0.0035, 0.0041, and 0.0017 for silent, synonymous, and nonsynonymous sites, respectively.
TABLE 1.
Nucleotide variation at 86 mapped loci in Boechera stricta
| Locus name | Linkage group | MAP_Position (CM) | No. of sequences | Sequence length | Sa | hb | θwc | πd | Tajima's D |
|---|---|---|---|---|---|---|---|---|---|
| Bst012863 | BstLG2 | 0.0 | 46 | 438 | 5 | 5 | 0.0026 | 0.0020 | −0.59 |
| Bst012428 | BstLG2 | 6.3 | 46 | 453 | 5 | 6 | 0.0026 | 0.0026 | 0.07 |
| Hypo_1 | BstLG2 | 29.4 | 45 | 756 | 6 | 7 | 0.0018 | 0.0008 | −1.41 |
| CAL | BstLG2 | 34.0 | 43 | 658 | 8 | 7 | 0.0029 | 0.0025 | −0.41 |
| Stzfp | BstLG2 | 39.7 | 45 | 584 | 3 | 4 | 0.0012 | 0.0008 | −0.72 |
| BstES0023 | BstLG2 | 42.4 | 44 | 594 | 4 | 5 | 0.0016 | 0.0014 | −0.19 |
| Bst004963 | BstLG2 | 45.6 | 41 | 451 | 7 | 8 | 0.0037 | 0.0023 | −1.03 |
| BstES0030 | BstLG2 | 60.7 | 46 | 475 | 5 | 5 | 0.0024 | 0.0020 | −0.44 |
| Bst011023 | BstLG2 | 62.2 | 45 | 449 | 19 | 9 | 0.0097 | 0.0078 | −0.64 |
| At131 | BstLG2 | 81.2 | 25 | 473 | 6 | 3 | 0.0033 | 0.0042 | 0.80 |
| Bst004807 | BstLG2 | 83.7 | 46 | 439 | 4 | 5 | 0.0021 | 0.0012 | −1.04 |
| MAF1 | BstLG2 | 102.5 | 42 | 751 | 3 | 4 | 0.0010 | 0.0005 | −1.08 |
| Bst011647 | BstLG2 | 114.2 | 46 | 416 | 4 | 4 | 0.0022 | 0.0019 | −0.36 |
| Bst027958 | BstLG3 | 0.0 | 44 | 445 | 3 | 4 | 0.0016 | 0.0008 | −0.97 |
| Bst003056 | BstLG3 | 7.4 | 46 | 461 | 10 | 8 | 0.0050 | 0.0054 | 0.21 |
| HRG | BstLG3 | 8.2 | 45 | 622 | 15 | 17 | 0.0058 | 0.0036 | −1.18 |
| Bst010608 | BstLG3 | 14.2 | 31 | 372 | 17 | 7 | 0.0116 | 0.0189 | 1.70 |
| Bst011191 | BstLG3 | 21.6 | 46 | 460 | 4 | 5 | 0.0022 | 0.0022 | −0.06 |
| Dex1 | BstLG3 | 25.2 | 42 | 1185 | 6 | 7 | 0.0012 | 0.0007 | −1.13 |
| FLD_1 | BstLG3 | 28.8 | 45 | 731 | 7 | 6 | 0.0022 | 0.0017 | −0.67 |
| SPY | BstLG3 | 32.0 | 44 | 745 | 6 | 7 | 0.0019 | 0.0007 | −1.63 |
| Bst001594 | BstLG3 | 41.9 | 46 | 499 | 6 | 7 | 0.0027 | 0.0021 | −0.61 |
| VRN1_1 | BstLG3 | 60.0 | 45 | 574 | 7 | 6 | 0.0028 | 0.0028 | −0.03 |
| Bst027506 | BstLG3 | 68.2 | 44 | 441 | 5 | 6 | 0.0026 | 0.0013 | −1.28 |
| Abi_3 | BstLG3 | 77.6 | 45 | 835 | 5 | 5 | 0.0014 | 0.0008 | −0.97 |
| MET1_1 | BstLG3 | 82.2 | 45 | 528 | 2 | 4 | 0.0009 | 0.0008 | −0.17 |
| Bst027135 | BstLG3 | 88.1 | 44 | 446 | 19 | 17 | 0.0098 | 0.0054 | −1.56 |
| Bst027974 | BstLG3 | 88.1 | 45 | 496 | 29 | 7 | 0.0137 | 0.0045 | −2.34 |
| BstES0049 | BstLG3 | 101.5 | 46 | 621 | 9 | 9 | 0.0033 | 0.0018 | −1.26 |
| Bst002609 | BstLG3 | 123.1 | 46 | 470 | 5 | 6 | 0.0025 | 0.0019 | −0.58 |
| Nph3 | BstLG3 | 134.3 | 45 | 1077 | 10 | 8 | 0.0021 | 0.0019 | −0.37 |
| Bst001650 | BstLG4 | 17.8 | 46 | 502 | 3 | 4 | 0.0014 | 0.0013 | −0.15 |
| Fdh_Song_1 | BstLG4 | 23.6 | 45 | 785 | 5 | 6 | 0.0015 | 0.0009 | −0.94 |
| BstES0026 | BstLG4 | 37.0 | 46 | 525 | 4 | 5 | 0.0017 | 0.0009 | −1.10 |
| TIGR3144_1 | BstLG4 | 46.1 | 45 | 537 | 5 | 4 | 0.0021 | 0.0016 | −0.64 |
| SNZ | BstLG4 | 70.6 | 43 | 467 | 4 | 5 | 0.0021 | 0.0010 | −1.24 |
| HOS1_1 | BstLG4 | 72.7 | 43 | 852 | 4 | 5 | 0.0011 | 0.0010 | −0.10 |
| Tigr1093 | BstLG4 | 82.8 | 43 | 486 | 5 | 6 | 0.0024 | 0.0020 | −0.42 |
| Fnr_Song_1 | BstLG5 | 18.2 | 44 | 559 | 4 | 5 | 0.0016 | 0.0014 | −0.41 |
| Bst028989 | BstLG5 | 23.5 | 45 | 452 | 7 | 5 | 0.0035 | 0.0030 | −0.41 |
| PhyB_1 | BstLG5 | 28.7 | 46 | 615 | 8 | 7 | 0.0030 | 0.0014 | −1.48 |
