

Abstract
Respiratory syncytial virus (RSV) infection has been shown to reduce Na+-driven alveolar fluid clearance in BALB/c mice in vivo. To investigate the cellular mechanisms by which RSV inhibits amiloride-sensitive epithelial Na+ channels (ENaC), the main pathways through which Na+ ions enter lung epithelial cells, we infected human Clara-like lung (H441) cells with RSV that expresses green fluorescent protein (rRA2). 3-6 days later patch clamp recordings showed that infected cells (i.e. cells expressing green fluorescence; GFP(+)) had significantly lower whole-cell amiloride-sensitive currents and single channel activity (NPo) as compared with non-infected (GFP(-)), non-inoculated, or cells infected with UV-inactivated RSV. Both α and β ENaC mRNA levels were significantly reduced in GFP(+) cells as measured by real-time reverse transcription-PCR. Infection with RSV increased expression of the inducible nitric-oxide synthase (iNOS) and nitrite concentration in the culture medium; nuclear translocation of NF-κB p65 subunit and NF-κB activation were also up-regulated. iNOS up-regulation in GFP(+) cells was prevented by knocking down IκB kinase γ before infection. Furthermore, pretreatment of H441 cells with the specific iNOS inhibitor 1400W (1 μm) resulted in a doubling of the amiloride-sensitive Na+ current in GFP(+) cells. Additionally, preincubation of H441 cells with A77-1726 (20 μm), a de novo UTP synthesis inhibitor, and 1400W completely reversed the RSV inhibition of amiloride-sensitive currents in GFP(+) cells. Thus, both UTP- and iNOS-generated reactive species contribute to ENaC down-regulation in RSV-infected airway epithelial cells.
Respiratory syncytial virus (RSV)2 is a member of the Pneumovirus genus within the family of the Paramyxoviridae, which is characterized by a linear, negative-sense, single-stranded RNA genome (1). It is the most common cause of lower respiratory tract disease in infants and children worldwide (2) and may also be under-diagnosed as a cause of community-acquired lower respiratory tract infections among adults (3). The primary targets of RSV infection are respiratory epithelial cells (4). Clinical manifestations may range from a mild cold syndrome to severe respiratory distress and failure (5). Respiratory tract fluid accumulation and rhinorrhea are common findings in most cases.
Our previous studies showed that infection of BALB/c mice with RSV impairs Na+-driven alveolar fluid clearance (AFC) across the distal lung and upper airways, respectively, resulting in increased levels of lung water and mild hypoxemia (6, 7). However, vectorial Na+ transport across epithelia involves the coordinated movement of both Na+ and Cl- ions through apically located transporters (such as amiloride-sensitive sodium selective (ENaC) and non-selective cation channels, Ca+2- and cAMP-activated Cl- channels, and a variety of basolateral transporters (such as the Na+/K-ATPase, K+ channels, and Na+, K+, Cl- co-transporters) (8, 9). Altering the activity of any of these transporters may affect vectorial Na+ transport. Furthermore, in vivo measurements cannot identify differences in Na+ transport among infected and non-infected cells and the effects of viral infection per se versus the inflammatory response. Thus, the cellular mechanisms by which RSV decreases epithelial Na+ transport cannot be elucidated in vivo.
