

Abstract
Members of the genus Trypanosoma, which include the pathogenic species Trypanosoma brucei and Trypanosoma cruzi, edit their post-transcriptional mitochondrial RNA via a multiprotein complex called the editosome. In T. brucei, the RNA is nicked prior to uridylate insertion and deletion. Following editing, nicked RNA is religated by one of two RNA-editing ligases (TbREL). This study describes a recent 70 ns molecular dynamics simulation of TbREL1, an ATP-dependent RNA-editing ligase of the nucleotidyltransferase superfamily that is required for the survival of T. brucei insect and bloodstream forms. In this work, a model of TbREL1 in complex with its full double-stranded RNA (dsRNA) substrate is created on the basis of the homologous relation between TbREL1 and T4 Rnl2. The simulation captures TbREL1 dynamics in the state immediately preceding RNA ligation, providing insights into the functional dynamics and catalytic mechanism of the kinetoplastid ligation reaction. Important features of RNA binding and specificity are revealed for kinetoplastid ligases and the broader nucleotidyltransferase superfamily.
The intricate processing of genetic information is central to biological complexity and to the replication processes that sustain life. The discovery of post-transcriptional RNA editing was a rich addition to our appreciation of the central dogma of biology and is still a vibrant area of research with many unanswered questions. Within the mitochondria of kinetoplastids, a unique form of RNA editing exists whereby uridylates are inserted or deleted (1). To carry out the editing process, kinetoplastids utilize a multiprotein complex known as the editosome. Though many details remain unresolved, the editosome is known to include a core 20S complex of 1.6 MDa containing 16−20 different proteins. Additionally, at least three distinct 20S complexes, distinguished by their constituent proteins and specificity for insertion or deletion editing, have been detected (2), suggesting that an inherent dynamism in the 20S core may accommodate the varied specificity requirements of different gene products.
The editosome is localized in the mitochondria, where mitochondrial DNA has a topologically linked network of thousands of minicircles, which encode guide RNAs (gRNAs),1 and dozens of maxicircles, which encode components of respiratory complexes and energy transduction systems. After transcription, gRNAs base pair with pre-mRNA from maxicircles via conserved “anchor sequences” (3,4). Following gRNA base pairing, an endonucleolytic component of the editosome cleaves the pre-mRNA at points of nucleotide mismatch between the pre-mRNA and the gRNA. Depending on the type of mismatch, uridylates are then inserted or removed from the nicked site of the pre-mRNA by a terminal uridydyl transferase or a U-specific 3′ exonuclease, respectively (5,6). One of two RNA-editing ligases, TbREL1 or TbREL2, then religates the nicked RNA, generally depending on whether the preceding mRNA processing was insertion or deletion, respectively. The religated, “mature” mRNA is then ready for translation. TbREL1 is relevant to structure-based rational drug design and associated structure−function studies both because its catalytically competent N-terminal adenyltransferase domain has been crystallized in complex with ATP at 1.2 Å resolution (7) and because it is required for the survival of Trypanosoma brucei insect and bloodstream forms (8,9).
An ATP-dependent RNA ligase, TbREL1 belongs to the nucleotidyltransferase superfamily, whose members include ATP and NAD+-dependent DNA ligases as well as eukaryotic mRNA capping enzymes (10). Each superfamily member shares five conserved active site structural motifs, along with a conserved catalytic mechanism in which a nucleotide is transferred to the 5′ end of a polynucleotide via a covalent enzyme−NMP intermediate. Catalysis of dsRNA by TbREL1 is initiated when a conserved catalytic lysine nucleophile attacks an ATP α-phosphate, displacing pyrophosphate and forming an enzyme−AMP intermediate. In the second step, nicked dsRNA binds and AMP is transferred from the enzyme to the 5′-PO4 end of the nick, forming a 5′−5′ phosphoanhydride linkage. In the third step, the 3′-OH end, which apposes the 5′-PO4, attacks the phosphoanhydride linkage, displacing AMP and forming the religated dsRNA product.
Though the three religation steps of TbREL1 are widely acknowledged, the exact dependence on divalent metal cofactors remains in doubt. For example, though the electron density map of the ATP-bound TbREL1 N-terminal domain clearly shows a Mg2+ coordinated between the nonbridging oxygen atoms of the ATP β- and γ-phosphates (7), nucleotide transfer has not yet occurred, suggesting that a single Mg2+ may be insufficient to trigger the first chemical step, though the orthogonal position of the lysine nucleophile relative to the α-phosphate electrophile, which hinders nucleophilic attack, is an alternative explanation (11). Additionally, kinetic evidence for related superfamily members suggests that the nucleotidyltransferase step proceeds via a two-Mg2+ mechanism (12), while crystal structure data from both the PBCV-1 capping enzyme and T4 RNA ligase 2 (Rnl2) show that a divalent metal cofactor in the vicinity of the α-phosphate remains after step 1 of catalysis (11,13) and imply that a two-metal mechanism may optimize the nucleotide transfer turnover rate. These contrasting results suggest that an allosteric signal, possibly from other protein members of the editosome, may lead to the uptake and coordination of a second Mg2+, thereby triggering nucleotide transfer (7). The plausibility of ATP-dependent allosteric communication between TbREL1 and other editosome members was demonstrated in molecular dynamics (MD) simulations of apo and ATP-bound TbREL1 (14). These simulations suggested that ATP binding induces increased mobility of a putative allosteric loop; similar phenomena may provide the impetus for the uptake of a second Mg2+.
Both sequence and structural phylogenetic analyses of superfamily members indicates that the closest known TbREL1 homologue is the T4 phage Rnl2 (7,14). Recently, the structure of full-length Rnl2, in complex with its nucleic acid substrate, was captured in two different conformations (13). These two unique conformations illustrate the active site conformational rearrangements that occur as the complex proceeds from the step 2 product, immediately following step 2 chemistry, to the step 3 substrate, immediately preceding step 3 chemistry. Additionally, comparing the crystal structure of human DNA ligase 1 (LIG1), in complex with its DNA substrate, to the Rnl2 nucleic acid complex reveals the salient features of substrate conformational requirements and substrate specificity. In both LIG1 and Rnl2, the nucleotides local to the nick are found in a RNA-like A-conformation. This conformation aligns the 3′-OH and apposing 5′-PO4 at the nick site and is likely required for step 3 chemistry. Normally in the canonical B-form, the DNA substrate of LIG1 is distorted into the required A-conformation via two phenylalanines that intercalate the minor grove. These phenylalanines, conserved in DNA ligases, are absent in RNA ligases like Rnl2 and TbREL1. Further details are presented in the original crystal structure paper (13) and are reviewed elsewhere in the context of other DNA and RNA ligases (15).
Motivated by the close homology between Rnl2 and TbREL1 and the desire to understand the functional dynamics of the kinetoplastid ligation reaction, we investigate the conformational dynamics of TbREL1 in complex with a nicked dsRNA substrate in its canonical A-form. On the basis of the homologous structural and functional relationships between TbREL1 and T4 Rnl2, we develop a model of a nicked dsRNA−TbREL1 complex (Figure 1) and perform an all-atom explicitly solvated molecular dynamics simulation of the system. The integration of RNA into MD simulations has benefited from recent improvements in both force field development and algorithms (16,17), allowing us to gain insights into the dynamics and catalytic mechanism. The simulated system contains AMP attached to the nick 5′-PO4 moiety (AppN), characteristic of the step 2 intermediate (13). The non-nicked strand represents the template, or gRNA, and the nicked strand represents the post-transcriptionally modified pre-mRNA. Fluctuations of conserved interactions between the enzyme and RNA substrate, as well as the dynamics of conserved active site residues, are examined and discussed in the context of step 3 of the religation mechanism. Furthermore, we explore the effect of a unique kinetoplastid insert region on dsRNA binding.
