

Abstract
P/Q-type calcium channels are known to form clusters at the presynaptic membrane where they mediate calcium influx, triggering vesicle fusion. We now report functional P/Q channel clusters in the axolemma of developing central axons that are also associated with sites of vesicle fusion. These channels were activated by axonal action potentials and the resulting calcium influx is well suited to mediate formation of a synaptic style SNARE complex involving SNAP-25, that we show to be located on the axolemma. Vesicular elements within axons were found to be the sole repository of vesicular glutamate in developing white matter. The axonal vesicular elements expressed the glutamate transporter V-ATPase, which is responsible for vesicular glutamate loading. The P/Q channel α1A subunit was found to be present within the axolemma at early nodes of Ranvier and deleterious mutations of the α1A subunit, or an associated α2δ-2 subunit, disrupted the localization of nodal proteins such as voltage-gated sodium channels, β IV spectrin and CASPR-1. This was associated with the presence of malformed nodes of Ranvier characterized by an accumulation of axoplasmic vesicles under the nodal membrane. The data are consistent with the presence of a vesicular signalling pathway between axons and glial cells that is essential for proper development of the node of Ranvier.
Myelinated axons are responsible for the rapid transmission of action potentials around the nervous system. A high conduction velocity of axons is achieved by restricting the generation of action potentials to nodes of Ranvier that are spaced at regular intervals along the axon, separated by stretches of insulating myelin laid down by oligodendrocytes. Formation of the node of Ranvier involves clustering of the Na+ channels responsible for action potential generation at node sites as myelin is deposited along the internodal region (Rasband & Shrager, 2000; Girault & Peles, 2002; Salzer, 2003). Na+ channel clustering is preceded by the clustering of several components of a Na+ channel–cytoskeletal complex, including ankyrin G and β IV spectrin (Lambert et al. 1997; Rasband et al. 1999; Jenkins & Bennett, 2002). Mediators of axon–glial interaction such as CASPR-1 also form clusters at early node sites before Na+ channels start to aggregate (Rasband et al. 1999), and the whole process is apparently dependent upon a soluble factor released by oligodendrocytes (Kaplan et al. 1997). It is currently unclear what determines where nodes will form or how the aggregation of the components of the node is controlled.
Intracellular Ca2+ ([Ca2+]i) regulates the outgrowth of axons (Henley & Poo, 2004; Conklin et al. 2005) and the formation of neuronal structures such as dendrites (Konur & Ghosh, 2005). Localized [Ca2+]i changes could also potentially coordinate the development of the node of Ranvier. Local Ca2+ changes could be produced by focal expression of voltage-gated Ca2+ channels along axons (Mackenzie et al. 1996; Forti et al. 2000). Ca2+ channels are heteromeric membrane proteins that play an important role in the regulation of numerous cellular processes and are classified according to their electrophysiological and pharmacological properties (L-, N-, P/Q-, R- and T-type). Each Ca2+ channel type is composed of a pore-forming α1 subunit and a number of accessory subunits. It is the α1 subunit that distinguishes Ca2+ channel subtype and there are currently 10 α1 subunit genes known (Catterall, 2000). The α1 subunit contains the elements responsible for voltage-dependent gating of the channels, which may be modulated by accessory subunits.
While the axons of mammalian neurons possess several proteins involved in Ca2+ homeostasis, evidence for the expression of functional Ca2+ channels on the axolemma has been slow to reveal itself. Past reports in the literature have suggested that axonal Ca2+ channels may, under physiological circumstances, play a part in the regulation of action potential frequency and the non-vesicular release of neurotransmitter substances at en passant synapses (Callewaert et al. 1996; Forti et al. 2000). More recently, vesicular release of glutamate has been demonstrated in both the corpus callosum and immature rat optic nerve (Kukley et al. 2007; Ziskin et al. 2007). Both studies independently reported activity-dependent activation of AMPA receptors on NG2(+) cells following vesicular glutamate release, predominantly from unmyelinated axons. Kukley et al. (2007) provided evidence that this synapse-like form of axon–glia communication was initiated by rapid Ca2+ signalling within Ca2+ microdomains in axons. Here we provide evidence that clustered axonal Ca2+ channels play a significant role in action potential conduction in neonatal central axons and make a pivotal contribution to the development of the node of Ranvier. The Ca2+ channels involved are largely of the synaptic P/Q-type and we provide additional evidence that they are involved in co-ordinating the fusion of vesicular elements from the axoplasm to the axolemma of developing axons.
Methods
Ethical approval
All animal procedures were approved by local ethical review and conformed to UK Home Office regulations. Animals were killed by vertebral dislocation or CO2 inhalation depending upon age.
General
Optic nerves were dissected from Lister-hooded rats and perfused with artificial cerebrospinal fluid (aCSF) of composition (mm): NaCl, 126; KCl, 3; NaH2PO4, 2; MgSO4, 2; CaCl2, 2; NaHCO3, 26; glucose, 10; pH, 7.45, bubbled with 5% CO2–95% O2 and maintained at 37°C. Bicarbonate, sulphates and phosphates were omitted from the high divalent cation solutions used to evoke divalent cation action potentials (osmolarity maintained with sucrose). These solutions contained 10 mm Hepes and were buffered to pH 7.45. Data are mean ±s.e.m., significance determined by ANOVA with Tukey's post test. Unless otherwise stated all chemicals were purchased from Sigma (UK).
Electrophysiology
Extracellular compound action potentials were evoked and recorded with suction electrodes. Peak-to-peak amplitude was used to assess changes in the number of unitary action potentials in the neonate since action potential area cannot be applied reliably to recordings from neonatal nerves. This is due to the large stimulus artifact produced by the longer stimulus times that are required to elicite a full neonatal compound action potential (Foster et al. 1982; Fern et al. 1998).
Nerves were maintained in an interface perfusion chamber (Medical Systems, Greenvale, New York, NY, USA), and continuously oxygenated with 5% CO2–95% O2. Individual nerves were electrically stimulated via a suction electrode at one end using square-wave constant current pulses of 150–600 μs (< P15) or 50 μs (nsd> P15) duration (Iso stim A320, WPI), and compound action potentials recorded via a second suction electrode at the other end (Cyber Amp 320, Axon Instruments). The recorded signal was subtracted from a parallel differential electrode, filtered (low pass: 800–10000 Hz, depending upon animal age), digitized (25000 Hz: 1401 mini, Cambridge Electronic Design) and displayed on a PC running Signal software (Cambridge Electronic Design) with positive (relative to the subtraction electrode) going up. Lower filter settings were used for neonatal nerves to prevent distortion of the wave form due to the slow time-course of the wave compared to those recorded from adult nerves.
Immuno-histochemistry
Optic nerves were dissected into 0.1 m PBS and fixed in 4% paraformaldehyde for 30 min. The optic nerves were subsequently incubated in 0.1 m PBS plus 20% sucrose w/v for 5 min prior to freeze-sectioning (20 μm sections) and subsequent blocking for 60 min in 0.1 m PBS, 10% fetal goat serum plus 0.5% Triton-X 100. Sections were then incubated in this solution plus primary antibody at 4°C overnight. Primary antibodies included: polyclonal α1A (Alomone; 1: 200), α1B, α1C, α1D (Sigma; 1: 200), and affinity-purified α2δ-2 (Brodbeck et al. 2002; 1: 200). In double-staining experiments, second primary incubations were at room temperature for 2 h: monoclonal antipan-Na+ channel (Sigma; 1: 200), anti-neurofilament-70 (NF-70) or anti-neurofilament-200 (NF-200) (Chemicon; 1: 100 and 1: 200, respectively), anti-GFAP (Glial Fibrillary Acidic Protein) (Sigma; 1: 500), anti-CNPase (2′, 3′-cyclic nucleotide 3′-phosphodiesterase) (Sigma; 1: 100), or anti-CASPR-1 (1: 1000) were used. The axonal markers NF-70 and NF-200 tend to label small diameter and larger diameter axons, respectively (Sanchez et al. 2000). Appropriate Alexa-conjugated secondary antibodies (Cambridge Bioscience; 1: 1000) were subsequently applied for 1 h and sections examined by scanning confocal microscopy using an Olympus FV300 Fluoview confocal image system.