To understand whether RSV infection of airway epithelial cells decreases ENaC levels and activity and identify putative mechanisms involved, we infected H441 cells, a human Clara cell line expressing native ENaC, with a recombinant RSV (rRA2) that expresses green fluorescent protein (GFP) during viral replication. 3-6 days later we measured the activity and mRNA levels of α, β, and γ ENaC in infected (i.e. H441 cells expressing green fluorescence; GFP(+)), non-infected (GFP(-)) as well as non-inoculated cells using whole-cell and single channel patch clamp recordings and real-time RT-PCR. Because of previous reports showing that RSV infection of epithelial cells up-regulates iNOS (10) and the well demonstrated inhibition of ENaC by nitric oxide (NO) and reactive oxygen nitrogen intermediates via cGMP-dependent and independent mechanisms (11, 12), we tested the hypothesis that increased levels of NO from iNOS were at least partially responsible for the observed decrease of ENaC function. To accomplish this goal we measured ENaC activity, NF-κB activity, iNOS expression, and levels of nitrite in the medium in RSV-infected cells pretreated with either 1400W (an iNOS inhibitor) or vehicle. Our results indicate that NO or reactive oxygen nitrogen species formed by the interaction of NO with reactive oxygen species are partially responsible for the down-regulation of ENaC activity in RSV-infected cells. Furthermore, because our in vivo experiments indicated that RSV inhibition of Na+-dependent AFC was mediated by UTP (6, 7), we also measured whole-cell amiloride-sensitive currents in RSV-infected cells pretreated either with A77-1726, an inhibitor of de novo UTP synthesis, or vehicle. Our results showed that down-regulation of ENaC activity in RSV-infected cells is due partly to increased levels of UTP as well. Thus, RSV infection down-regulates ENaC by multiple mechanisms.
MATERIALS AND METHODS
Cell Culture—H441 cells were obtained from ATCC and grown in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 1% l-glutamine, 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) as previously described (13). Cells were seeded in 75-cm2 flasks and incubated in a humidified atmosphere of 5% CO2, 95% O2 at 37 °C until they reached confluence and passaged weekly. For consistency, only cells between passages 57 and 73 were used in our studies. Cells were lifted from the flasks using 0.05% trypsin and 0.53 mm EDTA (Mediatech), seeded at a density of 1 × 106 cells/ml on round coverslips (10 mm in diameter, Fisher), and grown in RPMI 1640 medium supplemented with 200 nm dexamethasone to facilitate sodium channel differentiation (14). The medium was changed every other day.
Preparation of Viral Inocula—A recombinant respiratory syncytial virus (rRA2) that expressed green fluorescent protein in infected cells was recovered as previously described. The full-length cDNA of RSV strain A2, prsL1049N, contains all the RSV proteins with the exception of SH; the SH open reading frame was replaced with that of GFP (15, 16). Briefly, this plasmid consists of a bacteriophage T7 polymerase promoter sequence followed by a prsL1049N cDNA, RSV trailer, followed by a hepatitis delta virus ribozyme sequence which by autolytic cleavage produces an exact 3′ terminus, and finally a T7 polymerase termination sequence. The support plasmids expressing RSV proteins N, P, M2-1, and L (pN, pP, pm2ORF-1, and pL) have also been described previously (17, 18). HEp-2 cells (0.5 × 106 cells per well) were infected with modified vaccinia Ankara-T7 virus (MVA-T7) at a multiplicity of infection of 5 for 45 min and subsequently transfected with the RSV full-length cDNA plasmid and the support plasmids listed above using Lipofectamine 2000 (Invitrogen) (19). The cells were incubated at 37 °C overnight, and the supernatant was replaced with Dulbecco's modified Eagle's medium containing 2% fetal calf serum. 2-3 days after transfection the supernatants were harvested and used to inoculate Vero cells. The inoculum was replaced with fresh medium after 12 h at 37 °C, and cells were incubated for 5 days at 37 °C. Virus in the supernatants was amplified by additional growth in Vero cells. Viral titers were determined by serial dilution and plaque assay in Vero cells under agar (20). Virus titers were expressed in plaque-forming units/ml. In some cases RSV virus was purified by ultracentrifugation (12,000 rpm, 4 °C, overnight) through 35% sucrose as described before (21).