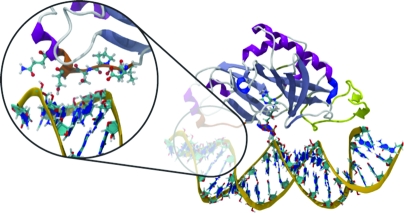
Figure 1.

Nicked dsRNA bound to TbREL1, poised to catalyze religation. A unique kinetoplastid insert is colored yellow, and a cluster of residues thought to be important in RNA recognition on the 5′-PO4 side of the nick is colored orange. The magnified inset shows protein residues that make stabilizing contacts with the RNA.
Materials and Methods
System Preparation
A model of the step 2 intermediate of TbREL1 in complex with A-form dsRNA was built in homology to chain B of the T4 Rnl2 crystal structure [Protein Data Bank (PDB) entry 2HVR], which captures T4 Rnl2 immediately preceding step 3 chemistry (13). A 12-mer of A-form dsRNA, with a sequence identical to that of the nicked DNA−RNA hybrid cocrystallized with T4 Rnl2, was generated using the NUCGEN module of Amber 9 (18), which creates accurate nucleotide models based on fiber diffraction studies. The dsRNA was then aligned with the nicked DNA−RNA hybrid by minimizing the root-mean-square deviation (rmsd) between the two nitrogenous bases of the DNA−RNA hybrid that flanked the nick and the commensurate nitrogenous bases of the model dsRNA. Next, the dsRNA was translated onto the crystal structure of the TbREL1 N-terminus (PDB entry 1XDN) by minimizing the rmsd between the AppN adenine moiety of the DNA−RNA hybrid and the adenine moiety of the ATP bound in the TbREL1 active site. A nick was then created in the model dsRNA identical to the nick of the DNA−RNA hybrid bound to T4 Rnl2. The β- and γ-phosphates of the TbREL1-bound ATP were subsequently deleted, and the O−PAMP−O−P5′PO4 dihedral angle was brought into agreement with the corresponding dihedral angle of the T4 Rnl2 crystal structure. The resulting AMP is linked through its phosphate to the 5′-PO4 nicked terminal nucleotide; we designate this AMP-phosphate-phosphate-nucleotide residue AppN according to the nomenclature of Nandakumar and co-workers (13).
To ensure that the active site of our model reflected the conformation of Rnl2 immediately preceding step 3 chemistry, TbREL1 active site residue R111 was given a conformation commensurate with its Rnl2 homologue, R55. Specifically, R111 was rotated away from the nonbridging oxygen of the AppN α-phosphate and placed within hydrogen bonding distance of the Y58 backbone carbonyl. The conformations of the other TbREL1 active site residues were in close agreement with the conformations of the homologous active site residues of T4 Rnl2 immediately preceding step 3 chemistry and so required no further manipulation. A Mg2+ cation was manually positioned approximately 2 Å from a nonbridging AMP α-phosphate on the basis of the assumptions that ion dissociation occurs neither during RNA binding nor during subsequent active site rearrangements and that the TbREL1 catalytic mechanism proceeds via a two-Mg2+ mechanism. This Mg2+ position was chosen on the basis of the crystal structure data of both the PBCV-1 capping enzyme (11) and the T4 Rnl2 step 1 product (13), as well as on a similarly positioned Ca2+ cation in the crystal structure of T4 Rnl1 bound to the unreactive analogue AMPCPP (19). Five additional water molecules, each approximately 2 Å from the Mg2+ cation, were manually positioned to duplicate the expected octahedral coordination complex.
All cocrystallized water molecules in the TbREL1 structure were retained in our model. Selenomethionines used for crystal structure refinement were replaced with methionines, and WHATIF (20) was used to assign histidine protonation states. Protonation state assignments were manually verified. With the exception of the nonstandard AppN residue, all RNA and protein hydrogens and force field parameters were assigned according to the Amber ff99SB force field (21) using the xleap module in Amber 9 (18). The nonstandard residue AppN is composed of an adenosine linked to a cytidine via a phosphoanhydride linkage; consequently, we assigned AppN charges using the RA5 and RC5 residues of the Amber ff99SB force field as a template. The charge of the oxygen bridging the phosphoanhydride linkage was assumed to be identical to that of the oxygen bridging the α- and β-phosphates in the Carlson ADP parameter set (22). All missing AppN force field parameters came from the GAFF force field (23). Following parameterization, the complex was immersed in a rectangular box of TIP3P waters (24) with a 10 Å buffer from the protein to the edge of the water box in each direction. Sodium and chloride ions were added to the system, bringing it to electric neutrality and yielding a 100 mM salt solution.
MD Simulations
Spurious contacts were removed through a four-tier set of 45000 energy minimization steps. The first 10000 steps were performed in two 5000-step cycles. Hydrogen atoms were relaxed during the first 5000 steps, all other atoms being held fixed. Hydrogen atoms, water molecules, and ions were relaxed during the next 5000 steps, the protein and nucleic acid being held fixed. Ten thousand steps of minimization followed in which only the protein backbone was fixed. No constraints were applied during the last 25000 steps of minimization.
To equilibrate our model, we performed a 1 ns restrained simulation using the minimized system as a starting point. The temperature was introduced at 310 K with harmonic restraints on the protein backbone. Through the gradual relaxation of the backbone restraints, the protein structure was preserved as kinetic energy was introduced. For the first 250 ps, a harmonic restraining force of 4.0 kcal mol−1 Å−2 was applied to the backbone atoms. Similarly, for the second, third, and fourth 250 ps segments, the harmonic restraints were consistently weakened to 3.0, 2.0, and 1.0 kcal mol−1 Å−2, respectively.
A 70 ns MD simulation followed in which unrestrained dynamics were performed with a 1 fs time step. The duration of the simulation was sufficient to equilibrate the system on the basis of rmsd values [overall α-carbon (Figure S1 of the Supporting Information) and the 3′-terminal sugar pucker phase angle (Figure 3)], while allowing us to sample functionally relevant conformations local to the step 2 intermediate. The temperature bath was maintained by Langevin dynamics at 310 K, and pressure was maintained with the hybrid Nose Hoover-Langevin piston method at 1 atm (25) using period and decay times of 100 and 50 fs, respectively. The particle mesh Ewald algorithm was used to treat long-range electrostatics (26). A multiple-time step algorithm was employed in which bonded interactions were computed at every time step, short-range nonbonded interactions were computed every two time steps, and full electrostatics were computed every two time steps. The water hydrogen−oxygen and hydrogen−hydrogen distances were constrained using the SHAKEH algorithm (27) to be within 0.0005 Å of the nominal lengths. All minimization and MD simulations were carried out with NAMD 2.6 (28) using the Amber ff99SB force field (21). Simulations were performed on the TeraGrid ABE cluster. A typical benchmark for the 57575-atom system on 128 processors was 0.21 days per nanosecond of simulation. System configurations were sampled every 50 ps, generating 1400 coordinate snapshots for subsequent analysis.