Images were analysed using either Metamorph (Universal Imaging Corporation) or Image J (NIH). Images of Na+ and Ca2+ channel clusters were analysed using Image J using the Cell Counter plug-in. Random single optical section fields of view (FOV) were taken from a minimum of three nerves with no more than three FOVs being taken from a single nerve, resulting in a sample number of at least nine. When such analysis was done using two or more different ages, or with either leaner or ducky mice and their respective controls, the individual performing the analysis (J.J.P.A.) was blinded to the images in order to avoid any potential bias. The degree to which Ca2+ channel clusters co-localized with cell-specific markers was assessed by counting the number of times the Ca2+ channel clusters either did, or did not, completely overlap with the marker within a field of view. The degree of co-localization was then reported as a percentage of the total number of Ca2+ channel clusters within the FOV. The degree of association of Ca2+ channel clusters with the paranodal marker CASPR-1 was assessed by observing the number of times clusters within a FOV either were co-localized (defined as complete overlap) or were immediately adjacent (defined as touching or partial overlap) to CASPR-1 immunoreactivity. The degree of association between these two markers is taken to be the degree of adjacent and co-localized markers combined. Finally, neurofilament intensity was calculated using Metamorph software.
Electron microscopy
Optic nerves from P23–P25 mice and P10 rats were post-fixed in 3% gluteraldehyde in Sorenson's phosphate buffer. Nerves from Leaner mice were obtained from Jackson Laboratories (MA, USA) and were fixed according to our instruction. Nerves were then post-fixed with 2% osmium tetroxide and dehydrated prior to infiltration in epoxy. Sections were counterstained with uranyl acetate and lead citrate and examined with a Jeol 100CX electron microscope (see Thomas et al. (2004) for further details). For post-embedding immuno-labelling, primary antibody was applied to the sections overnight. Antibodies raised against: α1A (Alomone; 1: 200), SNAP-25 (Sigma; 1: 50), V-ATPase (Santa Cruz Biochem; 1: 50), glutamate (Chemicon International; 1: 25–1: 50) were used and appropriate 20–30 nm gold particle secondary antibodies were applied following washing. Controls involved omitting primary antibodies, which resulted in zero staining. The anti-SNAP-25 antibody was used at the same concentration by Kolk et al. (2000), who demonstrated a synaptic pattern of staining in CNS grey matter, in addition to a lower level of axonal staining. The anti-glutamate antibody has been described in prior publications (e.g. Back et al. 2006), where a range of controls are described.
Results
Functional voltage-gated calcium channels on neonatal central axons
To probe for the presence of functional Ca2+ channels on developing axons we determined whether the axons can support a divalent cation-mediated compound action potential (DCAP) (Fatt & Ginsborg, 1958). Na+ channels were blocked by perfusion with zero-Na+ aCSF + 1 μm TTX, abolishing the normal action potential (Fig. 1A–C). Subsequently increasing the extracellular divalent cation concentration to 40 mm of both Ca2+ and Ba2+ (total divalent cation concentration = 80 mm) to potentiate current through Ca2+ channels produced an action potential from P2 to P12, but not at P22 (Fig. 1A–C). Between 10 and 20 min of perfusion with high concentrations of divalent cation solution was required before a DCAP appeared, consistent with the slow diffusion of the ions into the extracellular space (Fern & Harrison, 1988). The DCAP recorded from the neonatal optic nerve was similar to the Na+ action potential in amplitude but was relatively slower conducting (0.1–0.3 m s−1 compared to 1–3 m s−1). Slow action potential conduction will be partly due to differences in channel kinetics between Ca2+ channels and Na+ channels and partly to the surface negative charge screening effects of the divalent ions. High concentrations of Ca2+ and Ba2+ will increase the screening of fixed negative charge on the extracellular surface of the axon, effectively raising the activation threshold of voltage-gated channels (Brismar & Frankenhaeuser, 1972; Hille, 1998). Removal of extracellular Na+ will produce a degree of membrane depolarization that will partially counter this effect, while rapid depletion of axoplasmic Na+ will block any effect of Na–Ca exchange (NCX) upon membrane potential or ion fluxes (Stys et al. 1992; Leppanen & Stys, 1997). Although relatively slow, the rate of DCAP conduction precludes the possibility that the potential is conducted down glial cells in the optic nerve as a form of spreading depression. Spreading depression propagates at three to four orders of magnitude more slowly than the DCAP (between 30 and 50 μm s−1), travels only relatively short distances from a point stimulation (∼500 μm), lasts over four orders of magnitude longer (∼100 s), and does not operate in central white matter (e.g. Peters et al. 2003; Fabricius et al. 2006; Smith et al. 2006). A contribution from divalent cations passing through TTX-resistant Na+ channels can also be discounted since the Ca2+ permeability of Na+ channels is low, high Ca2+ concentrations block Na+ channels (Hille, 1998; Armstrong & Cota, 1999), and the TTX-insensitive Na+ channels in the axons are not capable of supporting an action potential (TTX completely blocks the normal Na+ action potential). Furthermore robust DCAPs were observed when Ba2+ alone was used as the charge carrier (see below), while Ba2+ permeability through Na+ channels is not measurable (Hille, 1998).
Figure 1. Divalent cation action potentials can be produced in neonatal rat optic nerve axons.

A, compound action potentials recorded at three developmental ages. Top traces show the normal, fast-conducting, action potential (arrow, following the stimulus artefact). Middle traces show block of this action potential in zero-Na+–TTX (1 μm). Lower traces show a large amplitude, slow-conducting, action potential recorded in the presence of 40 mm of both Ba2+ and Ca2+ (still in zero-Na+–TTX). Note the absence of a divalent cation action potential at P22. B, action potential amplitude recorded at P2 versus time. Zero-Na+–TTX blocks the action potential after ∼10 min and switching to Ba2+–Ca2+ produces a divalent cation action potential after 12 min that then declines slowly. C, similar plot of action potentials at P10 and P22. D, traces of normal action potential, block in zero-Na+–TTX and perfusion with Ba2+–Ca2+ (in zero-Na+–TTX) solution at P2 in the presence of the specific L-type blocker diltiazem (50 μm). No divalent cation action potential is apparent. E, similar data at P10, showing large divalent cation action potential in diltiazem. F, experiment as above demonstrating a much reduced divalent cation action potential in the presence of ω-agatoxin IVA (100 nm). G, amplitude of the divalent cation action potential relative to normal action potential at various ages. The divalent cation action potential was blocked by diltiazem at P2–P5 but not at P8–P12. ω-Agatoxin IVA significantly blocked the divalent cation action potential at P8–P12, as did the non-selective calcium channel blocker La3+ (100 μm). Error bars are s.e.m., ***P < 0.001 versus control P8–P12, ††P < 0.01 versus control P2–P5. Scale bars are 10 mV and 10 ms.
The Ca2+ channel-dependent nature of the DCAP was confirmed by application of selective Ca2+ channel blockers. The DCAP was blocked at P8–P12 by the broad-spectrum Ca2+ channel blocker La3+ (100 μm) (Fig. 1G). The selective L-type Ca2+ channel blocker diltiazem (50 μm) largely blocked the divalent cation action potential at P2–P5, but not at P8–P12 (Fig. 1D, E and G). The P8–P12 divalent cation action potential was largely blocked by the specific P/Q blocker ω-agatoxin IVA (Mintz et al. 1992) (100 nm) (Fig. 1F and G).
Perfusion with either divalent cation alone produced a DCAP (Fig. 2A–C). Once apparent, the DCAP declined over an ∼20 min period, irrespective of whether it was Ba2+ or Ca2+ in the divalent cation solution (Fig. 2A–C), or both (Figs 1B, C and 2E). This effect may be due to the run-down of concentration gradients in the small diameter, small volume, neonatal rat optic nerve axons. Axons will lack Na–Ca exchange under these zero-Na+ conditions (Stys et al. 1992; Leppanen & Stys, 1997), and divalent ions entering during an action potential are likely to remain in the axoplasm. The diameter of axons in the P8–P12 rat optic nerve ranges between ∼0.1–1.0 μm (Foster et al. 1982). The effect of a single 100 mV DCAP upon the divalent-cation reversal potential can be calculated using the equation n=CV/2F where n is the number of divalent ions entering the axon during one action potential (in moles), C is the membrane capacitance (1 μF cm−2), V is the action potential amplitude, and F is the Faraday constant. The DCAP amplitude is estimated at 100 mV since intra-axonal recording from optic nerve axons have never been achieved. The number of divalent cations within the resting axoplasm can be calculated from the cylinder volume and resting ion concentration, while the number of ions entering a given length of axon during a single action potential can be calculated from the voltage change across the membrane capacitance (Aidley, 1971). Assuming a resting axoplasmic divalent concentration of 100 nm, a single DCAP is predicted to result in a 20.2 μm rise in axoplasmic divalent cation concentration, producing a sharp decline in the divalent-cation reversal potential (Fig. 2D). The time-course of the DCAP was not significantly different in solutions with various divalent concentrations (Fig. 2E). However, the axoplasmic divalent cation concentration primarily determines the divalent cation reversal potential and these changes in extracellular divalent ion concentration are predicted to have only a small effect upon DCAP survival time (Fig. 2D). While these calculations are consistent with a collapse of divalent ion influx during repetitive stimulation, the associated rise in axoplasmic divalent-cation concentration may have other effects upon action potential conduction that might contribute to the relatively rapid conduction failure.