UV Inactivation of RSV—Aliquots of rRA2 stocks were inactivated by exposure to 1800 mJ of radiation in a Stratalinker UV cross-linker (Stratagene, La Jolla, CA), eliminating viral infectivity without altering the conformation of viral proteins and mediators (6).
rRA2 Infection of H441 Cells—Once H441 cells reached 70% confluence, they were infected with rRA2 stock to achieve a multiplicity of infection (m.o.i.) of 1 or 5. Viruses were allowed to adhere to the cells for 2 h, then the medium was removed; cells were washed and incubated with fresh medium without RSV for 2-8 days, at which time the cells had grown to 100% confluency. In some experiments cells were exposed to UV-inactivated rRA2 or wild type RSV strain A2. Non-inoculated control cells were exposed to viral stock medium only.
Assessment of Apoptosis—An annexin V-phycoerythrin (PE) apoptosis detection kit (Calbiochem) was utilized to evaluate whether infection of H441 cells with either wild type or GFP-tagged RSV (rRA2) resulted in apoptosis. H441 cells were infected with rRA2, UV-inactivated rRA2, or wild type RSV strain A2 (all m.o.i. = 1) as described above. 2, 4, and 6 days after infection cells were lifted from coverslips by gentle titration after brief exposure to 0.05% trypsin and 0.53 mm EDTA. Cell suspensions were centrifuged at 4000 rpm for 5 min, and pellets were re-suspended to a density of 1 × 106/ml in binding buffer; 10 μl/ml annexin V-PE were added, and cells were incubated at room temperature for 5 min in the dark. The extent of annexin V-PE binding to H441 cells was assessed by sorting them with a BD Biosciences (Franklin Lakes, NJ) FACS Aria cell sorter according to red fluorescence levels. Cells not incubated with annexin V-PE served as background. In some experiments rRA2-infected cells were first sorted by flow cytometry according to green fluorescence to obtain H441 populations that were infected by rRA2 (i.e. GFP(+) cells). The collected GFP(+) cells were then incubated with annexin V-PE as described above. H441 cells treated with actinomycin D (10 μg/ml, AG scientific, San Diego, CA) for 48 h served as the positive control.
Measurements of Whole-cell Currents—In previous in vivo experiments (6), we observed that the maximum inhibition of Na+-dependent alveolar fluid clearance in mice infected with RSV occurred at 48 h post infection. Because significant expression of GFP required at least 48 h, we opted to conduct patch clamp recordings beginning at 72 h post infection. Coverslips containing H441 cells were rinsed with phosphate-buffered saline (PBS (pH 7.4)) and placed onto a recording chamber mounted on the stage of an Olympus IMT-2 inverted fluorescent microscope. rRA2-infected cells were identified by the presence of green fluorescence. For whole-cell patch clamp recordings, cells were continuously perfused with external solution containing 145 mm sodium gluconate, 2.7 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2, 5.5 mm glucose, and 10 mm HEPES (pH 7.4). The osmolarity of this solution, measured with a vapor pressure osmometer (Vapro Wescor, Logan, UT), was 300 ± 5 mosmol/kg H2O. Pipettes were made from B150 capillary glass (Sutter, Novato, CA) with a two-stage vertical puller (HEKA PIP5, Pfalz, Germany). They were back-filled with the internal solution containing 135 mm potassium gluconate, 10 mm KCl, 6 mm NaCl, 2 mm MgCl2, 2 mm Mg2ATP, 2 mm Na3GTP, and 10 mm HEPES (pH 7.2). The pipette resistance varied from 5 to 10 megaohms when filled with this solution. The offset potential was corrected just before formation of a high resistance seal between the pipette and the cell surface (R > 10 gigaohms). The series resistance was about 80% compensated with an Axopatch 200B patch clamp amplifier (Axon Instruments, Foster City, CA). Currents were digitized with digital-to-analog and analog-to-digital converters (Digidata 1200A, Axon Instruments), filtered through an internal four-pole Bessel filter at 1 kHz, and sampled at 2 kHz.