| RGA1 | BstLG5 | 37.0 | 46 | 707 | 5 | 6 | 0.0016 | 0.0015 | −0.13 |
| Bst012271 | BstLG5 | 41.1 | 46 | 382 | 12 | 10 | 0.0076 | 0.0097 | 0.76 |
| Bst013659 | BstLG5 | 48.8 | 46 | 425 | 3 | 4 | 0.0016 | 0.0007 | −1.23 |
| Bst006119 | BstLG5 | 49.4 | 45 | 475 | 15 | 4 | 0.0074 | 0.0059 | −0.68 |
| Bst001200 | BstLG5 | 62.8 | 46 | 458 | 5 | 6 | 0.0025 | 0.0019 | −0.58 |
| Golm66 | BstLG5 | 68.5 | 45 | 471 | 4 | 5 | 0.0021 | 0.0007 | −1.56 |
| AtG1 | BstLG5 | 73.9 | 45 | 509 | 3 | 4 | 0.0014 | 0.0011 | −0.33 |
| Bst007412 | BstLG5 | 99.1 | 45 | 440 | 6 | 6 | 0.0031 | 0.0035 | 0.29 |
| LLox2_1 | BstLG5 | 112.5 | 43 | 733 | 2 | 3 | 0.0006 | 0.0006 | −0.02 |
| FRI_1 | BstLG6 | 0.0 | 44 | 758 | 14 | 10 | 0.0043 | 0.0033 | −0.73 |
| Golm73 | BstLG6 | 6.6 | 39 | 709 | 6 | 5 | 0.0020 | 0.0010 | −1.39 |
| GA1 | BstLG6 | 9.0 | 46 | 621 | 14 | 10 | 0.0056 | 0.0039 | −0.97 |
| AOP3 | BstLG6 | 11.7 | 46 | 629 | 6 | 7 | 0.0022 | 0.0012 | −1.21 |
| BstES0005 | BstLG6 | 20.0 | 45 | 512 | 24 | 7 | 0.0108 | 0.0155 | 1.36 |
| Bst030242 | BstLG6 | 21.9 | 39 | 486 | 24 | 15 | 0.0119 | 0.0060 | −1.75 |
| Det1 | BstLG6 | 27.7 | 45 | 687 | 4 | 5 | 0.0013 | 0.0015 | 0.35 |
| STK | BstLG6 | 28.2 | 43 | 482 | 5 | 5 | 0.0024 | 0.0027 | 0.32 |
| Bst004397 | BstLG6 | 28.3 | 46 | 457 | 3 | 4 | 0.0015 | 0.0007 | −1.12 |
| Bst002440 | BstLG6 | 29.6 | 38 | 492 | 11 | 11 | 0.0053 | 0.0028 | −1.23 |
| BstES0011 | BstLG6 | 32.5 | 46 | 590 | 3 | 4 | 0.0012 | 0.0010 | −0.25 |
| Bst030781 | BstLG6 | 39.3 | 41 | 471 | 20 | 11 | 0.0099 | 0.0095 | −0.20 |
| Rd22F | BstLG6 | 40.4 | 43 | 993 | 9 | 10 | 0.0021 | 0.0018 | −0.39 |
| Bst012781 | BstLG6 | 67.8 | 46 | 437 | 2 | 3 | 0.0010 | 0.0006 | −0.86 |
| TFL2_1 | BstLG6 | 69.3 | 43 | 526 | 7 | 6 | 0.0031 | 0.0017 | −1.19 |
| AtV8_1 | BstLG6 | 70.5 | 45 | 482 | 7 | 8 | 0.0033 | 0.0014 | −1.56 |
| CO | BstLG6 | 72.6 | 46 | 642 | 12 | 6 | 0.0044 | 0.0054 | 0.67 |
| Chs | BstLG6 | 78.6 | 45 | 725 | 3 | 3 | 0.0009 | 0.0002 | −1.71 |
| Bst007987 | BstLG6 | 107.5 | 45 | 449 | 29 | 4 | 0.0179 | 0.0278 | 1.48 |
| CYP79A2 | BstLG6 | 108.2 | 46 | 604 | 7 | 6 | 0.0026 | 0.0018 | −0.82 |
| Bst027123 | BstLG6 | 109.5 | 45 | 397 | 13 | 12 | 0.0076 | 0.0055 | −0.84 |
| Pul | BstLG6 | 109.8 | 43 | 653 | 6 | 7 | 0.0021 | 0.0016 | −0.70 |
| Bst006354 | BstLG6 | 115.1 | 45 | 491 | 2 | 3 | 0.0009 | 0.0004 | −1.14 |
| Golm23 | BstLG6 | 115.7 | 42 | 548 | 4 | 5 | 0.0017 | 0.0016 | −0.29 |
| BstES0012 | BstLG7 | 27.8 | 45 | 618 | 11 | 10 | 0.0041 | 0.0034 | −0.53 |
| Cyp83A_1 | BstLG7 | 30.6 | 27 | 1734 | 27 | 11 | 0.0041 | 0.0023 | −1.63 |
| BstES0007 | BstLG7 | 31.8 | 46 | 598 | 8 | 8 | 0.0030 | 0.0034 | 0.27 |
| BstES0008 | BstLG7 | 31.8 | 46 | 558 | 5 | 7 | 0.0020 | 0.0020 | −0.07 |
| FCA2_1 | BstLG7 | 42.2 | 44 | 570 | 8 | 9 | 0.0033 | 0.0023 | −0.85 |
| Bst010295 | BstLG7 | 50.4 | 46 | 475 | 12 | 12 | 0.0058 | 0.0052 | −0.28 |
| Bst001405 | BstLG7 | 65.0 | 46 | 466 | 6 | 7 | 0.0029 | 0.0028 | −0.10 |
| Cip7_1 | BstLG7 | 76.5 | 45 | 747 | 14 | 8 | 0.0044 | 0.0041 | −0.22 |
| VIN3_like | BstLG7 | 86.9 | 43 | 643 | 3 | 2 | 0.0011 | 0.0001 | −0.19 |
| Cyp83B1 | BstLG7 | 89.9 | 30 | 1335 | 6 | 7 | 0.0011 | 0.0011 | −0.22 |
| Bst004238 | BstLG7 | 98.5 | 45 | 452 | 1 | 2 | 0.0005 | 0.0008 | 0.75 |
| Bst027363 | BstLG7 | 117.4 | 45 | 438 | 3 | 4 | 0.0016 | 0.0026 | 1.42 |
Number of polymorphic sites.