Figure 3.

Equilibrated ribofuranose pseudorotation angle for the 3′-OH nucleotide as depicted within the active site (left panel) and in polar coordinates (right panel; equilibration data colored gray and dynamics data black).
Pucker Analysis
The pseudorotational formalism proposed by Rao, Westhof, and Sundaralingam (29) was used to characterize the puckering of the nick 3′-ribose ring. We first defined the following Fourier sums:
where j ranges from 0 to 4 and θ0, θ1, θ2, θ3, and θ4 are the torsion angles about the C2′−C3′, C3′−C4′, C4′−O4′, O4′−C1′, and C1′−C2′ bonds, respectively. The pucker phase angle, P, identifies which part of the ring is furthest from planarity. Ribose configurations generally take on the 2′-endo pucker typical of DNA or the 3′-endo pucker typical of RNA. The possible values of P, which range from 0 to 360, correspond to the stages along the so-called pseudorotation pathway. The ribose ring has five atoms, each of which can be above or below the plane, yielding 10 distinct stages. The envelope conformations correspond to P values of 0°, 36°, 72°, etc., and alternate with the twist conformations at P values of 18°, 54°, 90°, etc. In the Rao−Westhof−Sundaralingam pseudorotational formalism
The maximal torsion angle or puckering amplitude, θm, measures the magnitude of the deviation from planarity (29).
Polar plots are generally used to represent sugar pucker, with the puckering amplitude and phase angle assigned as the radial and angular coordinate, respectively. Analysis of experimental structures shows that the 2′-endo ribose configuration typical of DNA has a P between 137° and 194°, and the 3′-endo ribose configuration typical of RNA has a P between −1° and 34° (30).
Principal Component Analysis
Principal component analysis, or PCA, is a common statistical data analysis technique whose application to the field of biomolecular simulation was pioneered by Berendsen (31) and Garcia (32). By diagonalizing a covariance matrix constructed of molecular coordinates sampled over the duration of a MD trajectory and listing the resulting eigenvectors in order of descending eigenvalue magnitude, PCA separates large, low-frequency conformational changes from small, high-frequency motions. By considering only the conformational fluctuations of the first few modes, researchers frequently use PCA to filter out large isomerization events and to reduce the complexity of trajectory analysis. In this work, PCA was used to explore the large, low-frequency motions of the dsRNA substrate. Following trajectory equilibration, principal component analysis was carried out on the phosphate atoms of the dsRNA substrate, using the ptraj module in Amber 9 (18). Prior to the performance of PCA, translational and rotational motions were removed by fitting the sampled coordinates to the first equilibrated frame of the trajectory.
Results and Discussion
Equilibration
A 70 ns constraint-free simulation was performed on the dsRNA-bound TbREL1 system to examine TbREL1 dynamics. A plot of the time evolution of the rmsd of the protein α-carbons (Figure S1 of the Supporting Information), in which each trajectory frame was aligned with the initial frame to remove rotational and translational motion, suggested that our preliminary efforts to equilibrate the system prior to our 70 ns simulation were successful. However, subsequent examination of the distance between the nick 3′-OH and the phosphorus atom of the 5′-phosphoanhydride linkage suggested that equilibration was not complete until after 38.75 ns. During the initial phase (0−38.75 ns), these two atoms, between which a bond forms following step 3, were distant (7.16 ± 1.20 Å). During the second phase (38.75−70 ns), however, the nick 3′-OH and the nick 5′-phosphorus moved closer together (3.95 ± 0.29 Å), as required for catalysis. Correspondingly, the trajectory time from 0 to 38.75 ns is called the “equilibration phase” and from 38.75 to 70 ns the “dynamics phase”. Unless otherwise specified, all analysis was performed on the dynamics phase.
Active Site Interactions
The atomic interactions within the active site can be divided into three categories: interactions between TbREL1 and the adenine moiety of AppN, interactions between TbREL1 and the ribofuranose of AppN, and interactions between TbREL1 and the pyrophosphate of AppN. Throughout our discussion, we adopt the naming scheme of Nandakumar and co-workers, naming the nicked strands on the basis of their active site ends (i.e., the 5′-PO4 strand and the 3′-OH strand) (13).
Deep within the active site, F209, E86, and V88 interact with the adenine moiety of AppN and stabilize its motion (Figure 2). F209 acts as a conserved hydrophobic platform that forms π−π interactions with the aromatic adenine moiety of AppN, preventing large lateral fluctuations of this base within the binding pocket. Hydrophobic stabilization in this position is essential for step 3 catalysis in other type II RNA ligases, as demonstrated by the partial recovery of religation activity when a leucine residue was substituted for the F209 homologue in an alanine scan of T4 Rnl2 (33). Our simulation is consistent with this mutagenesis study; F209 exhibits minor fluctuations, with an average distance of 4.14 Å between the centers of mass of its phenyl group and the AppN adenine moiety (Table 1). The movement of AppN is also restrained by E86, which accepts a hydrogen bond from the adenine N6 amine that holds O1 (or O2) of E86 within 2.97 Å, on average (Table 1). Conserved among type II RNA ligases, E86 is essential in mediating step 3 catalysis in T4 Rnl2 (33). The observed distances and fluctuations of both F209 and E86 are similar to those observed in a previous simulation of the ATP−TbREL1 complex (14). These results, along with the T4 Rnl2 mutational analysis (33), reflect the importance of F209 and E86 in mediating the religation of the adenylated dsRNA substrate and the formation of the adenylated enzyme intermediate.
Figure 2.

Equilibrated structure illustrating the close contacts between key active site residues (shown in licorice), the AppRNA (shown in ball and stick), and the 3′-OH RNA strand.
Table 1. Active Site Interactions, Including Protein−AppN Interactions, Protein−Nick Interactions, and Interactions with the Mg2+-Coordinated Water Molecules.
| length (Å)[standard deviation (Å)] | |
|---|---|
| AppN adenine−F209 phenyl group | 4.14 (0.22) |
| E86 O−AppN adenine N6 | 2.97 (0.20) |
| E159 OE1−AppN ribose O2′ | 2.55 (0.08) |
| N92 ND2−AppN ribose O2′ | 3.30 (0.41) |
| K87 NZ−AppN O1P | 3.39 (0.42) |
| K307 NZ−AppN O2P | 3.87 (0.56) |
| R111 NH1−AppN O1P | 2.80 (0.11) |
| nick 3′-HO−nearest nonbridgingnick 5′-PO4 oxygen | 1.90 (0.64) |
| R111 CZ−nick O3′ | 3.95 (0.34) |
| H89 ND1−WAT O | 2.85 (0.21) |
| E159 CD−WAT O | 3.49 (0.27) |
| E283 CD−WAT O | 3.45 (0.24) |
| E283 CD−WAT O | 3.44 (0.21) |
| nick O2′−WAT O | 3.30 (0.53) |
Stabilizing interactions between the protein and the AppN ribofuranose complement the contacts formed between the adenine moiety and residues deep within the binding pocket. In particular, E159, conserved across all members of the superfamily, and N92, conserved across all type II RNA editing ligases (33), make critical hydrogen bonds with the ribofuranose O2′ hydroxyl group (Figure 2 and Table 1). The close, stable contact made by E159 was observed in a previous simulation of the ATP-bound complex (14) and is consistent with the conservative mutational analysis, which identified E159 as an essential mediator of steps 1 and 3 of catalysis (33). Similarly, an alanine scan of T4 Rnl2 revealed that the N92 homologue plays a critical role in the religation of the preadenylated dsRNA substrate (33). No isomerization of the AppN ribofuranose moiety was observed during the course of catalysis in the T4 Rnl2 homologue (13); consequently, the mutational analysis results, coupled with the short, stable contacts observed during our simulation, suggest that the N92−ribofuranose interaction helps maintain the ribofuranose conformation throughout the catalytic cycle.