Figure 2. Features of the DCAP in P8–P12 rat optic nerve.

A, high concentrations of a single divalent cation can produce a DCAP. Top, a normal compound action potential recorded in aCSF. Middle, block of this action potential in zero-Na+–TTX solution. Bottom, a large-amplitude, slow-conducting DCAP is recorded in the presence of 95 mm Ba2+. B, action potential amplitude is plotted against time, demonstrating the decline in the DCAP over an ∼10 min time course. C, a similar protocol showing the time-course of a DCAP recorded in 60 mm Ca2+. Switching to zero-Na+–TTX solution produced block of the normal action potential and changing to 60 mm extracellular Ca2+ resulted in a short-lasting DCAP. D, the divalent cation reversal potential in axons calculated assuming a starting axoplasmic concentration of 100 nm, an extracellular divalent cation concentration of 90 mm, a resting membrane potential of –70 mV, a DCAP amplitude of 100 mV and a membrane capacitance of 1 μF cm−2. It is also assumed that there is no significant extrusion of divalent cations entering during the DCAP and that there is no influx into the axon when the axolemma is at rest. Given these assumptions, it is apparent that a small number of divalent cation action potentials have a dramatic affect upon the divalent cation reversal potential, in particular for the smaller diameter axons (0.1 μm, filled squares). Reducing the extracellular divalent cation concentration to 40 mm has minimal effect upon the time course of the collapse of the reversal potential (grey circles, plotted for a 1 μm axon). E, the time between the first appearance of a DCAP and its failure (survival time) in various high divalent cation conditions, showing that varying the total divalent cation concentration between 40 and 100 mm had no significant affect, as predicted in D.
Calcium channel activation during action potential conduction
We examined the possibility that the calcium channels expressed by developing central axons might contribute to normal action potential conduction. Compound action potentials recorded from neonatal rat optic nerves were stable under control conditions of perfusion with normal aCSF (Fig. 3A), and exhibited a reversible fall in amplitude at P2–P5 (Fig. 3D and F) and P8–P12 (Fig. 3B, E and F) upon perfusion with the broad-spectrum calcium channel blocker La3+ (100 μm). This effect of La3+ was lost by P20 (Fig. 3F). The selective L-type calcium channel blocker diltiazem (50 μm) mimicked the effect of La3+ upon compound action potential amplitude at P2–P5 but not at later developmental stages (Fig. 3C–F). Diltiazem did slow action potential conduction at P8–P12, however (Fig. 3C), but had no comparable effect in P18–P25 optic nerves (data not shown). A similar partial conduction block was produced by nifedepine (5 μm) at P2, reducing amplitude by 44.5%± 12.8% (n= 3, data not shown). Nifedepine is not the L-type channel blocker of choice in this preparation, however (Fern et al. 1995). Block of P/Q-type channels with ω-agatoxin IVA (100 nm) at P8–P12 produced a significant fall in action potential amplitude, a result also obtained with the N- and P/Q-type blocker ω-conotoxin MVIIC (1 μm) (Fig. 3G, I and J). In contrast, specific block of N-type channels with ω-conotoxin GVIA (1 μm) had no effect (Fig. 3H–J). These data suggest that L-type calcium channels contribute to normal action potential conduction at P2–P5 (prior to the appearance of compact myelin in this preparation), that P/Q-type channels have a similar effect at P10 (when myelination is getting underway), and that no significant contribution to action potential amplitude from calcium channels is apparent at P18–P25.
Figure 3. Ca2+ channels contribute to normal action potentials in developing axons.

A, P12 control showing the compound action potential recorded at T= 0 min (control start) and T= 150 min (control end). B, non-selective Ca2+ channel block with La3+ reduced action potential amplitude at P12. C, block of L-type channels with diltiazem at P12 did not reduce action potential amplitude (slowing is evident). D, La3+ and diltiazem produced a similar, reversible, decline in the compound action potential at P2. E, La3+ but not diltiazem produced a reversible decline in the action potential at P10. F, summary showing the effect of La3+ and diltiazem upon compound action potential amplitude at different ages. G, block of P/Q-type channels with ω-agatoxin IVA (100 nm) reduced action potential amplitude at P12. H, block of N-type channels with ω-conotoxin GVIA (1 μm) had no significant affect at P12. I, time course of effect of ω-agatoxin IVA, ω-conotoxin GVIA and ω-conotoxin MVIIC on action potential amplitude at P12. J, summary of the effects of ω-agatoxin IVA or ω-conotoxins upon action potential amplitude at P12. *P < 0.05, ***P < 0.001.
Synaptic-type calcium channels are clustered on developing central axons
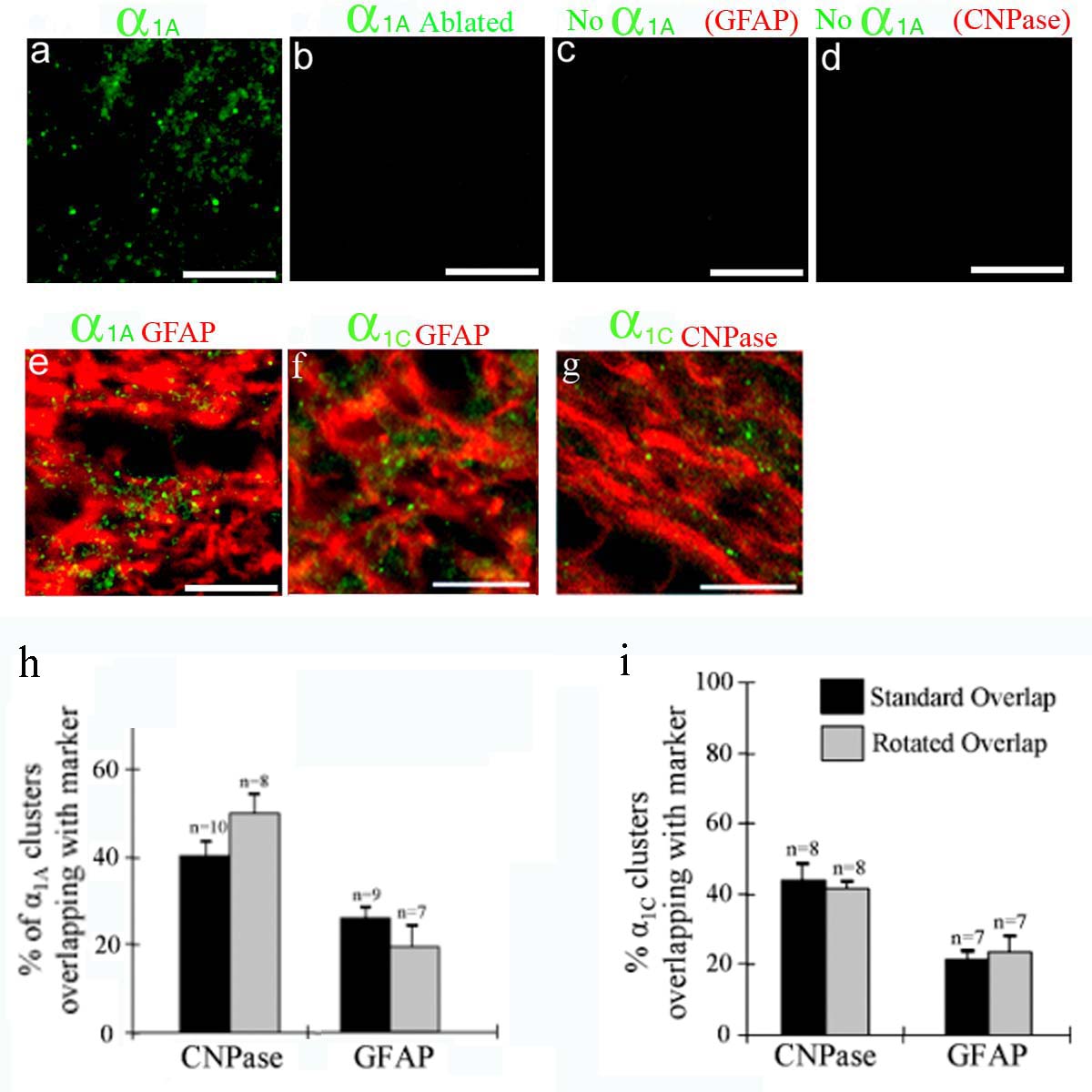
We examined the expression of various calcium channel α1 subunits in developing white matter and detected a clustered expression patern of α1A (P/Q-type: Figs 4A and B, and 5A) and α1C (L-type: Figs 4A and 5B) subunits (α1B and α1D were not detected, data not shown). While α1C clusters were present at a relatively stable level between P2–P20, α1A clusters were almost absent at P2, reached a maximum at P12 and declined by P20 (Figs 4A, and 5A and B). Calcium channel clusters were co-localized with small NF-70(+) axons (Fig. 4B), and were less numerous on larger NF-200(+) axons (data not shown). Small diameter developing axons are close-packed and as a result single NF-70(+) axons are difficult to resolve at the light level. To test whether overlap of the α1A clusters with the NF-70 immuno-staining was significant rather than a random event, we blind-counted the extent of α1A–NF-70 overlap in normal images and in images with the α1A rotated by 90 deg, revealing 63.4 ± 5.1% (n= 8) co-localization in rotated compared to 87.1 ± 2.6% (n= 9, P < 0.001) in the non-rotated images (Fig. 4D). Similar levels of co-localization were detected for α1C–NF-70 (P < 0.001, Fig. 4E). Little co-localization was apparent between either α1A or α1C and the glial markers CNPase (Fig. 4C) or GFAP (Supplemental Fig. 1e–g), and no significant difference was found in the degree of overlap between normal and rotated images (P > 0.05 in all cases, Supplemental Fig. 1h and i). In control experiments, α1A clusters were abolished by pre-incubating the antibody with a blocking peptide, or by omitting the primary antibody during co-labelling experiments (Supplemental Fig. 1b–d). Confirmation of the clustered α1A staining of axons was obtained using a second α1A antibody from a different source (Sigma UK; data not shown).