Haplotype diversity (Nei 1987).
Sequence diversity (Watterson 1975).
Pairwise sequence diversity (Lynch and Crease 1990).
The site frequency spectrum can indicate whether patterns of polymorphism are compatible with equilibrium neutral expectations. Our results show that the distribution of polymorphism (θw) for silent sites shows an excess of rare alleles (Figure 2A), in comparison to equilibrium neutral expectation. We also investigated the frequency spectrum of nucleotide polymorphisms using Tajima's D (Tajima 1989). The frequency distribution of Tajima's D is skewed toward negative values (Figure 2B). The mean value of Tajima's D for all sites was −0.46 (Table 1), which is significantly <0 (t = −6.43, d.f. = 85, P < 0.001), showing a departure from neutral equilibrium expectations. The Tajima's D for silent sites was also significantly <0 (t = −2.647; d.f. = 71; P = 0.01). The Tajima's D value is slightly less negative in noncoding than in coding regions (−0.144 and −0.521, respectively), which is compatible with the hypothesis that nonsynonymous polymorphisms occur at relatively lower frequencies due to weak purifying selection (Mitchell-Olds et al. 2007; Fox et al. 2008).
Figure 2.—

The distribution of nucleotide diversity and Tajima's D among loci. (A) The frequency distribution of diversity. (B) The frequency distribution of Tajima's D.
Population structure:
To verify independence of the 86 loci assumed by INSTRUCT (Gao et al. 2007), levels of LD were tested using haplotype data. We used sequential Bonferroni (Rice 1989) to control for multiple statistical tests, providing a less conservative test than the standard Bonferroni procedure. Using this approach for genomewide tests on haplotype data, we found no significant pairwise LD. Although closely linked SNPs show association (below), the haplotype data used for INSTRUCT are compatible with the assumption of independent marker loci. INSTRUCT identified three population clusters for the 46 B. stricta ecotypes, henceforth referred to as the NORTHERN, SOUTHERN, and WESTERN groups (Figure 1B). STRUCTURE, which can accommodate disequilibrium among linked markers (Pritchard et al. 2000; Falush et al. 2003), gave very similar results, as did PCA and tree-based analyses. Only the results from STRUCTURE are shown here. The NORTHERN group is geographically located in the northern Rocky Mountains, while the SOUTHERN group is centered in Colorado. The WESTERN group corresponds to the Monida group found previously (Song et al. 2006), which is now seen to extend westward to the Cascades and Sierra Nevada. Three individuals from Utah (UT02, UT05, and UT07) and one accession from Arizona (AZ01) showed admixture between NORTH and SOUTH, and one accession from Idaho (ID89) showed admixture between NORTH and WEST. Aside from admixed individuals, there are 6, 12, and 23 individuals in the WESTERN, SOUTHERN, and NORTHERN groups, respectively. The PCA analysis of 46 B. stricta individuals and 2 B. holboellii individuals showed similar patterns of genetic differentiation (see supplemental Figure 2), as did a neighbor-joining tree using genetic distance (not shown). Despite their very different assumptions and modeling approaches, INSTRUCT, STRUCTURE, PCA, and tree-based analyses all provided very similar results, giving confidence in the robustness of these inferred population groups.
We observed a clear pattern of isolation by distance: individuals are more similar to individuals that grow nearby (Figure 1A). The Mantel test showed significant isolation by distance across the sampled range (r = 0.35; P = 0.001; 10,000 randomizations; see supplemental Figure 3). In comparison, most analyses in A. thaliana have found a weaker pattern of isolation by distance (Sharbel et al. 2000; Nordborg et al. 2005), which may reflect a homogenization of allele frequencies resulting from thousands of years of human disturbance across Eurasia.
High differentiation was detected between the three groups identified by structure analyses. Average FST among STRUCTURE groups for the 86 mapped loci is 0.45. This result is similar to a previous microsatellite study (Song et al. 2006), although population sampling was rather different between these two analyses. The pairwise FST estimates between the three STRUCTURE groups averaged 0.24, 0.53, and 0.56 for NORTH vs. SOUTH, WEST vs. SOUTH, and WEST vs. NORTH, respectively, on the basis of sequence data (Figure 3). There is much higher differentiation between WEST and EAST (= NORTH + SOUTH), while relatively lower differentiation is found between the NORTHERN and SOUTHERN genotypes. Pairwise Snn values showed a pattern similar to FST analyses: Snn values are 0.69, 0.86, and 0.92 for NORTH vs. SOUTH, WEST vs. SOUTH, and WEST vs. NORTH, respectively. The analysis of molecular variance (AMOVA) implemented in GENALEX confirmed high levels of differentiation between the WESTERN group vs. the other two lineages (38% of total variation occurs between WESTERN vs. EASTERN groups) (see supplemental Figure 4).
Figure 3.—

Population differentiation along linkage groups. Pairwise genomewide distribution of FST between the three historical lineages identified by STRUCTURE and INSTRUCT.