During whole-cell recording the cells were always held on -40 mV. Inward and outward whole-cell currents were elicited by a step-pulse protocol from -80 to +120 mV in 20-mV increments every 10 s for 500 ms duration. Current-voltage (I-V) relationships were constructed by averaging the steady state current values between 300 and 400 ms from the start of the recording using the Clampfit Program (Axon), then plotting these currents to their corresponding driving potentials using Origin Software (Microcal Software, Northampton, MA). These measurements were repeated after perfusing cells with the external solution containing 10 μm amiloride. Amiloride-sensitive currents were then calculated by digitally subtracting the currents in the presence of amiloride from its corresponding control value. Recordings from cells with unstable base-line currents or series resistance were discarded.
Single Channel Currents—We recorded single channel Na+ currents from H441 cells using the cell-attached configuration. Cells were perfused with a solution containing 143 mm sodium gluconate, 5.4 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2, 10 mm glucose, and 10 mm HEPES (pH 7.4). Pipettes were filled with the same solution. Cell attached patches were formed as described in the previous section. Currents were measured at a membrane potential of about -100 mV. Single-channel currents were filtered at 1 kHz, sampled at 2 kHz, and analyzed with Clampfit (Axon Instruments). The amplitude and open probability (Po) of single channel currents were calculated from all event histograms, constructed from at least 10 min of recordings as previously described (13, 14). Recordings were either continuous or appended to each other to satisfy this condition. The product of the number of channels (N) in a patch multiplied by the open probability (NPo), which reflects the activity of open channels, was calculated from single-channel recordings as described before (22). Calculation of NPo does not involve any assumptions about either term.
Real-time RT-PCR of Human ENaC mRNAs—3-5 days after rRA2 infection, H441 cells were lifted from coverslips by 0.05% trypsin and 0.53 mm EDTA, washed, and resuspended in 0.5 ml of PBS. They were then sorted based on green fluorescence with a BD Biosciences FACS Aria cell sorter according to manufacturer's instructions. Equal numbers of control (non-inoculated), GFP(+), and GFP(-) cells were collected. Total RNA was purified from each group of cells with TRI reagent (Sigma-Aldrich). First-strand cDNA was synthesized using Taqman reverse transcription reagents (Applied Biosystems, Foster City, CA). Levels of α, β, and γ ENaC mRNAs were quantified with real-time PCR using a Lightcycler 480 SYBR Green I Master system (Roche Applied Science) according to manufacturer's instructions. Levels of human hypoxanthine phosphoribosyltransferase (HPRT) were used as an internal control. The sequences of the primers used were: ENaC α forward (5′-CTT TGG CAT GAT GTA CTG GCA-3′) and reverse (5′-GGA AGA CGA GCT TGT CCG AGT-3′); ENaC β forward (5′-GAG CCC GGC AAC TAC CGG A-3′) and reverse (5′-GCC GAA GGA AGT GCC TTC TC-3′); ENaC γ forward (5′-GCC CTG AAG TCC CTG TAT GG-3′) and reverse (5′-CGG TGG GAG AAT CTA GGC TG-3′); HPRT forward (5′-TCA GGC AGT ATA ATC CAA AGA TCC T-3′) and reverse (5′-AGT CTG GCT TAT ATC CAA CAC TTC G-3′). All primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). The relative levels of α, β, or γ ENaC mRNAs were presented as ΔCt, which was obtained by subtraction of the Ct value of the internal control (HPRT) from those of α, β, or γ ENaC. The fold changes in expression of α, β, or γ ENaC mRNA after rRA2 infection was obtained by calculation of 2ΔΔCt, in which ΔΔCt was obtained by subtraction of the ΔCt value of cells of control non-inoculated group from the ΔCt value of GFP(-) or GFP(+) cells as previously described (23).