Charged residues at the periphery of the binding site, K87, K307, and R111, each interact with the AppN pyrophosphate and stabilize the step 3 substrate conformation (Figure 2). The orthogonal orientation of the K87 nucleophile to the α-phosphate is typical of the step 3 substrate conformation and persists throughout the 31.25 ns dynamics phase. The system does not undergo isomerization away from the step 3 substrate conformation, indicating that the network of interactions captured in the homologous T4 Rnl2 crystal structure is a reasonable model of the TbREL1 active site architecture prior to step 3 of catalysis. Interestingly, K87 remains within hydrogen bonding distance of a nonbridging pyrophosphate oxygen during the dynamics phase of the simulation (Table 1). This result suggests that the K87 nucleophile helps maintain the protein in the step 3 conformation once isomerization into that conformation has occurred. Consequently, K87 seems to perform a dual role during catalysis: during step 1, it plays the central chemical role as the nucleophile, and during step 3, it plays a peripheral stabilizing role as a hydrogen bond donor. To the best of our knowledge, no experimental evidence has been published supporting this hypothesis, but the putative dual role of K87 could be probed using the methods Yin and co-workers employed during their structure−function analysis of T4 Rnl2 (33). Nearby K307 also donates a hydrogen bond to a nonbridging pyrophosphate oxygen, remaining close throughout the dynamics phase of the simulation (Table 1). The AppN stabilization that this interaction provides is in agreement with mutational analysis, which has shown that a positively charged hydrogen bond donor at this position is most efficient for religating preadenylated nicked dsRNA. Along with K87 and K307, R111 also donates a hydrogen bond to one of the nonbridging pyrophosphate oxygens and remains in close contact throughout the dynamics phase of the simulation (Table 1).
Insights into Catalysis
A requirement for step 3 catalysis is that the O3′ nucleophile and the departing, bridging oxygen of the AppN pyrophosphate take on an apical, or “in-line”, arrangement. This apical requirement is achieved only when the terminal ribofuranose of the nucleotide on the 3′-OH side of the nick takes on a 3′-endo conformation (34). The 3′-endo conformation is the native conformation of dsRNA, explaining why T4 Rnl2 requires that the strand on the 3′-OH side of the nick be RNA while showing no preference for nucleic acid type on the 5′-PO4 side (34). In vitro religation experiments with TbREL1 alone have likewise demonstrated that the strand on the 3′-OH side of the nick must be RNA (35), though inclusion of other components of the editosome complex alters substrate specificity. Here, we assume on the basis of the close homology between TbREL1 and T4 Rnl2 that the RNA requirement on the 3′-OH side of the nick arises from the same 3′-endo requirement observed in T4 Rnl2. The equilibrium distribution of sugar conformations observed during our simulation should be a reasonable approximation of those available for catalysis during step 3 religation.
To gain insight into the temporal fluctuations of the required 3′-endo conformation, we monitored the ribofuranose pseudorotation angle of the nucleotide at the 3′-OH terminus of the nick over the dynamics phase of the simulation (Figure 3). The pseudorotation angle is a measure of the sugar conformational space and includes two stable conformations corresponding to the 2′-endo and 3′-endo conformations of DNA and RNA, respectively. As noted before, the 2′-endo conformation is characterized by a pseudorotation angle between 137° and 194°, and the 3′-endo conformation is characterized by a pseudorotation angle between −1° and 34° (30). Consistent with the A-form dsRNA structure, the ribofuranose of the nick terminal 3′-hydroxyl nucleotide remains almost exclusively in the 3′-endo conformation (15.2 ± 10.9°).
Visual inspection of the O3′ nucleophile and the bridging oxygen leaving group of the AppN pyrophosphate demonstrated that while the two atoms remain parallel to one another, they are not strictly in-line; the attacking O3′ nucleophile is vertically displaced from the plane of the leaving group (Figure 3). The vertical displacement is a consequence of relaxing steric conflicts in the initial model during minimization and equilibration and implies that the active site conformation is suboptimal for religation. Despite this, the time evolution of active site topography still provides a useful model from which mechanistic insight may be inferred.
The nicked 3′-hydroxyl nucleophile must be deprotonated either prior to or following reaction with the β-phosphate of the AppN intermediate. Determining the lowest-energy pathway of proton transfer is complicated and highly dependent on the electrostatic environment surrounding the acid. However, proton transfer is restrained by the requirement that a new covalent bond form between the acidic hydrogen and the base. This requirement allows simple hypotheses of proton transfer pathways to be made on the basis of the proximity of the acidic hydrogen to potential general bases. Our simulation demonstrates that the acidic hydrogen of the nicked 3′-hydroxyl nucleophile resides an average distance of 1.90 Å from the nearest negatively charged nonbridging pyrophosphate oxygen (Table 1). Furthermore, protein fluctuations bring these two atoms within 1.40 Å of each other, causing significant overlap of their van der Waals radii and suggesting possible proton transfer between these two atoms. As the acidic hydrogen lies between the putative basic oxygen and the nucleophilic O3′, it is likely that proton transfer either precedes nucleophilic attack or occurs concertedly with phosphodiester formation. Additionally, the positively charged guanidinium moiety of R111 is proximal to the nick O3′ hydroxyl (Table 1) and may play a role in stabilizing the O3′ as it acquires negative charge during the proton transfer process. This is consistent with mutational analysis carried out on T4 Rnl2, which demonstrated that a lysine mutant in the position of the R111 homologue was nearly as efficient in religating preadenylated nicked dsRNA as the wild-type enzyme but that the alanine mutant lacked religation ability (33). The idea that a nonbridging oxygen of a nucleotide phosphate may serve as a general base has also been proposed as a step in the catalytic mechanism of the PBCV-1 mRNA capping enzyme (36).
Mg2+ Coordination
The exact dependence of nicked dsRNA religation on divalent metal cofactors remains uncertain. In our model of the step 3 substrate, we assume that TbREL1 step 1 chemistry proceeds via a two-Mg2+ cofactor mechanism. This assumption, along with homology to the crystal structures of other nucleotidyltransferase superfamily members, suggests the Mg2+ cation placement in our model (Figure 4). The first Mg2+ cation, which is initially coordinated by the nonbridging oxygens of the AppN β- and γ-phosphates, dissociates with pyrophosphate following the formation of the enzyme nucleotide intermediate in step 1. The second Mg2+ cation, which is coordinated in the vicinity of the α-phosphate, remains bound within the active site to mediate later steps of catalysis. With the model so developed, analysis of the network of interactions mediated by the presence of the divalent magnesium, as well as the proximity of the cation to the AppN substrate, suggests possible functional roles for the bound metal in the context of step 3 chemistry.