The distribution of FST along the linkage groups may identify chromosome regions showing unusually high or low FST values, which might reflect selective differentiation among population clusters. However, we found no clear trend of FST values along the linkage groups (Figure 3). High levels of FST in most chromosomal regions indicated high differentiation among populations, especially for WEST vs. NORTH and WEST vs. SOUTH. Loci that show high FST within B. stricta are different from those showing high differentiation between B. stricta and B. holboellii; hence, the processes influencing FST evidently differ within vs. between species (data not shown). However, loci with high FST in the WEST vs. NORTH comparison also tend to show high differentiation between WEST and SOUTH (Figure 3).
We also examined sequence polymorphism and differentiation among these groups using A. thaliana to identify ancestral polymorphisms. The WESTERN group has 14 fixed mutations and 37 exclusive polymorphisms, indicating its high differentiation from the rest of the species. The SOUTHERN group has no fixed mutations, although it has 87 exclusive polymorphisms, indicating its high diversity. The NORTHERN group is intermediate, and has 2 fixed mutations and 61 exclusive polymorphisms (Table 2). WEST shows 28 and 22 shared polymorphisms with SOUTH and NORTH separately, while SOUTH and NORTH shared 97 polymorphisms, indicating high differentiation of the WESTERN group from the EASTERN group.
TABLE 2.
Fixed differences and shared polymorphisms in each group (A. thaliana as outgroup)
Fixed biallelic mutations in the studied group.
Polymorphic exclusive biallelic mutations in the studied group.
Shared biallelic mutations.
Polymorphic in the studied group but fixed in the other group.
Polymorphism within populations:
Contrasting results for the entire species vs. populations (three groups identified in the structure analyses) showed a significantly higher level of diversity at specieswide sampling (see supplemental Figure 5; paired t-test, all P < 0.001). A similar pattern has also been found in maize (Moeller et al. 2007). Also, polymorphism levels within the three groups showed notable differences. At silent sites, the SOUTHERN group showed the highest diversity levels (for silent sites, θSOUTH = 0.0024, θNORTH = 0.0017, θWEST = 0.0018; for synonymous sites, θSOUTH = 0.0030, θNORTH = 0.0024, θWEST = 0.0023) (see supplemental Tables 2 and 3), in agreement with an earlier study based on chloroplast sequences (Dobes et al. 2004).
Linkage disequilibrium:
Intralocus LD:
The average value of r2 is 0.66 for all samples, with a range from 0.0 to 1.0. The decay of intralocus LD is shown in the plot of r2 against distance in base pairs between SNPs (Figure 4; see supplemental Figure 6). Nonlinear regression shows clear and rapid decline of LD with distance (correlation coefficient, r = −0.48, P < 0.001), with LD declining to background levels within ∼1 kb. The intralocus LD pattern found in this study is consistent with results for inbreeding barley accessions, as well with outcrossing maize (Morrell et al. 2005). Nevertheless, within-group analyses of intralocus LD show a much higher level of LD within each geographical structure group (Figure 4; see supplemental Figure 6).
Figure 4.—

The decay of short-range linkage disequilibrium within 86 loci for all samples and within each group. The curves are nonlinear regressions of r2 onto distance in base pairs.
Interlocus LD:
The average interlocus r2 value is 0.17 for all samples based on SNPs. Average genomewide LD decays within ∼10 kb for all samples, with similar values within the NORTHERN and SOUTHERN groups (Figure 5). These results are comparable to A. thaliana (Nordborg et al. 2005; Kim et al. 2007). In contrast, elevated r2 persists within the WESTERN group among loosely linked markers separated by >10 cM (∼1 Mb). As with A. thaliana, the rapid decay of LD at a genome level seems surprising considering the high level of inbreeding in these two species. The average r2 calculated within population groups is 0.15 (SOUTH) and 0.22 (NORTH), but shows a higher average of 0.34 in the WESTERN group (Figure 5). The relatively high LD value in the WEST may reflect the limited sample size of this population, or perhaps a historical population bottleneck. In addition, most of the linkage groups showed a significant negative correlation between LD and map distance (P < 0.01 for LG3, -5, and -6; P < 0.05 for LG7), except for LG2 and LG4, which have fewer SNPs (not shown).
Figure 5.—

Linkage disequilibrium as a function of distance for 86 mapped loci for the specieswide sample and within each structure group.
LD partitioning:
Partitioning variance components of LD showed that the variances of disequilibrium among subpopulations (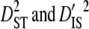 ) are much greater than those within subpopulations (
) are much greater than those within subpopulations (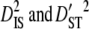 ) for each linkage group (Table 3). Moreover, since the total variance of disequilibrium =
) for each linkage group (Table 3). Moreover, since the total variance of disequilibrium = 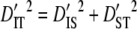 ,
, 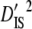 contributed a much larger portion to
contributed a much larger portion to 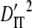 (Table 3). These results suggest that genetic drift plays an important role in shaping observed patterns of LD.
(Table 3). These results suggest that genetic drift plays an important role in shaping observed patterns of LD.
TABLE 3.
OHTA's variance components of linkage disequilibrium in each linkage group of Boechera stricta
| Linkage groups | 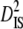 |
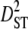 |
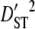 |
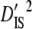 |
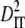 |
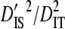 |
|---|---|---|---|---|---|---|
| LG2 | 0.0016 | 0.1020 | 0.0086 | 0.5757 | 0.8753 | 0.6579 |
| LG3 | 0.0043 | 0.1114 | 0.0117 | 0.6493 | 0.8048 | 0.8068 |
| LG4 | 0.0014 | 0.2046 | 0.0130 | 1.0730 | 1.2924 | 0.8302 |
| LG5 | 0.0006 | 0.2071 | 0.0131 | 1.0644 | 1.2666 | 0.8404 |
| LG6 | 0.0056 | 0.0835 | 0.0066 | 0.3827 | 0.4700 | 0.8142 |
| LG7 | 0.0050 | 0.1252 | 0.0283 | 0.7757 | 1.0582 | 0.7330 |
Population recombination parameters:
We examined LD for specieswide samples, as well as for each structure group using the population recombination parameter (ρ = 4Nr). We found no significant difference for ρ in the specieswide sample vs. within each population. The comparisons still remain nonsignificant when controlling for diversity (ρ/θ, not shown). Comparing the two populations with similar sample sizes, the SOUTH shows a higher population recombination rate than the NORTHERN group (ρsouth = 16.3, while ρnorth = 10.1). These estimates are influenced by demographic factors (Beaumont et al. 2001; Przeworski and Wall 2001), complicating interpretation in these spatially structured real-world populations.