Figure 4. The evolution of ion channel clusters in optic nerve during development.

A, the density of sodium channel (black), α1A (red) and α1C (blue) clusters during development. B, α1A subunits (green) are clustered on NF-70(+) axons (red). C, few α1A subunits (green) co-localized with CNPase(+) oligodendroglia (red). Some overlap is seen due to the close apposition of axons and myelinating oligodendrocyte processes. Scale bars, 10 μm. D and E, rotating the NF-70 and NF-200 images by 90 deg significantly reduced the degree of co-localization, indicating a greater association than would be due to chance. A relatively high degree of association was still observed due to the ubiquitous nature of neurofilament staining. ***P < 0.001.
Figure 5. Voltage-gated Ca2+ channel clusters on developing central axons are precursors of nodes of Ranvier.

A, α1A subunits (green) are present in clusters in P2 optic nerve, increase in number at P12 but are largely absent at P20 (although diffuse staining remains). Na+ channels (NaV) clusters (red) are present at P12 and P20. B, α1C subunits (green) are present at a low density throughout the postnatal ages studied. NaV (red) clusters are visible at P12 and P20. C, α1A clusters (green) are associated with CASPR-1 clusters (red) at P9 (two left panels) and P12 (right panel). D, α1C clusters (green) are associated with CASPR-1 immunoreactivity (red) at P9 (two left panels) and P12 (right panel). E, rotating the CASPR-1 images by 90 deg relative to the α1A images (left) or α1C (right) significantly reduced the association between α1 and CASPR-1 clusters (***P < 0.001, normal versus rotated), indicating a greater association than would be due to chance. All scale bars, 10 μm.
The antibody staining indicated that Ca2+ channels appear in clusters on central axons before Na+ channel clusters first appear (Figs 4A, and 5A and B). We therefore co-stained for α1A and α1C subunits and CASPR-1, an early component of the node of Ranvier (Rasband & Shrager, 2000). α1A clusters were co-localized with 12.2 ± 0.5% of CASPR-1 clusters and were adjacent with a further 43.5 ± 0.8% (16 fields of view, 5 nerves) at P9 (Fig. 5C and E). By P12, 22.2 ± 1.6% of α1A clusters were co-localized with CASPR-1 clusters and 35.2 ± 1.7% were adjacent (12 fields of view, 4 nerves) (Fig. 5C and E). A similar pattern of expression was seen for clusters of α1C subunits (Fig. 5D and E). In all cases, the degree of association was significantly greater than that found in rotated controls (P < 0.001, Fig. 5E). In contrast to CASPR-1, only 22.5 ± 3.3% of Na+ channel clusters were associated (overlap + adjacent) with α1A clusters at P12 (4 fields of view, 4 nerves) with very little actual overlap (2.8%). α1A clusters therefore appear several days before Na+ channel clusters are evident at sites on central axons where nodes are forming, and disperse once Na+ channel clusters appear.
α1A subunits are localized to the axolemma
At the ultrastructural level, white matter α1A protein was largely restricted to the axolemma and axoplasm of developing axons. Immuno-gold particles were particularly common at zones where glial processes were making contact with axons (Fig. 6), areas that were also rich in sites where the vesiculotubular complex appeared to be fusing or approaching the axolemma. Blinded counting of all the gold particles within eight randomly selected whole grid sections revealed that 69.0% of the axolemma particles were located at sites where vesicular fusion was occurring, defined as regions where axoplasmic vesicles or tubules were closely associated with the axolemma (n= 54 gold particles in total). The mean distance to the nearest fusion zone for the remaining 31.0% of particles was 0.42 μm. Analysis revealed that 30.9 ± 2.8% of axolemma area was associated with vesicle fusion sites at P8–P12 (47 axons examined in the same 8 grid sections), and the high coincidence of α1A particles and vesiculotubular complex fusion sites is not due to chance alone (P < 0.001). Of the particles that were located at or near fusion sites, 93.0% were found at points where glial cell processes were directly apposed to the axolemma. In 37.0% of these cases the glial process was myelinating the axon, and the remaining 63.0% of the peri-axonal glial processes were either non-myelinating oligodendrocyte or astrocyte processes, which are often hard to differentiate.
Figure 6. α1A subunits are present on the axolemma of developing axons.

A, immuno-gold labelling of α1A protein reveals gold particles on the intracellular face of the axolemma of axons in P10 rat optic nerve (arrows, shown at higher gain to the right). α1A immunoreactivity is present on an unmyelinated axon that is being contacted by an astrocyte (Ast: identified using standard ultrastructural criteria). Note the large vesiculotubular complex vacuole (asterisk) in the axon (ax). B, an unmyelinated axon (ax) with a glial process running parallel to the axolemma (arrow heads) has immuno-gold labelling of α1A protein at a point where an element of the vesiculotubular complex appears to be fusing with the axolemma (shown at higher gain in the box). A prominent component of the vesiculotubular complex present in the centre of the axon is indicated by an asterisk. C, vesicle elements of the vesiculotubular complex (asterisk) appear to be approaching and fusing with the axolemma at a point where α1A protein is detected by immuno-gold labelling (arrow). This axon is being myelinated and has prominent oligodendrocyte interior and exterior tongue processes (itp, etp). Scale bars: A, 500 nm; B and C, 200 nm.