Figure 4.
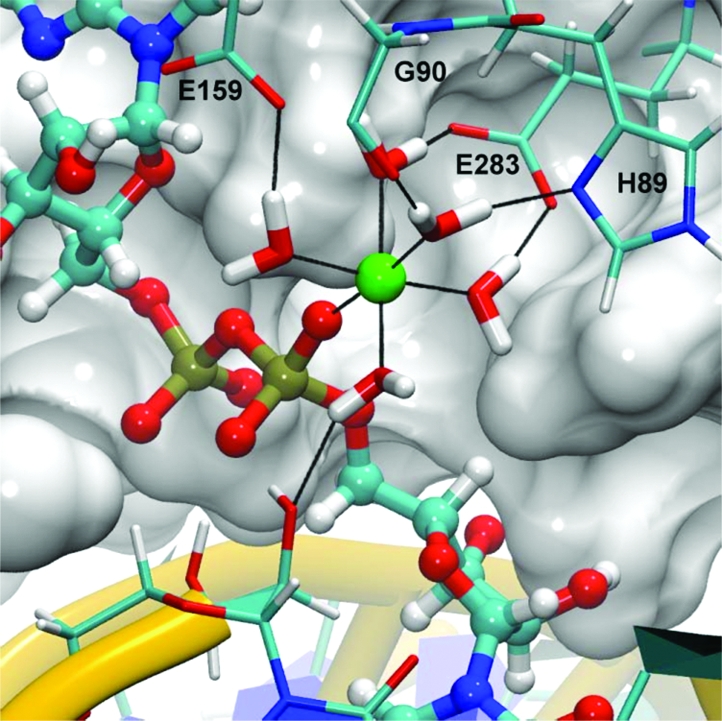
Equilibrated coordination of the active site magnesium ion.
In our model, the magnesium cation is octahedrally coordinated by five water molecules and a nonbridging α-phosphate oxygen atom of the AppN pyrophosphate (Figure 4). None of these water molecules exchanged with bulk solvent during the simulation. Four of the five water molecules participate in a hydrogen bond network with conserved residues in motifs I, III, and IV, while the fifth water donates a hydrogen bond to the 2′-oxygen of the terminal nucleotide on the nicked 3′-hydroxyl strand. This interaction network provides bridging hydrogen bond interactions to conserved active site residues and serves to maintain the relative position of the magnesium within the active site, which may be important in transition state stabilization as the reaction proceeds.
In phosphoryl transfer reactions, there are three mechanistically distinct nucleophilic substitution pathways: dissociative, concerted, and addition−elimination (37). The addition−elimination and concerted pathways are SN2-like; as the nucleophile approaches the phosphate electrophile, the reacting phosphate passes through a planar, pentacoordinate transition state in which one of the nonbridging oxygens acquires an additional negative charge (37,38). In the dissociative, E1-like pathway, the leaving group first departs, forming a planar metaphosphate, phosphocation intermediate before the nucleophile reacts. In our model of the step 3 substrate, the Mg2+ cation coordinates one of the nonbridging oxygens of the phosphate electrophile. The magnesium resides an average distance of 1.83 Å from the nonbridging oxygen atom, opposite the O3′ nucleophile across the pyrophosphate backbone (Figure 4). The proximity of the divalent magnesium cation and the nonbridging oxygen atom of the phosphate electrophile may provide electrostatic stabilization of the excess transition state negative charge typical of the concerted and addition−elimination pathways, enhancing the turnover rate. Conversely, if step 3 chemistry proceeds via a dissociative pathway, the proximity of the positively charged divalent magnesium cation would destabilize the phosphocation of the metaphosphate intermediate, thereby attenuating the turnover rate. While these considerations are qualitative and nonrigorous, they suggest that the presence of a divalent metal during step 3 chemistry favors either a concerted or an addition−elimination pathway.
Having considered the effects of a divalent metal on the turnover rate of the third chemical step, we now consider the network of interactions provided by conserved residues, which stabilize the magnesium within the active site. Within motif I, both the unprotonated δ-nitrogen of H89 and the carbonyl oxygen of G90 accept hydrogen bonds from two unique water molecules, each coordinating Mg2+ (Figure 4 and Table 1). Within motif III, one of the negatively charged carboxyl oxygen atoms of E159 accepts a hydrogen bond from a coordinating water molecule. The carboxyl oxygen atoms of E283 in motif IV likewise accept hydrogen bonds from two coordinating water molecules in a similar manner (Table 1). When the E159 or E283 homologues are substituted with either aspartate or glutamine in T4 Rnl2, religation activity is completely abolished, indicating a strict glutamate requirement in these positions (33). Although the cause of this dependence is not immediately apparent, we speculate that neither glutamine, which is neutrally charged, nor aspartate, whose side chain is one methylene group shorter, provides stable hydrogen bonding partners for the magnesium-coordinating water molecules.
In addition to the E159 and E283 interactions, one of the Mg2+-coordinating water molecules also donates a hydrogen bond to the 2′-oxygen atom of the terminal nucleotide on the nicked 3′-hydroxyl strand (Figure 4 and Table 1). This interaction indirectly links the terminal nucleotide of the nicked 3′-hydroxyl strand to the AppN moiety and may play a role in properly orienting the terminal nucleotide for step 3 chemistry.
RNA Binding Footprint
While the nucleotide specificity determinants for the T4 Rnl2 and human DNA ligase I superfamily members have been clearly defined (15), the specificity determinants of TbREL1 have not. Available experimental data demonstrate that, in vitro, TbREL1 cannot religate nicked nucleic acid in complex with gRNA unless both of the constituent fragments are RNA (35). On the basis of studies of the homologous T4 Rnl2, we hypothesize that the RNA requirement on the 3′-OH side of the nick may arise at least in part from the need to confer a 3′-endo conformation at the 3′-OH nicked terminal nucleotide (34). The absence of religation when DNA is present on the 5′-side of the nick suggests that specific TbREL1−RNA interactions play important roles in mediating substrate stability during steps 2 and 3 of the religation mechanism.
To identify molecular specificity determinants that mediate dsRNA binding, we generated a RNA binding footprint based on a set of protein−RNA interaction persistence times (Figure 5). This analysis allows us to identify primary contact sites between the RNA and the protein and to discriminate longer-lived interactions from more transient ones. The footprint-like mapping of the protein−RNA interaction persistence times onto the protein and RNA surfaces shows that more extensive interactions occur on the 5′-PO4 side of the nick. Although this contact pattern contrasts with the results of Nandakumar and co-workers, who report that the closest homologue of TbREL1, T4 Rnl2, makes more extensive contacts with the strands on the 3′-OH side of the nick, it supports the experimental observations that TbREL1 requires RNA on the 5′-PO4 strand of the nick for in vitro polynucleotide religation. A large, contiguous set of interactions, as well as various individual interactions, makes up the more extensive contacts on the 5′-PO4 side of the nick. Specifically, D62, L63, P64, S65, S67, and Q70 collectively form a contiguous surface, herein called the “recognition sequence”, which intercalates the minor grove of the dsRNA on the 5′-PO4 side of the nick (Figures 1 and 5). Although the recognition sequence is not conserved at the superfamily level, it is conserved among the Trypanosomatidae type I mitochondrial RNA-editing ligase sequences available in the SWISS-PROT database (39). As the depth and width of the minor grooves differ between the canonical RNA and DNA forms, this recognition sequence may play a key role in polynucleotide discrimination.