DISCUSSION
Levels of LD in B. stricta:
Linkage disequilibrium decays rapidly in B. stricta, declining to background levels (r2 < 0.2) in ∼10 kb or less. For tightly linked SNPs separated by <1 kb, LD is dependent on the reference population: LD is lower in the specieswide sample than within population groups (Figure 4; see supplemental Figure 6), due to the shorter coalescence time for recombination in regional population groups. Nevertheless, loosely linked polymorphisms show comparable levels of LD in the NORTHERN, SOUTHERN, and specieswide samples (Figure 5), reflecting the relatively high rate of recombination among loosely linked markers. However, the WESTERN group had higher LD and lower levels of polymorphism, perhaps due to a historical population bottleneck. Except in the WESTERN group, LD declines rapidly on a scale of a few genes, then shows an asymptotic decline among loosely linked markers. The fine-scale LD pattern among tightly linked markers may be influenced by gene conversion on a local scale (Przeworski and Wall 2001; Haubold et al. 2002; Morrell et al. 2006; Plagnol et al. 2006).
Inbreeding species have low levels of heterozygosity, which causes reduced rates of effective recombination, and therefore high levels of LD are expected in inbreeding species. In contrast to this prediction, two recent studies of inbreeding species, A. thaliana (Kim et al. 2007) and barley (Morrell et al. 2005), have found surprisingly low levels of LD. Low levels of LD also were found in the current study among tightly (intralocus) and loosely linked (interlocus) markers (Figures 4 and 5). Thus, our results show that low levels of LD in specieswide samples of inbreeding organisms are not exceptional. Our findings shed light on several hypotheses, which previously have been suggested to explain the unexpectedly low levels of LD in inbreeding species.
First, low LD might result from a specieswide scale of sampling, which incorporates the entire history of polymorphism and recombination within a species over thousands of generations (Morrell et al. 2005; Kim et al. 2007). Our data support this sample-scale explanation, which predicts that LD should be higher within populations or local regions, as we observe for r2 within loci (Figure 4; see supplemental Figure 6). However, the scale of sampling is less important among loosely linked SNPs (Figure 5), indicating that correlations among distant polymorphisms have had time to decay to background levels within the relatively large NORTHERN and SOUTHERN populations. The analysis of components of LD further supports this sample-scale hypothesis, showing that population subdivision and genetic drift play important roles in shaping the patterns of LD in B. stricta. Similar results also were found in Marsilera strigosa (Vitalis et al. 2002).
Second, inbreeding species may have higher levels of recombination per kilobase, which may reduce levels of LD in these species. Within the limits of available data, our comparative studies support this hypothesis. The genome size of inbreeding B. stricta is only 5% larger than outcrossing A. lyrata (Johnston et al. 2005; Windsor et al. 2006; Oyama et al. 2008), with similar numbers of chromosomes (7 vs. 8, respectively), but B. stricta has a linkage map that is ∼50% longer than A. lyrata (Kuittinen et al. 2004; Yogeeswaran et al. 2005; Schranz et al. 2007b). Likewise, the DNA content of A. lyrata is ∼50% greater than A. thaliana, but the linkage maps of both Arabidopsis species are of similar length (Kuittinen et al. 2004; Yogeeswaran et al. 2005). Thus inbreeding B. stricta and A. thaliana have ∼45% higher recombination per kilobase than outcrossing A. lyrata. Future work is needed to verify the generality of this trend in additional species comparisons.
Third, low LD might result from selection that favors recombinant genotypes with new combinations of parental traits (Bakker et al. 2006). This explanation is plausible for organisms growing in spatially or temporally heterogeneous environments, or as a mechanism to eliminate deleterious mutations (Keightley and Otto 2006; Martin et al. 2006). Experiments are needed from natural populations to verify such fitness advantages for recombinant genotypes.
Finally, low LD may result from ancient recombination events that took place in an outcrossing ancestor (Morrell et al. 2005; Bechsgaard et al. 2006; Tang et al. 2007). However, outcrossing species are rare or absent in the genus Boechera (Schranz et al. 2005); hence, there is little evidence for a recent transition to inbreeding. Rather, it appears that B. stricta has been predominantly inbreeding throughout the history of existing nucleotide polymorphisms.
These analyses suggest that the surprisingly low levels of LD in inbreeding species are influenced by the rangewide geographic sampling that spans heterogeneous genetic groups. In contrast, analyses conducted within population groups result in higher LD estimates among tightly linked polymorphisms, as expected for inbreeding species. This poses a substantial challenge for association studies: low levels of LD (which are desirable for analyses of association) occur in broad genetic and geographic samples, where population structure may be confounded with phenotypic differences among groups, and where statistical adjustment for population structure may remove the very effects of interest (Remington et al. 2001). In contrast, more local samples of homogeneous populations may be simpler to analyze, but are characterized by higher levels of LD and partial confounding of polymorphisms in nearby loci. These concerns further emphasize that the full toolset for analysis of complex trait variation should include both association studies and QTL mapping, especially in species where experimental crosses are feasible.