Vesiculotubular complex is a poorly defined but significant component of developing central axons (Hildebrand & Waxman, 1984), and probably contains elements of tubulovesicular organelles transported from the somata (Nakata et al. 1998), in addition to ‘axoplasmic reticulum’ (Lindsey & Ellisman, 1985), but it is probably not limited to these. The vesiculotubular complex is thought to be involved in trafficking of components to the axolemma during axonal diameter expansion, and is less apparent in mature axons (Hildebrand & Waxman, 1984). An analysis of axons in the P8–P12 optic nerve revealed that prior to the initiation of glial contact 20.3 ± 4.0% (n= 13 axons) of the axolemma was associated with apparent vesiculotubular complex fusion sites. This proportion significantly increased to 31.5 ± 4.2% in more mature axons that had glial processes aligned along the axolemma (n= 15, P < 0.001), and to 38.0 ± 4.5% in axons with layers of compact myelin (n= 19, P < 0.001). By P20 the proportion of the axolemma exhibiting fusion sites in myelinated axons fell to 23.1 ± 3.1%, which was significantly lower than that in myelinating P8–P12 axons (n= 32, P < 0.001), confirming the relationship between the onset of myelination, axon diameter expansion and fusion of the vesiculotubular complex to the axolemma. Tubular elements of this structure can range in diameter between 20 and 40 nm and measure up to 25 μm in length, making them difficult to distinguish from microtubules. Vesicular elements can range between tens to several hundreds of nanometres across (e.g. Fig. 6 and Supplemental Fig. 2), and in the current study we found that both vesicular and tubular elements appear to fuse to the axolemma in places (Fig. 6).
Since the α1A protein is located at regions where vesiculotubular complex elements are fusing with the axolemma, we probed for other components of synaptic vesicle targeting in developing central axons. Using immuno-electron microscopy, we found SNAP-25 expression in a similar pattern to α1A (Fig. 7A and B), with 71.2% of gold particles located on the interior axolemma surface (25% on vesiculotubular complex, 3.8% in the cytoplasm; 49 micrograph fields analysed). In contrast, expression of the vesicular ATPase (V-ATPase) was a mirror image of this pattern (Fig. 7C–E) with 75.8% present in the vesiculotubular complex (12% on the interior axolemma, 10.5% in the cytoplasm; 54 micrograph fields). V-ATPase is involved in glutamate loading of synaptic vesicles and glutamate was found to be concentrated in the vesiculotubular complex (Fig. 7F and G), including within vesiculotubular complex elements fusing with the axolemma (Fig. 7G). Vesiculotubular complex was highlighted in micrographs by intensity thresholding, revealing that 33.1% of the axoplasmic area was taken up by vesiculotubular complex measured in this way (which does not effectively exclude all the microtubules; 49 micrograph fields). Of the gold particles in glutamate immuno-labelled sections 94.4% were in direct contact with vesiculotubular complex highlighted in this fashion, consistent with significant glutamate enrichment in the vesiculotubular complex. Glutamate reactivity was also detected in glial somata and in their primary processes close to the somata (not shown), but glutamate staining was infrequent in glial processes neighbouring developing axons, and glutamate was not seen in vesicular structures in any other cellular compartment within white matter other than the vesiculotubular complex. SNAP-25, V-ATPase and glutamate gold-labelling of this type was seen in small diameter yet-to-be-myelinated axons through to large diameter axons with multiple myelin wraps (e.g. SNAP-25 staining in Fig. 7A and B).
Figure 7. Elements involved in vesicular docking are present in developing axons.

A and B, SNAP-25 localization in P10 rat optic nerve axons detected with immuno-gold labelling. Gold particles (arrows) are concentrated on the interior face of the axolemma, in particular at regions where elements of the vesiculotubular complex are close to, or appear to have docked with, the axolemma. Note in A that gold staining is apparent both in a region where a tubular element is contacting the axolemma (left arrows) and in a region where vesicles appear to be fusing with the axolemma (right arrow). Note in B that a large axoplasmic vesicle is apposed to a region with two gold particles on the axolemma (compare this micrograph with E below). C–E, V-ATPase is densely expressed in elements of the vesiculotubular complex. Note in C the intense gold labelling of a tubular element, shown in greater magnification in D (box) indicated by an arrow head and having a diameter and morphology that distinguishes it from a microtubule. Note in E that staining is apparent on what appears to be a large vesicle in the process of fusing with the axolemma. F, glutamate reactivity in a vesicular element of the vesiculotubular complex (arrowheads). G, glutamate reactivity in vesicular elements of the vesiculotubular complex (arrow heads) that are in the process of fusing with the axolemma, as shown at higher magnification in the box where vesicular and axolemma membranes can be seen to be contiguous. The vesicles are fusing in an area tightly apposed to a glial process (asterisk) and the immuno-gold particle is located in the extracellular space where the vesicles contents are likely to be released (arrows). Scale bar, 100 nm throughout.
Calcium channel mutants have disrupted nodes of Ranvier
As P/Q-type channels were the most abundant channel subtype in developing optic nerve axons and were maximally expressed at a crucial point in maturation we sought evidence of their potential role from two mice strains carrying mutations affecting P/Q-type voltage-gated calcium channels. Ducky (du) and ducky-2J (du2J) mice carry functionally null mutations in the Cacna2d2 gene encoding the α2δ-2 calcium channel subunit, resulting in reduced P/Q currents in cells expressing these subunits (Barclay et al. 2001; Brodbeck et al. 2002). Expression of α2δ-2 was examined in P12 rat optic nerve and showed a similar clustered expression on NF-70(+) axons to that seen for the α1A and α1C subunits (Fig. 8A). The α2δ-2 clusters also had a similar association with CASPR-1 to that seen for α1A clusters at P9 and P12 in the rat (Fig. 8B). At P9 41.9 ± 0.4% (n= 10) of CASPR-1 clusters were associated with α2δ-2 clusters and 51.2 ± 2.6% (n= 9) of CASPR-1 clusters were associated with α2δ-2 clusters at P12. Similar clustered expression of α1A and α2δ-2 was also common in NF-70(+)+/+ mouse optic nerve axons (Fig. 8C), while the number of α1A clusters was significantly reduced in du2J/du2J optic nerve compared to their +/+ littermates (P < 0.01, Fig. 8D). As the α2δ-2 subunit is thought mainly to enhance α1A expression with only minor effects on gating, this down-regulation of α1A clusters in du2J/du2J is in line with the ∼30% loss of P/Q conductance in Purkinje cells of both du/dμ (Barclay et al. 2001; Brodbeck et al. 2002) and in du2J/du2J (Donato et al. 2006).
Figure 8. α2δ-2 subunits are present in clusters on developing central axons.

A, α2δ-2 (green) and NF-70 (red) expression in P12 rat optic nerve. Note that α2δ-2 clusters are present along NF-70 axons. B, α2δ-2 clusters (green) are associated with CASPR-1 clusters (red) in P9 (left) and P12 (right) rat optic nerve. C, α1A (green, left) and α2δ-2 (green, right) co-localize with NF-70+) axons (red) in P9+/+ mouse optic nerve. D, fewer α1A clusters (left, green) are apparent in axons (red) in the du2J mouse, which was statistically significant (right). **P < 0.01. Scale bars, 10 μm.
Since α1A clusters were reduced in du2J/du2J we examined the morphology of the optic nerve nodes of Ranvier in these animals at ∼P25. No obvious differences were found in cross-sections of control +/+ littermates and du2J/du2J (not shown), and the +/+ animals displayed normal nodal morphology for this point in development when examined in long section, with end-loops of the myelinating sheath framing a clear nodal gap (Fig. 9A, n= 43 nodes examined). In contrast, the nodes of Ranvier of du2J/du2J animals (n= 42 nodes examined) had a significantly extended nodal length (Fig. 9D; 1.07 ± 0.05 μm in wild-type versus 1.2 ± 0.05 μm in du2J/du2J, P < 0.05), and a significantly expanded nodal width, as determined by measuring the diameter of the node versus the diameter of the paranode to account for differences in axon size (Fig. 9E; 111 ± 4% in wild-type versus 139 ± 5% in du2J/du2J, P < 0.001). No significant difference in axon or fibre diameter were found between +/+ and du2J/du2J; however, a significant decrease in the g value (axon diameter/fibre diameter) was found (0.79 ± 0.005, n= 70, in wild-type versus 0.76 ± 0.006 in du2J/du2J, n= 119, P < 0.01). The aberrant nodes of the du2J/du2J animals typically contained large vesicular inclusions under the nodal membrane (Fig. 9B and C), while the remaining nodes exhibited a range of malformations from swelling of the nodal membrane to the presence of vacuoles and other inclusions within the nodal axoplasm.
Figure 9. Ultrastructural correlates of P/Q channel dysfunction in developing central axons.