Figure 5.
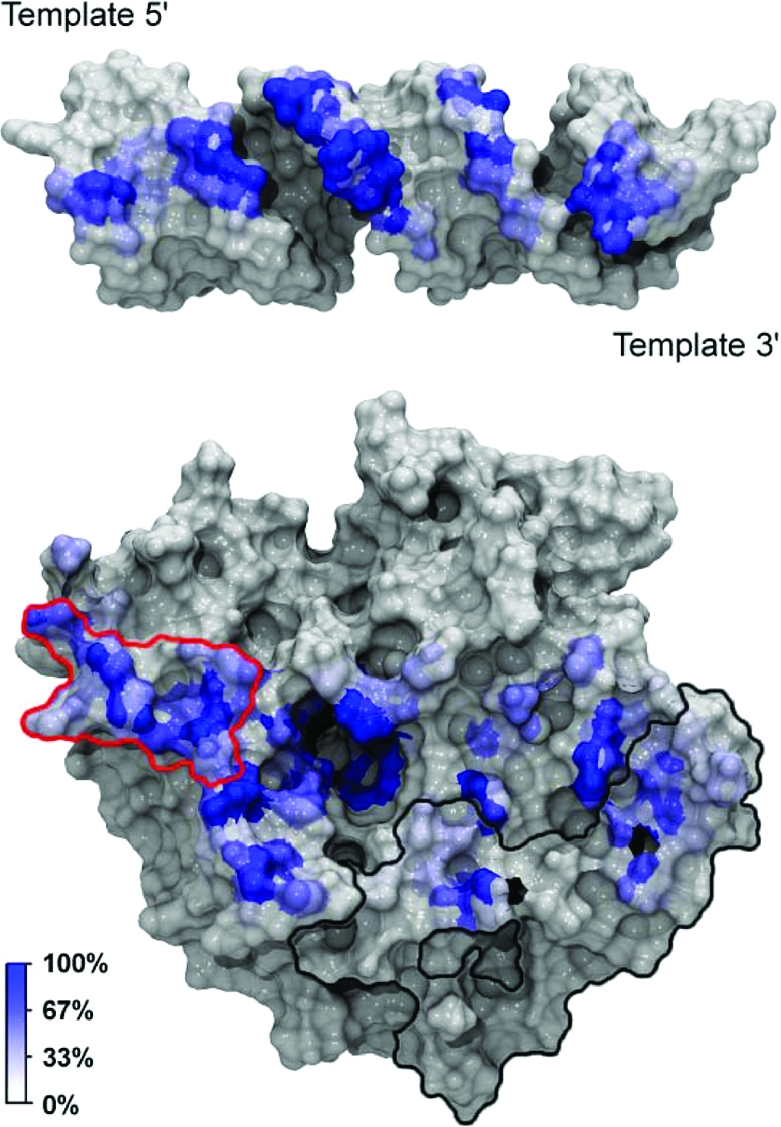
RNA binding footprint. In the top panel, the protein dynamic contact footprint is projected onto the molecular surface of the RNA. In the bottom panel, the RNA dynamic contact footprint is projected onto the molecular surface of TbREL1. Both the protein and RNA are shown in gray surface representation with the contacts colored by percent occupancy over the dynamics portion of the simulation. A unique kinetoplastid insert is outlined in black, and a cluster of residues thought to be important in RNA recognition on the 5′-PO4 side of the nick is outlined in red.
In addition to the geometric complementarity conferred by these residues, several specific atomic interactions within this sequence may contribute to polynucleotide discrimination (Table 2). The carboxyl group of D62, the carbonyl backbone of P64, the side chain hydroxyl group of S65, and the side chain amino group of Q70 each make contact with a ribofuransose 2′-OH. Two of these four RNA-specific contacts are shared between D62 and S65; roughly 19 ns into the dynamics portion of the simulation, the interaction of 2′-OH with S65 exchanges with D62 and persists throughout the remainder of the simulation. In DNA, the lack of the 2′-OH precludes hydrogen bond interactions with D62, P64, S65, and Q70. These interactions may partly explain why the nucleic acid on the 5′-PO4 side of the nick must be RNA for religation to occur in vitro.
Table 2. RNA−Protein Interactionsa.
| protein | RNA | length (Å) [standard deviation (Å)] | % time bond <3.5 Å |
|---|---|---|---|
| Leu63 N | Cg-7 O1P | 3.84 (0.99) | 55.50% |
| Gln70 NE2 | Un-11 O1P | 5.34 (1.78) | 19.62% |
| Ser67 OG | An-10 N3 | 4.67 (1.29) | 24.4% |
| Ser65 OG | Cg-8 O2′ | 4.86 (1.43) | 18.34% |
| Pro64 O | Ug-9 O2′ | 4.14 (0.68) | 19.62% |
| Asp62 OD1 | Cg-8 O2′ | 5.49 (1.64) | 20.73% |
| Met314 O | An-1 O2′ | 3.57 (1.22) | 71.29% |
| Glu119 OE1 | Cn+12 O5′ | 6.37 (4.17) | 25.84% |
| Asn120 ND2 | An+11 O1P | 3.36 (1.74) | 88.20% |
| Phe121b | Ug+4 O2′ | 7.85 (0.99) | 0.00% |
| Phe122b | Gg3 O2′ | 5.19 (0.48) | 0.00% |
| Arg194 NH2 | Gg+3 O1P | 3.09 (0.45) | 82.46% |
| Arg309 NH1 | An-1 O1P | 2.82 (0.12) | 99.84% |
| Arg309 NH1 | Cn-2 O2P | 2.94 (0.33) | 94.90% |
Distances and occupancy percentages are calculated over the dynamics phase only.
Side chain ring center of mass.
The stability conferred by the recognition sequence is immediately apparent upon examination of the root-mean-square fluctuation (rmsf) values and the porcupine plots describing the motion along the first three principal modes (Figure 6). The recognition sequence makes contact with the unnicked gRNA strand at positions −8 and −7, causing a 2-fold decrease in root-mean-square fluctuation relative to that observed at the 5′-PO4 terminus of the gRNA (position −10). At positions −7 to 2, where the gRNA nucleotides are not in contact with the protein, there is a 1.6-fold decrease in rmsf. Additionally, the porcupine plots illustrate that the magnitudes of atomic displacements along the first three modes are attenuated in positions −8 and −7 (Figure 6). The relatively low interaction persistence times observed for the recognition sequence (Table 2) are readily explained by the relatively large positional fluctuations of the dsRNA at the terminal ends on the 5′-PO4 side of the nick (Figure 6).
Figure 6.

RNA flexibility as shown by both the rmsf per residue of the RNA strands and PCA analysis. In both panels, the template strand is colored orange, the 3′-OH strand gray, and the 5′-PO4 strand purple. PCA vectors were multiplied by a factor of 25 for easier visualization.
Two additional independent TbREL1−RNA interactions play a role in mediating substrate stability on the 5′-PO4 side of the nick. The carbonyl group of M314 forms a hydrogen bond with the 2′-OH of the penultimate nucleotide of the nicked strand, contributing to RNA specificity (Table 2). Additionally, the guanidinium moiety of R309 bridges the penultimate and antepenultimate RNA residues on the 5′-PO4 side of the nick by forming two stable hydrogen bonds with two nonbridging phosphate oxygen atoms. The lack of RNA-specific interactions with R309 suggests that this residue may confer stability on the bound state without affecting substrate specificity.