Nucleotide polymorphism in B. stricta:
Genomewide nucleotide diversity in B. stricta averages π = 0.003 for all loci, and 0.0035, 0.0041, and 0.0017 for silent, synonymous, and nonsynonymous sites, respectively. One explanation for the relatively low level of polymorphism in B. stricta is its predominately self-pollinating breeding system, leading to reduction in effective population size. Microsatellite data suggest that the inbreeding coefficient is ∼0.9 in this species (Song et al. 2006). Mating system is a major factor that affects polymorphism levels and the distribution of genetic variation within and among populations (Charlesworth 2003). Contrasting patterns of genetic variation are found in the outcrossing relative, A. lyrata, with FIS ≈ 0.01, FST ≈ 0.2–0.5, and π ≈ 0.01 (Ramos-Onsins et al. 2004; Clauss and Mitchell-Olds 2006; Wright et al. 2006; Ross-Ibarra et al. 2008). Within-population polymorphism in A. lyrata is even higher than the specieswide level of diversity in A. thaliana. However, the levels of polymorphism in B. stricta are lower than those observed in A. thaliana. This might be explained by the broad Eurasian distribution of A. thaliana, while B. stricta has a relatively restricted distribution in western North America. The levels of diversity in B. stricta are also lower than other well-studied wild species: wild barley (Hordeum vulgare ssp. spontaneum, inbreeding, π = 0.0075; Morrell et al. 2005), teosinte (Zea mays ssp. parviglumis, outcrossing, π = 0.0134; Moeller et al. 2007), and wild sunflower (Helianthus annuus, outcrossing, π = 0.0128; Liu and Burke 2006). Founder effects and extinction of local metapopulations also may have reduced genetic variation in our species.
As in A. thaliana (Nordborg et al. 2005; Schmid et al. 2005), we also observed genomewide deviation from a standard equilibrium neutral model in B. stricta. Demographic factors such as population structure, changes in population size, and low levels of gene flow from related species are possible factors that might explain these patterns of variation. Analyses with more flexible demographic models (Schmid et al. 2005; Caicedo et al. 2007) and increased population sampling may help elucidate evolutionary influences on the patterns of polymorphism in this wild species (Nordborg et al. 2005; Heuertz et al. 2006).
Genetic differentiation among lineages:
Patterns of genomewide neutral variation can be used to infer historical lineages contributing to current populations and may help interpret natural selection at ecologically important genes. Sequence polymorphism from these 86 mapped loci assayed on 46 individuals across the species range identified three lineages corresponding to SOUTHERN and NORTHERN groups, as well as a divergent WESTERN lineage. These results support and expand upon an earlier microsatellite study of 15 populations ranging from Arizona to Montana (Song et al. 2006). Moreover, the sampling of plants in this study spans the whole species range in the United States, and samples loci across the genome. This provides more statistical power in understanding genomewide population structure. The high levels of FST along each linkage group showed no evidence for heterogeneous patterns of differentiation among chromosomal regions (Figure 3), hence no suggestion of variable introgression of chromosomal regions among population lineages.
We also found that the WESTERN group is quite distinct from the rest of the species. Average FST between the WESTERN group and its eastern counterparts is about twofold higher than levels of NORTH–SOUTH differentiation in the eastern part of the range (Figure 3). The WESTERN group is widely dispersed in North America (California, Washington, Idaho, and Montana), and more studies are needed to define the distribution of this group (Figure 1A). The WESTERN group also showed high levels of LD (Figure 5D), although sample size is low within this lineage. Finally, the WESTERN group shows noticeable morphological divergence from the EASTERN individuals, with significantly larger rosette size, growth rates, and trichome density (B.-H. Song, A. Manzaneda, and T. Mitchell-Olds, unpublished data).
Finally, it is striking that the structure of historical lineages in B. stricta (Figure 1B) is simpler than in A. thaliana (with three vs. eight or more genetic lineages), and that B. stricta individuals show less admixture among groups than is found in A. thaliana [compare our Figure 1B with Figure 2A of Nordborg et al. (2005)]. These differences may result from contrasting Pleistocene migrations, or widespread human disturbance in Europe, or a combination of these and other factors.
A model system for ecological genomics:
The genus Boechera is closely related to Arabidopsis, providing access to information and techniques from this model plant species. B. stricta is an inbreeding-tolerant sexual diploid, which is very important for development of near-isogenic lines and positional cloning of QTL. Although it is unknown whether our study populations are at ecological or genetic equilibrium, they have not been impacted by habitat destruction and introduced genotypes, which complicates evolutionary inferences in weedy ephemerals such as A. thaliana and Capsella rubella. We have shown that Boechera populations are locally adapted to some of the ecological differences among these diverse habitats (Knight et al. 2006). To understand the evolutionary processes that influence ecologically important trait variation in natural populations requires positional cloning, genetically undisturbed populations, and the opportunity to compare the fitness of QTL alleles in their natural environments. In the current study we have documented genomewide patterns of genetic variation and linkage disequilibrium, which are essential for evolutionary interpretation of nucleotide polymorphisms at genes of interest (Mitchell-Olds et al. 2007). This combination of ecological characteristics and genetic information is the reason why we have developed B. stricta as a model organism for ecological genomics.
Acknowledgments
We thank B. Charlesworth, M. Johnson, S. Kumagai, A. Manzaneda, C. Olson-Manning, M. Rausher, J. Willis, and several anonymous reviewers for helpful discussion and comments. We thank K. Eberhardt, D. Schnabelrauch, and K. Weniger for technical assistance. We also greatly thank the subject editor O. Savolainen and three anonymous reviewers for helpful comments. This work was supported by the National Science Foundation (award EF-0723447), the National Institutes of Health (award R01 GM086496-01), Duke University, and the Max-Planck Society.