A–E, du2J/du2J, F–J, tgla/tgla. A, long-section of a P25+/+ optic nerve axon (ax) showing a typical node of Ranvier. Note the normal nodal membrane that separates the myelin end-loops and the typical appearance of the nodal and paranodal glial processes. B, two nodes are present in this lower power micrograph of P25 du2J/du2J optic nerve. In both cases, vacuoles of various sizes are present in the nodal axoplasm (arrow heads). C, long section of a node from P25 du2J/du2J optic nerve. Note the presence of large vacuoles within the nodal axoplasm adjacent to the axolemma (indicated by arrowheads). D, comparison of the node length in +/+ (WT) and du2J/du2J optic nerve. E, comparison of the node diameter ((node width/paranodal width) × (100) in +/+ and du2J/du2J. F and G, long sections of nodes of Ranvier in tgla/tgla. Note the presence of vacuoles of various sizes and shapes within the nodal axoplasm (arrowheads). The nodal axolemma was generally wider in the mutant and in some cases had become extruded into the extra-nodal space (H, axon shaded blue). Paranodal glial processes were often abnormal (e.g. H: shaded red), and paranodal end loops appeared swollen in places (yellow arrows). I, comparison of the node length in +/tgla and tgla/tgla optic nerve. J, comparison of the node diameter in +/tgla and tgla/tgla. Scale bars: A, C, F and G, 2 μm; B, 5 μm.
We also examined node morphology in the leaner (tgla/tgla) mutant, which has a mutation in the Cacna1a gene encoding the α1A subunit. tgla/tgla has more severely compromised P/Q channel function than du2J/du2J, corresponding to a 60–70% reduction in peak P-type current density in Purkinje cells (Lorenzon et al. 1998; Fletcher et al. 1996; Dove et al. 1998; Wakamori et al. 1998). While optic nerves from control (+/tgla) littermates (used as control since leaner is bred on oligosyndactylism (Os) background, and Os/Os is fatal) appeared normal, tgla/tgla optic nerves had regions of mild and severe malformation throughout. Regions of severe injury were infiltrated with activated microglia (Supplemental Fig. 3), indicating an ongoing pathology. We examined regions of mild injury to look for the initial changes in axons before wide-scale pathology was evident. Nodes of Ranvier in tgla/tgla axons were often malformed and contained numerous vesicular inclusions (Fig. 9F–H). Malformations ranged from a marked distension of the nodal membrane (Fig. 9F), to complete herniation of inclusion-loaded axoplasm through the paranodal glial processes (Fig. 9H, shaded blue). Swollen and electron lucent peri-nodal glial processes were often seen in these malformed nodes (Fig. 9H, shaded red), and while there was not detachment or disintegration of the myelin end-loops, they often appeared swollen. Compared to controls, nodes of tgla/tgla animals were significantly longer (Fig. 9I; 1.25 ± 0.07, n= 19, versus 1.6 ± 0.1 μm, n= 26, P < 0.05) and wider (Fig. 9J; 1.06 ± 0.07, n= 19 in +/tgla versus 1.77 ± 0.02, n= 27, P < 0.01). Morphometric analysis of myelinated axons also revealed significant decreases in axon diameter (0.96 ± 0.03 μm +/tgla, n= 70, versus 0.82 ± 0.03 μm in tgla/tgla, n= 105, P < 0.05) and fibre diameter (1.18 ± 0.03 μm +/tgla, n= 70, versus 1.01 ± 0.03 μm in tgla/tgla, n= 105, P < 0.01) in tgla/tgla animals. The calculated g value was not significantly affected by the mutation (0.81 ± 0.005, n= 70, versus 0.79 ± 0.006, n= 105, P > 0.05).
The distribution of nodal proteins was examined in du2J/du2J and tgla/tgla, revealing a significant reduction in sodium channel clusters and a marked displacement of CASPR-1 along axons in tgla/tgla animals compared to control (Fig. 10A). Regions of elongated CASPR-1 expression were sometimes punctuated by sodium channel clusters, but not on all occasions. While β IV spectrin and sodium channel clustering appeared normal on the +/tgla controls, there was no detectable β IV spectrin co-localized with the remaining sodium channel clusters in tgla/tgla (Fig. 10B). Less marked changes were apparent in du2J/du2J and included a significant increase in the sodium channel cluster length (Fig. 10C–E).
Figure 10. P/Q channel dysfunction is associated with a disruption in the localization of nodal proteins.

A and B, tgla/tgla; C–E, du2J/du2J. A, top, left: NaV (red) and CASPR-1 (green) localization in control (+/tgla) optic nerve. Bottom, left: the density of NaV clusters is significantly lower in tgla/tgla optic nerve than in +/tgla. Middle: NaV (red) and CASPR-1 (green) localization in tgla/tgla optic nerve. Note the grossly elongated regions of CASPR-1 localization and the sparse, ectopic nature of the NaV clusters (the box is shown in higher magnification to the right). B, β IV-spectrin (green) and NaV (red) clusters are co-localized in control (+/tgla) optic nerve (top) but no β IV-spectrin reactivity is found in tgla/tgla optic nerve (bottom). Overlaid images are shown to the right. C, left, NaV (red) and CASPR-1 (green) localization in control (+/+) optic nerve. Middle: NaV (red) and CASPR-1 localization in du2J/du2J optic nerve. Note the similarity in the staining patterns. Right: two control images showing the absence of CASPR-1 reactivity when the CASPR-1 antibody is omitted from the staining protocol (red, NaV staining remains), and the absence of NaV reactivity when the NaV antibody is omitted from the staining protocol (green, CASPR-1 staining remains). D, the length of NaV clusters (left), the distance between CASPR-1 clusters at nodes (middle, measured from the juxta-paranodal end of one CASPR-1 cluster to the juxta-paranodal end of its companion cluster), and the density of NaV clusters (right) in +/+ and du2J/du2J optic nerve. E, spectrin (green) and NaV (red) staining is co-localized in both +/+ (top) and du2J/du2J (bottom) optic nerve. Overlaid images are shown to the right. *P < 0.05, ***P < 0.001. Scale bars, all 5 μm, apart from middle panel in A, 20 μm.
Discussion
The results presented here show that developing central axons express functional voltage-gated calcium channels that, since they contribute to action potential propagation, must mediate a significant inward calcium current during the action potential. The L-type and P/Q-type channels involved are expressed in a clustered pattern along immature axons, and the α1A subunits that form P/Q channels are transiently clustered in the axolemma at sites where the underlying vesiculotubular complex is fusing with the axolemma, which includes regions where node of Ranvier formation is underway. Vesiculotubular complex is a poorly defined axoplasmic component of developing central axons and includes both tubular and vesicular elements (Hildebrand & Waxman, 1984; Lindsey & Ellisman, 1985; Nakata et al. 1998). In the current study, both tubular and vesicular elements appeared to fuse with the axolemma in regions rich in α1A expression, regions that also had a high coincidence with the presence of glial processes aligned to the extracellular surface of the axolemma. While the vesiculotubular elements are larger and lack the classic features of synaptic vesicles, they did express some of the molecular components of synaptic vesicle fusion such as V-ATPase and the excitatory neurotransmitter glutamate, while the axolemma was shown to contain a high density of SNAP-25, a component of the vesicular docking machinery. Mutations of the α1A subunit, or the α2δ-2 subunit also shown to be present at early node sites, resulted in the malformation of nodes of Ranvier featuring loss of nodal protein localization, disruption of the nodal membrane and a build-up of vesicles under the nodal axolemma. The presence of the various components of vesicular fusion throughout the development of myelination is consistent with a role for vesicular glutamate release in the co-ordination of myelination and node of Ranvier formation.