Aside from the hypothesis that the 3′-OH nucleic acid adjacent to the nick must take on the RNA-like 3′-endo conformation for catalysis to proceed, the obligate presence of RNA on the 3′-OH side of the nick may also stem from specific RNA−protein interactions that possibly impart stability to the bound complex. The 2′-OH of the terminal nucleotide on the 3′-OH side of the nick interacts with the carboxyl group of E119 and attenuates fluctuations of the terminal dsRNA, thereby conferring stability to the bound complex (Table 2). N120 also provides stabilizing interactions with the RNA phosphate backbone contributing to RNA stability, though probably not specificity (Table 2). Two phenylalanine residues, F121 and F122, contribute nonspecific van der Waals interactions with adjacent ribonucleotides in the gRNA strand on the 3′-OH side of the nick.
Mutational religation assays carried out on T4 Rnl2 suggest important roles for both F121 and F122; the F121 homologue is an important contributor to nonspecific polynucleotide stabilizing interactions and the F122 homologue is important in conferring RNA specificity. Additionally, the DNA/RNA−T4 Rnl2 crystal structure shows both the F121 and F122 homologues intercalating the minor groove (13). In light of the experimentally determined importance of the F121 and F122 homologues and their crystallographically determined positions, which our initial homology model mimicked, it is appropriate to explore why F121 and F122 were not found intercalating the minor groove during the equilibrated, dynamics phase of the trajectory. Roughly 21 ns into the equilibration phase of the simulation, the dsRNA on the 3′-OH side of the nick moved away from the protein surface. When it returned, approximately 2 ns later, F121 and F122 no longer penetrated the minor groove. Instead, they made nonspecific van der Waals contacts with the two adjacent ribofuranose residues that border the 3′-OH side of the minor groove in which F121 and F122 were originally positioned. While these new positions of F121 and F122 are conserved throughout the remainder of the simulation, approximately 29 ns into the dynamics phase of the simulation the dsRNA on the 3′-OH side of the nick again moved away from the protein. These structural fluctuations indicate that the dsRNA undergoes occasional “breathing” motions, moving toward and away from the protein on the nanosecond time scale. During these breathing motions, F121 and F122 may move from positions where they intercalate the minor groove to positions where they make nonspecific van der Waals interactions with the ribofuranose moieties flanking the minor groove. This suggestion is consistent with polynucleotide temperature (Debye−Waller) factors reported for the DNA−RNA hybrid in the T4 Rnl2 complex (13) (Figure S2 of the Supporting Information).
RNA Binding through a Unique Kinetoplastid Insert Region
The kinetoplastids share a unique insert region that roughly spans TbREL1 residues 165−200, depending on the exact alignment (7,13,14) (Figure 1). Two regions of this insert lack defined secondary structure and contain positively charged residues that were postulated to be involved in RNA binding (7). On the basis of a homology model of the T4 Rnl2 structure, Nandakumar et al. (13) hypothesized that the loops may rearrange or adopt secondary structure and help stabilize the 3′-OH side of the nicked dsRNA. More recently, ensemble-averaged electrostatic calculations over a 20 ns molecular dynamics simulation of the ATP-bound TbREL1 protein without RNA indicated a large lobe of positive electrostatic potential near this region, further substantiating the suggested RNA binding role (14). The electrostatic calculations also suggested that charged residues K166, K172, K175, and/or R194 could play important dsRNA stabilizing roles (14).
In this work, we directly monitor the dynamics of this unique TbREL1 insert region and assess its dsRNA binding capacity. Through a set of transient interactions, the insert region significantly impacts the mobility of the gRNA strand, thus providing a secondary stabilization effect on the nicked 3′-OH strand (Figures 5 and 6). Despite the prediction that K166, K172, and K175 would play important dsRNA stabilizing roles, the current simulation shows that none of these residues directly contact the RNA. However, because they are commensurate with a large lobe of positive potential (14), they most likely participate in providing the electrostatic environment responsible for directing the diffusional association of the RNA substrate into a productive bimolecular association complex. R194, on the other hand, directly contacts the RNA, forming a persistent salt bridge with the nonbridging phosphate oxygen atoms of the gRNA strand backbone (Table 2). Despite their positive charge, K183 and K184, two residues positioned adjacent to the terminal end of the 3′-OH strand, do not make persistent salt bridges or hydrogen bonds with the dsRNA. The RNA−DNA hybrid bound to T4 Rnl2, which was used to construct the simulated model, likewise does not take advantage of the high positive electrostatic potential immediately adjacent to this region; thus, we note that the observed behavior may be an artifact of our homology model, yet the stability conferred by the kinetoplastid insert on the bound RNA is apparent (Figure 6). The kinetoplastid insert contacts the gRNA sequence from position 2 to 4, corresponding to a 0.5 Å drop in rmsf, and contacts the nicked 5′-PO4 strand at positions 10 and 11, attenuating fluctuations at the 3′-OH terminus.
Conclusions
This molecular dynamics simulation of TbREL1 in complex with a dsRNA substrate has clarified the roles of active site residues during step 3 of catalysis and allowed us to suggest a possible deprotonation pathway involving a nonbridging oxygen of the AppN intermediate. In an effort to help clarify the number of metals required for catalysis, a Mg2+ cation was positioned within the active site on the basis of the homology with other superfamily members. By examining the relation of the Mg2+ cation to the reactive residues, we inferred that Mg2+ is likely to confer transition state stabilization during nicked strand religation and may also help stabilize the ribofuranose of the AppN intermediate. Both of these functional aspects have the potential to impart greater catalytic efficiency; as a result, we conclude that the TbREL1 mechanism likely requires two Mg2+ cations for optimal efficiency. Additionally, by examining the persistence contact times between the protein and RNA during our simulation, we were able to explain, at least in part, why TbREL1 requires RNA on the 5′-PO4 side of the nick in vitro (35) and to clarify the role of the unique kinetoplastid insert in RNA binding.
Our analysis allows us to make several predictions that may motivate future experimental work, adding to a growing body of knowledge concerning the structure−function relationship of RNA-binding residues within this enzyme. The side chains of D62, S65, S67, and Q70 make RNA-specific contacts; as a result, we predict that mutations to these residues will adversely affect the religation rate by disrupting RNA binding. Additionally, the recognition sequence formed by D62, L63, P64, S65, S67, and Q70 collectively forms a smooth surface that intercalates the minor grove; consequently, we predict that mutating any of these residues to a large, bulky residue such as tryptophan will perturb the protein surface continuity and attenuate the religation rate by lowering RNA affinity. Positively charged residues K166, K172, and K175 reside within the unique kinetoplastid insert loop but do not directly contact the RNA. Instead, they are predicted to contribute to the electrostatic environment responsible for mediating RNA association. As a result, we predict that mutating K166, K172, and K175 to alanine will lower the religation activity by decreasing the total charge on the protein. We note that while step 1 autoadenylation requires ATP binding, the net charge on ATP is significantly lower than that carried by dsRNA, and as a result, step 1 activity will be significantly less influenced by the decrease in total protein charge.