References
- Aranzana, M. J., S. Kim, K. Y. Zhao, E. Bakker, M. Horton et al., 2005. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PloS Genet. 1 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker, E. G., E. A. Stahl, C. Toomajian, M. Nordborg, M. Kreitman et al., 2006. Distribution of genetic variation within and among local populations of Arabidopsis thaliana over its species range. Mol. Ecol. 15 1405–1418. [DOI] [PubMed] [Google Scholar]
- Barrett, J. C., B. Fry, J. Maller and M. J. Daly, 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263–265. [DOI] [PubMed] [Google Scholar]
- Beaumont, M., E. M. Barratt, D. Gottelli, A. C. Kitchener, M. J. Daniels et al., 2001. Genetic diversity and introgression in the Scottish wildcat. Mol. Ecol. 10 319–336. [DOI] [PubMed] [Google Scholar]
- Bechsgaard, J. S., V. Castric, D. Charlesworth, X. Vekemans and M. H. Schierup, 2006. The transition to self-compatibility in Arabidopsis thaliana and evolution within S-haplotypes over 10 Myr. Mol. Biol. Evol. 23 1741–1750. [DOI] [PubMed] [Google Scholar]
- Black, W. C., and E. S. Krafsur, 1985. A FORTRAN program for analysis of genotypic frequencies and description of the breeding structure of populations. Theor. Appl. Genet. 70 484–490. [DOI] [PubMed] [Google Scholar]
- Bonnet, E., and Y. Van de Peer, 2002. ZT: a software tool for simple and partial Mantel tests. J. Stat. Soft. 7 1–12. [Google Scholar]
- Buckler, E. S., and J. M. Thornsberry, 2002. Plant molecular diversity and applications to genomics. Curr. Opin. Plant Biol. 5 107–111. [DOI] [PubMed] [Google Scholar]
- Caicedo, A. L., S. H. Williamson, R. D. Hernandez, A. Boyko, A. Fledel-Alon et al., 2007. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PloS Genet. 3 e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., 2003. Effects of inbreeding on the genetic diversity of populations. Phil. Trans. R. Soc. Lond. Ser. B Biol. Sci. 358 1051–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss, M. J., and T. Mitchell-Olds, 2006. Population genetic structure of Arabidopsis lyrata in Europe. Mol. Ecol. 15 2753–2766. [DOI] [PubMed] [Google Scholar]
- Dobes, C. H., T. Mitchell-Olds and M. A. Koch, 2004. Extensive chloroplast haplotype variation indicates Pleistocene hybridization and radiation of North American Arabis drummondii, A. divaricarpa, and A. holboellii (Brassicaceae). Mol. Ecol. 13 349–370. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia, S. A., J. M. Thornsberry and E. S. Buckler, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54 357–374. [DOI] [PubMed] [Google Scholar]
- Fox, A., B. Tuch and J. Chuang, 2008. Measuring the prevalence of regional mutation rates: an analysis of silent substitutions in mammals, fungi, and insects. BMC Evol. Biol. 8 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadberry, M. D., S. T. Malcomber, A. N. Doust and E. A. Kellogg, 2005. Primaclade: a flexible tool to find conserved PCR primers across multiple species. Bioinformatics 21 1263–1264. [DOI] [PubMed] [Google Scholar]
- Gao, H., S. Williamson and C. D. Bustamante, 2007. An MCMC approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 176 1635–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, D. B., and M. E. Weale, 2001. Population genomics: linkage disequilibrium holds the key. Curr. Biol. 11 R576–R579. [DOI] [PubMed] [Google Scholar]
- Goudet, J., 2001. Fstat, a program to estimate and test gene diversities and fixation indices (v. 2.9. 3). http://www2.unil.ch/popgen/softwares/fstat.htm.
- Gracey, A., and A. Cossins, 2003. Application of microarray technology in environmental and comparative physiology. Annu. Rev. Physiol. 65 231–259. [DOI] [PubMed] [Google Scholar]
- Hartl, D. L., and A. G. Clark, 2007. Principles of Population Genetics. Sinauer Associates, Sunderland, MA.
- Haubold, B., J. Kroymann, A. Ratzka, T. Mitchell-Olds and T. Wiehe, 2002. Recombination and gene conversion in a 170-kb genomic region of Arabidopsis thaliana. Genetics 161 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuertz, M., E. De Paoli, T. Kallman, H. Larsson, I. Jurman et al., 2006. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce. [Picea abies (L.) Karst] Genetics 174 2095–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2000. A new statistic for detecting genetic differentiation. Genetics 155 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Slatkin and W. P. Maddison, 1992. Estimation of levels of gene flow from DNA sequence data. Genetics 132 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J., A. Pepper, A. Hall, Z. Chen, G. Hodnett et al., 2005. Evolution of genome size in Brassicaceae. Ann. Bot. 95 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantama, L., T. F. Sharbel, M. E. Schranz, T. Mitchell-Olds, S. de Vries et al., 2007. Diploid apomicts of the Boechera holboellii complex display large-scale chromosome substitutions and aberrant chromosomes. Proc. Natl. Acad. Sci. USA 104 14026–14031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and S. P. Otto, 2006. Interference among deleterious mutations favours sex and recombination in finite populations. Nature 443 89–92. [DOI] [PubMed] [Google Scholar]
- Kim, S., V. Plagnol, T. T. Hu, C. Toomajian, R. M. Clark et al., 2007. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 39 1151–1155. [DOI] [PubMed] [Google Scholar]
- Knight, C. A., H. Vogel, J. Kroymann, A. Shumate, H. Witsenboer et al., 2006. Expression profiling and local adaptation of Boechera holboellii populations for water use efficiency across a naturally occurring water stress gradient. Mol. Ecol. 15 1229–1237. [DOI] [PubMed] [Google Scholar]
- Kremer, A., and A. Zanetto, 1997. Geographical structure of gene diversity in Quercus petraea (Matt.) Liebl. II: Multilocus patterns of variation. Heredity 78 476–489. [Google Scholar]
- Kuittinen, H., A. A. de Haan, C. Vogl, S. Oikarinen, J. Leppala et al., 2004. Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics 168 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C. E., and T. Mitchell-Olds, 2006. Preface to the special issue: ecological and evolutionary genomics of populations in nature. Mol. Ecol. 15 1193–1196. [DOI] [PubMed] [Google Scholar]
- Liu, A., and J. M. Burke, 2006. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and T. J. Crease, 1990. The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 7 377–394. [DOI] [PubMed] [Google Scholar]