The effects of P/Q channel mutation upon node of Ranvier development is either a direct effect associated with a disruption in vesicle docking at the nodal axolemma, or an indirect effect possibly following a reduction in axonal excitability. Thus, a reduction in current through P/Q channels might produce a decline in the rate of action potential discharge down optic nerve axons, either due to a loss of inward calcium current during action potential propagation, or following a reduction in synaptic input to the retinal ganglion cell somata. While a loss of axon excitability such as this might affect axon development, it seems unlikely to underlie the disruption of node of Ranvier formation seen in the current study. It has been shown, for example, that complete block of electrical activity in the optic pathway has no comparable effect upon the development of the node, although it does have profound effects upon peri-axonal glial cell processes (Friedman & Shatz, 1990), that were not seen in the current study. It is more likely, therefore, that the effects of P/Q channel mutation upon node development arise from disruption of tubular–vesicular docking and the subsequent interruption in the supply of membrane proteins necessary for node of Ranvier development, and/or from the failure of a signalling pathway mediated by vesicular release from the nodal axolemma. The presence of V-ATPase and glutamate in the vesiculotubular complex, and of SNAP-25 in the axolemma, indicate that vesicular fusion triggered by calcium influx through calcium channels will liberate glutamate onto the closely associated oligodendrocyte processes. Recently, Kukley et al. (2007) and Ziskin et al. (2007) have reported vesicular release of glutamate from both developing and mature central axons, leading to the activation of AMPA/kainate receptors on neighbouring NG2(+) cells. Ziskin et al. (2007) suggest that these AMPA/kainate receptor-mediated currents may have a role in stimulating differentiation of the cells into oligodendrocytes, while Kukley et al. (2007) speculate that glutamate release may provide a signal capable of directing oligodendrocytes to axons. Indeed, it is known that oligodendrocyte migration, differentiation and survival are regulated by non-NMDA-type glutamate receptors (Gallo & Ghiani, 2000), and recent findings have highlighted the presence of NMDA-type glutamate receptors on early oligodendrocyte processes and within myelin itself (Káradóttir et al. 2005; Salter & Fern, 2005; Micu et al. 2006). Such receptors are ideally located to receive vesicular glutamate released from developing axons, and indeed glutamatergic postsynaptic potentials are observed in white matter oligodendrocytes (Káradóttir et al. 2005). White matter contains no synaptic boutons, and taken together, the data strongly suggest that axons are the synaptic partners of oligodendrocytes, at least during myelination.
Signalling pathways between axons and oligodendrocytes are known to be important in myelination (Demerens et al. 1996; Stevens et al. 1998). The glutamatergic signalling pathways described by Ziskin et al. (2007) and Kukley et al. (2007) are consistent with the current findings of glutamate-rich vesicular elements within developing axons, synaptic proteins on these structures and on the axolemma, and the requirement of functional axonal synaptic-type calcium channels for proper node of Ranvier formation. A pathway of this type may act in parallel to the recently reported leukaemia inhibitory factor-mediated (LIF) signalling pathway involving axons and astrocytes (Ishibashi et al. 2006). Interruption of a glutamatergic pathway in leaner can be expected to influence the relationship between axons and their myelinating oligodendrocyte processes. Indeed, dysmyelinated oligodendrocyte phenotypes such as the shiverer (lacking myelin basic protein) and myelin-deficient (md) mutants exhibit a similar disruption in CASPR-1 expression to that found in leaner in the current study, and have a similar reduction in sodium channel clusters (Kaplan et al. 1997; Rasband et al. 1999; Tait et al. 2000; Boiko et al. 2001; Arroyo et al. 2002). A direct effect of calcium channel dysfunction upon protein targeting to the node can not be discounted, since axons lacking scaffold elements such as ankyrin G and β IV spectrin have reduced sodium channel clusters and the presence of vacuoles in the nodal axoplasm (e.g. Komada & Soriano, 2002; Yang et al. 2004).
Several other spontaneous mutations with compromised P/Q channel function have been identified in addition to du2J/du2J and tgla/tgla. Of these, ducky (du/dμ), tottering (tg/tg) and tottering/leaner (tg/tgla) show marked malformation of axon tracts (Meier, 1968; Meier & MacPike, 1971; Rhyu et al. 1999a). While abnormal axons have also been described in P/Q null mice (Jun et al. 1999) and the rolling (tgrol/tgrol) mutation (Rhyu et al. 1999b), gross disruption of white matter has not been reported in these or other P/Q mutants. The phenotype of P/Q mutants is highly variable (Fletcher & Frankel, 1999), which is presumably a product of differences in the effects upon channel function and the degree of compensatory changes in other Ca2+ channel types. Of the mutant mice, tgla/tgla is reported to have the most compromised function and shows no compensatory increases in other high-voltage-activated Ca2+ channels, possibly due to the unaffected level of subunit protein expression in this mutant (Dove et al. 1998; Wakamori et al. 1998). Large compensatory increases in other high-voltage-activated Ca2+ channels have been reported in P/Q null mice (Jun et al. 1999; Fletcher et al. 2001), which can completely counter the loss of P/Q current in presynaptic nerve terminals (Inchauspe et al. 2004). Similar compensatory changes may overcome the loss of axonal P/Q channels in null animals, allowing formation of the node to proceed adequately.
Alongside the important role in nodal development, we have shown that Ca2+ channels contribute to action potential conduction at a point when the axon diameter is increasing rapidly (Hildebrand & Waxman, 1984). This is a period when Na+ channels are being re-distributed to mediate saltatory conduction (Rasband & Shrager, 2000), but before sufficient compact myelin is present to allow saltatory conduction to proceed. It has been unclear how axons retain the ability to conduct action potentials as they undergo this transformation but it is essential for proper CNS development that they do so (Shatz, 1996). By boosting inward current during the action potential, Ca2+ channels are well placed to counter the temporary biophysical constraints that will inhibit axon function during early myelination. In particular, the P/Q channel expression pattern is ideal to assist with action potential conduction during the period of axon diameter expansion, while the L-type channels may be involved primarily before diameter expansion begins, having a relatively high expression level in P2–P5 axons. Calcium channels may therefore have multiple functions in developing central axons. Two earlier studies failed to report the presence of a significant calcium conductance in developing central axons. Foster et al. (1982) did not record a DCAP in the neonatal rat optic nerve following perfusion with isotonic Ba2+. It appears that the rate of stimulation used in this study was 0.5 Hz and our analysis indicates that any DCAP evoked under these conditions would fail after several seconds (see Fig. 2D). Since the recordings collected in this study were averages of eight sweeps, a short-lived DCAP would have been near-impossible to detect. Sun & Chiu (1999) recorded Ca2+ influx into regions between neonatal rat optic nerve glia following axonal stimulation. They report effects of L-type calcium channel blockers that did not reach significance given the small number of trials, but may have done with a larger sample size.
Finally, in addition to physiological and developmental roles, axonal calcium channels are likely to be significant for a variety of pathological conditions. The axons of immature white matter are the focus of injury in a number of developmental white matter injuries including periventricular leukomalacia, the main pathology associated with cerebral palsy (Back & Rivkees, 2004). Cerebral palsy is the most common human birth disorder and is thought to be at least partly ischaemic in origin. Since ischaemic axonal injury is largely calcium mediated (Stys et al. 1992; Fern et al. 1995), calcium channels must represent a major pathway for ischaemic calcium influx into developing central axons.
Acknowledgments
This work was supported by the National Institutes of Neurological Disorders and Stroke grant NS 44875 to R.F. and an MRC programme grant to A.C.D. We wish to thank Dr Peles (Weizmann Institute of Science, Israel) for the generous gift of ant-CASPR-1 and Dr Komada (Tokyo Institute of Technology) for kindly providing anti-βIV spectrin. We acknowledge the excellent technical assistance of Natalie Allcock and Stuart Martin, and thank Abbie Fairs for her excellent electron microscopy work (Supplemental Fig. 4).