Acknowledgments
The National Center for Supercomputing Applications, the San Diego Supercomputing Center, the W. M. Keck Foundation, Accelrys, Inc., the National Biomedical Computational Resource, and the Center for Theoretical Biological Physics are gratefully acknowledged.
Supporting Information Available
Additional figures and a representative PDB file of the protein−RNA complex. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: TbREL, T. brucei RNA-editing ligase; MD, molecular dynamics; gRNA, guide RNA; LIG1, DNA ligase 1; AppN, AMP attached to the nick 5′-PO4 moiety; rmsd, root-mean-square deviation; rmsf, root-mean-square fluctuation; dsRNA, double-stranded RNA.
Supplementary Material
References
- Stuart K.; Allen T. E.; Heidmann S.; Seiwert S. D. (1997) RNA editing in kinetoplastid protozoa. Microbiol. Mol. Biol. Rev. 61, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi A. K.; Ernst N. L.; Domingo G. J.; Fleck M.; Salavati R.; Stuart K. D. (2006) Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B.; Simpson L. (1992) Formation of guide RNA/messenger RNA chimeric molecules in vitro, the initial step of RNA editing, is dependent on an anchor sequence. Proc. Natl. Acad. Sci. U.S.A. 89, 11944–11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert S. D.; Heidmann S.; Stuart K. (1996) Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell 84, 831. [DOI] [PubMed] [Google Scholar]
- Simpson L.; Sbicego S.; Aphasizhev R. (2003) Uridine insertion/deletion RNA editing in trypanosome mitochondria: A complex business. RNA 9, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K. D.; Schnaufer A.; Ernst N. L.; Panigrahi A. K. (2005) Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 30, 97. [DOI] [PubMed] [Google Scholar]
- Deng J.; Schnaufer A.; Salavati R.; Stuart K. D.; Hol W. G. (2004) High resolution crystal structure of a key editosome enzyme from Trypanosoma brucei: RNA editing ligase 1. J. Mol. Biol. 343, 601–613. [DOI] [PubMed] [Google Scholar]
- Schnaufer A.; Panigrahi A. K.; Panicucci B.; Igo R. P. Jr.; Wirtz E.; Salavati R.; Stuart K. (2001) An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science 291, 2159–2162. [DOI] [PubMed] [Google Scholar]
- Panigrahi A. K.; Gygi S. P.; Ernst N. L.; Igo R. P. Jr.; Palazzo S. S.; Schnaufer A.; Weston D. S.; Carmean N.; Salavati R.; Aebersold R.; Stuart K. D. (2001) Association of Two Novel Proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA Editing Complex. Mol. Cell. Biol. 21, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S.; Lima C. D. (2004) The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin. Struct. Biol. 14, 757–764. [DOI] [PubMed] [Google Scholar]
- Håkansson K.; Doherty A. J.; Shuman S.; Wigley D. B. (1997) X-Ray Crystallography Reveals a Large Conformational Change during Guanyl Transfer by mRNA Capping Enzymes. Cell 89, 545–553. [DOI] [PubMed] [Google Scholar]
- Cherepanov A. V.; de Vries S. (2002) Kinetic Mechanism of the Mg2+-dependent Nucleotidyl Transfer Catalyzed by T4 DNA and RNA Ligases. J. Biol. Chem. 277, 1695–1704. [DOI] [PubMed] [Google Scholar]
- Nandakumar J.; Shuman S.; Lima C. D. (2006) RNA ligase structures reveal the basis for RNA specificity and conformational changes that drive ligation forward. Cell 127, 71. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Swift R. V.; McCammon J. A. (2007) Functional and Structural Insights Revealed by Molecular Dynamics Simulations of an Essential RNA Editing Ligase in Trypanosoma brucei. PLoS Neglected Trop. Dis. 1, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal J. M. (2008) DNA and RNA ligases: Structural variations and shared mechanisms. Curr. Opin. Struct. Biol. 18, 96–105. [DOI] [PubMed] [Google Scholar]
- Norberg J.; Nilsson L. (2002) Molecular dynamics applied to nucleic acids. Acc. Chem. Res. 35, 465–472. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Nilsson L. (2008) Molecular dynamics simulations of nucleic acid-protein complexes. Curr. Opin. Struct. Biol. 18, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham Iii T. E., Simmerling C. L., Wang J., Duke R. E., Luo R., Merz K. M., Pearlman D. A., Crowley M., Walker R. C., Zhang W., Wang B., Hayik S., Roitberg A., Seabra G., Wong K. F., Paesani F., Wu X., Brozell S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Beroza P., Mathews D. H., Schafmeister C., Ross W. S., Kollman P. A. (2006) Amber 9.
- El Omari K.; Ren J.; Bird L. E.; Bona M. K.; Klarmann G.; LeGrice S. F.; Stammers D. K. (2006) Molecular architecture and ligand recognition determinants for T4 RNA ligase. J. Biol. Chem. 281, 1573–1579. [DOI] [PubMed] [Google Scholar]
- Vriend G. (1990) WHAT IF: A molecular modeling and drug design program. J. Mol. Graphics 8, 52–56. [DOI] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher K. L.; Redman L. T.; Carlson H. A. (2003) Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 24, 1016–1025. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An N,ãÖlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089. [Google Scholar]
- Kale L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. (1999) NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312. [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. T.; Westhof E.; Sundaralingam M. (1981) Exact method for the calculation of pseudorotation parameters Ptm and their errors. A comparison of the Altona-Sundaralingam and Cremer-Pople treatment of puckering of five-membered rings. Acta Crystallogr. A37, 421–425. [Google Scholar]
- Arora K.; Schlick T. (2003) Deoxyadenosine sugar puckering pathway simulated by the stochastic difference equation algorithm. Chem. Phys. Lett. 378, 1–8. [Google Scholar]
- Amadei A.; Linssen A.; Berendsen H. (1993) Essential dynamics of proteins. Proteins: Struct., Funct., Genet. 17, 412–425. [DOI] [PubMed] [Google Scholar]
- Garcia A. E. (1992) Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 68, 2696–2699. [DOI] [PubMed] [Google Scholar]
- Yin S.; Ho C. K.; Shuman S. (2003) Structure-function analysis of T4 RNA ligase 2. J. Biol. Chem. 278, 17601–17608. [DOI] [PubMed] [Google Scholar]
- Nandakumar J.; Shuman S. (2004) How an RNA ligase discriminates RNA versus DNA damage. Mol. Cell 16, 211–221. [DOI] [PubMed] [Google Scholar]
- Palazzo S. S.; Panigrahi A. K.; Igo R. P.; Salavati R.; Stuart K. (2003) Kinetoplastid RNA editing ligases: Complex association, characterization, and substrate requirements. Mol. Biochem. Parasitol. 127, 161–167. [DOI] [PubMed] [Google Scholar]
- Håkansson K.; Wigley D. B. (1998) Structure of a complex between a cap analogue and mRNA guanylyl transferase demonstrates the structural chemistry of RNA capping. Proc. Natl. Acad. Sci. U.S.A. 95, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. N.; Dunaway-Mariano D. (2004) Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 29, 495–503. [DOI] [PubMed] [Google Scholar]
- Fersht A. (1999) Structure and Mechanism in Protein Science, W. H. Freeman and Company, New York. [Google Scholar]
- Bairoch A. (2000) The ENZYME database in 2000. Nucleic Acids Res. 28, 304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.