- Martin, G., S. P. Otto and T. Lenormand, 2006. Selection for recombination in structured populations. Genetics 172 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather, K. A., A. L. Caicedo, N. Polato, K. M. Olsen, S. McCouch et al., 2007. The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177 2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Olds, T., 2001. Arabidopsis thaliana and its wild relatives: a model system for ecology and evolution. Trends Ecol. Evol. 16 693–700. [Google Scholar]
- Mitchell-Olds, T., and J. Schmitt, 2006. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 441 947–952. [DOI] [PubMed] [Google Scholar]
- Mitchell-Olds, T., J. H. Willis and D. B. Goldstein, 2007. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat. Rev. Genet. 8 845–856. [DOI] [PubMed] [Google Scholar]
- Moeller, D. A., M. I. Tenaillon and P. Tiffin, 2007. Population structure and its effects on patterns of nucleotide polymorphism in Teosinte (Zea mays ssp. parviglumis). Genetics 176 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell, P. L., D. M. Toleno, K. E. Lundy and M. T. Clegg, 2005. Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc. Natl. Acad. Sci. USA 102 2442–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell, P. L., D. M. Toleno, K. E. Lundy and M. T. Clegg, 2006. Estimating the contribution of mutation, recombination and gene conversion in the generation of haplotypic diversity. Genetics 173 1705–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PloS Biol. 3 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and H. Innan, 2002. Molecular population genetics. Curr. Opin. Plant Biol. 5 69–73. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1982. Linkage disequilibrium due to random genetic drift in finite subdivided populations. Proc. Natl. Acad. Sci. USA 79 1940–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama, R. K., N. Formanova, M. J. Clauss, J. Kroymann, K. J. Schmid et al., 2008. The shrunken genome of Arabidopsis thaliana. Plant Syst. Evol. 273 257–271. [Google Scholar]
- Peakall, R. O. D., and P. E. Smouse, 2006. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol, V., B. Padhukasahasram, J. D. Wall, P. Marjoram and M. Nordborg, 2006. Relative influences of crossing over and gene conversion on the pattern of linkage disequilibrium in Arabidopsis thaliana. Genetics 172 2441–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., and J. D. Wall, 2001. Why is there so little intragenic linkage disequilibrium in humans? Genet. Res. 77 143–151. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins, S. E., B. E. Stranger, T. Mitchell-Olds and M. Aguade, 2004. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics 166 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington, D. L., J. M. Thornsberry, Y. Matsuoka, L. M. Wilson, S. R. Whitt et al., 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, W., 1989. Analyzing tables of statistical tests. Evolution 43 223–225. [DOI] [PubMed] [Google Scholar]
- Ross-Ibarra, J., S. I. Wright, J. P. Foxe, A. Kawabe, L. DeRose-Wilson et al., 2008. Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PloS One 3: e2411. [DOI] [PMC free article] [PubMed]
- Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rozen, S., and H. Skaletsky, 2000. Primer3 on the WWW for general users and for biologist programmers, pp. 365–386 in Bioinformatics Methods and Protocols: Methods in Molecular Biology, edited by S. Krawetz and S. Misener. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genomewide departure from a neutral model of DNA sequence polymorphism. Genetics 169 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K. J., T. R. Sorensen, R. Stracke, O. Torjek, T. Altmann et al., 2003. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 13 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schranz, M. E., C. Dobes, M. A. Koch and T. Mitchell-Olds, 2005. Sexual reproduction, hybridization, apomixis, and polyploidization in the genus Boechera (Brassicaceae). Am. J. Bot. 92 1797–1810. [DOI] [PubMed] [Google Scholar]
- Schranz, M. E., B. H. Song, A. J. Windsor and T. Mitchell-Olds, 2007. a Comparative genomics in the Brassicaceae: a family-wide perspective. Curr. Opin. Plant Biol. 10 168–175. [DOI] [PubMed] [Google Scholar]
- Schranz, M. E., A. J. Windsor, B.-H. Song, A. Lawton-Rauh and T. Mitchell-Olds, 2007. b Comparative genetic mapping in Boechera stricta, a close relative of Arabidopsis. Plant Physiol. 144 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbel, T. F., B. Haubold and T. Mitchell-Olds, 2000. Genetic isolation by distance in Arabidopsis thaliana: biogeography and post-glacial colonization of Europe. Mol. Ecol. 9 2109–2118. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 2008. Linkage disequilibrium: understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, B., and T. Mitchell-Olds, 2007. High genetic diversity and population differentiation in Boechera fecunda, a rare relative of Arabidopsis. Mol. Ecol. 16 4079–4088. [DOI] [PubMed] [Google Scholar]
- Song, B. H., M. J. Clauss, A. Pepper and T. Mitchell-Olds, 2006. Geographic patterns of microsatellite variation in Boechera stricta, a close relative of Arabidopsis. Mol. Ecol. 15 357–369. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24 1596–1599. [DOI] [PubMed] [Google Scholar]
- Tang, C. L., C. Toomajian, S. Sherman-Broyles, V. Plagnol, Y. L. Guo et al., 2007. The evolution of selfing in Arabidopsis thaliana. Science 317 1070–1072. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis, R., M. Riba, B. Colas, P. Grillas and I. Olivieri, 2002. Multilocus genetic structure at contrasted spatial scales of the endangered water fern Marsilea strigosa Willd. (Marsileaceae, Pteridophyta). Am. J. Bot. 89 1142–1155. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Pop. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]
- Windsor, A. J., M. E. Schranz, N. Formanova, S. Gebauer-Jung, J. G. Bishop et al., 2006. Partial shotgun sequencing of the Boechera stricta genome reveals extensive microsynteny and promoter conservation with Arabidopsis. Plant Physiol. 140 1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., J. P. Foxe, L. DeRose-Wilson, A. Kawabe, M. Looseley et al., 2006. Testing for effects of recombination rate on nucleotide diversity in natural populations of Arabidopsis lyrata. Genetics 174 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., and B. S. Gaut, 2005. Molecular population genetics and the search for adaptive evolution in plants. Mol. Biol. Evol. 22 506–519. [DOI] [PubMed] [Google Scholar]
- Yogeeswaran, K., A. Frary, T. L. York, A. Amenta, A. H. Lesser et al., 2005. Comparative genome analyses of Arabidopsis spp.: inferring chromosomal rearrangement events in the evolutionary history of A. thaliana. Genome Res. 15 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]