Supplementary material
Online supplemental material for this paper can be accessed at:
{kind=link}
{kind=link}
{kind=link}
http://jp.physoc.org/cgi/content/full/jphysiol.2008.155077/DC1
References
- Aidley DJ. The Physiology of Excitable Cells. Cambridge: Cambridge University Press; 1971. [Google Scholar]
- Armstrong CM, Cota G. Calcium block of Na+ channels and its effect on closing rate. Proc Natl Acad Sci U S A. 1999;96:4154–4157. doi: 10.1073/pnas.96.7.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E, Scherer SS. Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci. 2002;22:1726–1737. doi: 10.1523/JNEUROSCI.22-05-01726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK, Allcock N, Fern R. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab. 2006;27:334–347. doi: 10.1038/sj.jcbfm.9600344. [DOI] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez-Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Brismar T, Frankenhaeuser B. The effect of calcium on the potassium permeability in the myelinated nerve fibre of Xenopus laevis. Acta Physiol Scand. 1972;85:237–241. doi: 10.1111/j.1748-1716.1972.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M, Dolphin AC. The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated α2δ2 protein with abnormal function. J Biol Chem. 2002;277:7684–7693. doi: 10.1074/jbc.M109404200. [DOI] [PubMed] [Google Scholar]
- Brown AM, Westenbroek RE, Catterall WA, Ransom BR. Axonal L-type Ca2+ channels and anoxic injury in rat CNS white matter. J Neurophysiol. 2001;85:900–911. doi: 10.1152/jn.2001.85.2.900. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Eilers J, Konnerth A. Axonal calcium entry during fast ‘sodium’ action potentials in rat cerebellar Purkinje neurones. J Physiol. 1996;495:641–647. doi: 10.1113/jphysiol.1996.sp021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Conklin MW, Lin MS, Spitzer NC. Local calcium transients contribute to disappearance of pFAK. focal complex removal and deadhesion of neuronal growth cones and fibroblasts. Dev Biol. 2005;287:201–212. doi: 10.1016/j.ydbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, Zalc B, Lubetzki C. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci U S A. 1996;93:9887–9892. doi: 10.1073/pnas.93.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto-Rostro M, Foucault I, Davies A, Wilkinson T, Rees M, Edwards FA, Dolphin AC. The ducky2J mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci. 2006;26:12576–12586. doi: 10.1523/JNEUROSCI.3080-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- Fatt P, Ginsborg BL. The ionic requirements for the production of action potentials in crustacean muscle fibres. J Physiol. 1958;142:516–543. doi: 10.1113/jphysiol.1958.sp006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R, Davis P, Waxman SG, Ransom BR. Axon conduction and survival in CNS white matter during energy deprivation: a developmental study. J Neurophysiol. 1998;79:95–105. doi: 10.1152/jn.1998.79.1.95. [DOI] [PubMed] [Google Scholar]
- Fern R, Harrison PJ. Conduction slowing and conduction block induced by high extracellular calcium concentration in isdlated nerve fibres of the frog. rana temporaria. J Physiol. 1988;406:108. P. [Google Scholar]
- Fern R, Ransom BR, Waxman SG. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol. 1995;74:369–377. doi: 10.1152/jn.1995.74.1.369. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Frankel WN. Ataxic mouse mutants and molecular mechanisms of absence epilepsy. Hum Mol Genet. 1999;8:1907–1912. doi: 10.1093/hmg/8.10.1907. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, Hosford DA, Tessarollo L, McEnery MW, Pietrobon D, Copeland NG, Jenkins NA. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–1290. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- Forti L, Pouzat C, Llano I. Action potential-evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurones. J Physiol. 2000;527:33–48. doi: 10.1111/j.1469-7793.2000.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RE, Connors BW, Waxman SG. Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Brain Res. 1982;255:371–386. doi: 10.1016/0165-3806(82)90005-0. [DOI] [PubMed] [Google Scholar]
- Friedman S, Shatz CJ. The effects of prenatal intracranial infusion of tetrodotoxin on naturally occurring retinal ganglion cell death and optic nerve ultrastructure. Eur J Neurosci. 1990;2:243–253. doi: 10.1111/j.1460-9568.1990.tb00416.x. [DOI] [PubMed] [Google Scholar]
- Gallo V, Ghiani CA. Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. 2000;21:252–258. doi: 10.1016/s0165-6147(00)01494-2. [DOI] [PubMed] [Google Scholar]
- Girault JA, Peles E. Development of nodes of Ranvier. Curr Opin Neurobiol. 2002;12:476–485. doi: 10.1016/s0959-4388(02)00370-7. [DOI] [PubMed] [Google Scholar]
- Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–330. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand C, Waxman SG. Postnatal differentiation of rat optic nerve fibers: electron microscopic observations on the development of nodes of Ranvier and axoglial relations. J Comp Neurol. 1984;224:25–37. doi: 10.1002/cne.902240103. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- Inchauspe CG, Martini FJ, Forsythe ID, Uchitel OD. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of held presynaptic terminal. J Neurosci. 2004;24:10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci U S A. 2002;99:2303–2308. doi: 10.1073/pnas.042601799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MR, Meyer-Franke A, Lambert S, Bennett V, Duncan ID, Levinson SR, Barres BA. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386:724–728. doi: 10.1038/386724a0. [DOI] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolk SM, Nordquist R, Tuinhof R, Gagliardini L, Thompson B, Cools AR, Roubos EW. Localization and physiological regulation of the exocytosis protein SNAP-25 in the brain and pituitary gland of Xenopus laevis. J Neuroendocrinol. 2000;12:694–706. doi: 10.1046/j.1365-2826.2000.00500.x. [DOI] [PubMed] [Google Scholar]
- Komada M, Soriano P. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol. 2002;156:337–348. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–405. doi: 10.1016/j.neuron.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci. 1997;17:7025–7036. doi: 10.1523/JNEUROSCI.17-18-07025.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen L, Stys PK. Ion transport and membrane potential in CNS myelinated axons I. Normoxic conditions. J Neurophysiol. 1997;78:2086–2094. doi: 10.1152/jn.1997.78.4.2086. [DOI] [PubMed] [Google Scholar]
- Lindsey JD, Ellisman MH. The neuronal endomembrane system. III. The origins of the axoplasmic reticulum and discrete axonal cisternae at the axon hillock. J Neurosci. 1985;5:3135–3144. doi: 10.1523/JNEUROSCI.05-12-03135.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzon NM, Lutz CM, Frankel WN, Beam KG. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18:4482–4489. doi: 10.1523/JNEUROSCI.18-12-04482.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie PJ, Umemiya M, Murphy TH. Ca2+ imaging of CNS axons in culture indicates reliable coupling between single action potentials and distal functional release sites. Neuron. 1996;16:783–795. doi: 10.1016/s0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Meier H. The neuropathology of ducky, a neurological mutation of the mouse. A pathological and preliminary histochemical study. Acta Neuropathol (Berl) 1968;11:15–28. doi: 10.1007/BF00692792. [DOI] [PubMed] [Google Scholar]
- Meier H, MacPike AD. Three syndromes produced by two mutant genes in the mouse. Clinical, pathological, and ultrastructural bases of tottering, leaner, and heterozygous mice. J Hered. 1971;62:297–302. doi: 10.1093/oxfordjournals.jhered.a108176. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters O, Schipke CG, Hashimoto Y, Kettenmann H. Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. J Neurosci. 2003;23:9888–9896. doi: 10.1523/JNEUROSCI.23-30-09888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Shrager P. Ion channel sequestration in central nervous system axons. J Physiol. 2000;525:63–73. doi: 10.1111/j.1469-7793.2000.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyu IJ, Abbott LC, Walker DB, Sotelo C. An ultrastructural study of granule cell/Purkinje cell synapses in tottering (tg/tg), leaner (tgla/tgla) and compound heterozygous tottering/leaner (tg/tgla) mice. Neuroscience. 1999a;90:717–728. doi: 10.1016/s0306-4522(98)00518-1. [DOI] [PubMed] [Google Scholar]
- Rhyu IJ, Oda S, Uhm CS, Kim H, Suh YS, Abbott LC. Morphologic investigation of rolling mouse Nagoya (tgrol/tgrol) cerebellar Purkinje cells: an ataxic mutant, revisited. Neurosci Lett. 1999b;266:49–52. doi: 10.1016/s0304-3940(99)00254-2. [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Salzer JL. Polarized domains of myelinated axons. Neuron. 2003;40:297–318. doi: 10.1016/s0896-6273(03)00628-7. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatz CJ. Emergence of order in visual system development. Proc Natl Acad Sci U S A. 1996;93:602–608. doi: 10.1073/pnas.93.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Bradley DP, James MF, Huang CL. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81:457–481. doi: 10.1017/S1464793106007081. [DOI] [PubMed] [Google Scholar]
- Stevens B, Tanner S, Fields RD. Control of myelination by specific patterns of neural impulses. J Neurosci. 1998;18:9303–9311. doi: 10.1523/JNEUROSCI.18-22-09303.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun BB, Chiu SY. N-type calcium channels and their regulation by GABAB receptors in axons of neonatal rat optic nerve. J Neurosci. 1999;19:5185–5194. doi: 10.1523/JNEUROSCI.19-13-05185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Salter MG, Wilke S, Husen A, Allcock N, Nivison M, Nnoli AN, Fern R. Acute ischemic injury of astrocytes is mediated by Na-K-Cl cotransport and not Ca2+ influx at a key point in white matter development. J Neuropathol Exp Neurol. 2004;63:856–871. doi: 10.1093/jnen/63.8.856. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lacas-Gervais S, Morest DK, Solimena M, Rasband MN. βIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J Neurosci. 2004;24:7230–7240. doi: 10.1523/JNEUROSCI.2125-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.