iNOS Imaging with Indirect Immunofluorescence—Cover-slips with H441 cell monolayers were rinsed in cold PBS three times, fixed with 3% formaldehyde in PBS for 45 min at room temperature, then permeabilized by the addition of 0.5% Triton X-100 at room temperature for 3-5 min. After blocking cellular nonspecific sites with 3% (w/v) bovine serum albumin in PBS for 30 min at room temperature, cells were incubated with a rabbit polyclonal anti-iNOS antibody (10 μg/ml, catalog no. SC-651, Santa Cruz Biotechnology Inc., Santa Cruz, CA) in blocking buffer for 1 h at room temperature. Non-immune normal rabbit IgG (10 μg/ml, Santa Cruz Biotechnology) in blocking buffer served as the negative control. After washing with PBS 4 times, cells were incubated with 3% bovine serum albumin for 10 min followed by incubation with goat anti-rabbit IgG conjugated to AlexaFluor 594 (1:100, Molecular Probes, Eugene, OR) in the dark at room temperature for 1 h. Coverslips were washed and imaged with an inverted epifluorescence microscope (Olympus IX70, Olympus). Images were captured and analyzed with IPLab software (BD Sciences). For fluorescence quantification the mean fluorescence intensity was calculated from at least four fields (under 10× objective lens) from each sample. H441 cells treated with 1 μg/ml lipopolysaccharide (LPS, Sigma-Aldrich) and 200 ng/ml human interferon γ (IFN-γ, Peprotech, Rocky Hill, NJ) for 48 h served as the positive control.
Real-time RT-PCR of Human iNOS mRNAs—Two days after rRA2 infection, H441 cells were sorted using a flow cytometry cell sorter to collect equal numbers of control non-inoculated, GFP(+) and GFP(-) cells; levels of iNOS mRNAs were quantified with real-time PCR using a Lightcycler 480 SYBR Green I Master system (Roche Applied Science). The sequences of the primers used were: iNOS forward (5′-ACA GGA GGG GTT AAA GCT GC-3′) and reverse (5′-TTG TCT CCA AGG GAC CAG G-3′)(24) and HPRT forward (5′-TCA GGC AGT ATA ATC CAA AGA TCC T-3′) and reverse (5′-AGT CTG GCT TAT ATC CAA CAC TTC G-3′). All primers were purchased from Integrated DNA Technologies. The relative levels of iNOS mRNA was presented as ΔCt, which was obtained by subtraction of the Ct value of the internal control, HPRT, from that of iNOS. The -fold change in expression of iNOS mRNA after rRA2 infection was obtained by calculation of 2 ΔΔCt, in which ΔΔCt was obtained by subtraction of the ΔCt value of cells of the control non-inoculated group from the ΔCt value of GFP(-) or GFP(+) cells as previously described (23).
Chemiluminescence Measurement of Nitrite
(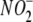 )
Concentrations—
)
Concentrations—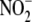 concentrations in the culture medium of non-inoculated cells as well as that
of the cells 48 h post-rRA2 or UV-inactivated rRA2 infections were measured as
an index of NO steady state levels. Cells treated with 1 μg/ml LPS and 200
ng/ml human IFN-γ for 48 h served as the positive control.
concentrations in the culture medium of non-inoculated cells as well as that
of the cells 48 h post-rRA2 or UV-inactivated rRA2 infections were measured as
an index of NO steady state levels. Cells treated with 1 μg/ml LPS and 200
ng/ml human IFN-γ for 48 h served as the positive control.
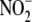 was determined by tri-iodide
(
was determined by tri-iodide
(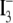 ) reductive chemiluminescence
according to the method of Feelisch et al.
(25) using an Ecomedics CLD 88
sp chemiluminescence analyzer (Eco-Physics, Ann Arbor, MI) with reference to
) reductive chemiluminescence
according to the method of Feelisch et al.
(25) using an Ecomedics CLD 88
sp chemiluminescence analyzer (Eco-Physics, Ann Arbor, MI) with reference to
 standards. Scavenging
standards. Scavenging
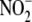 from samples with sulfanilamide
(0.5%, 20 min) before injection into the analyzer eliminated the signal,
indicating that other NO metabolites such as nitrosothiol, nitrosamine, or
metal nitrosyl did not contribute to the nitrite signal. To measure protein
concentrations in culture wells, coverslips containing H441 cells were washed
with ice-cold PBS, and 100 μl of cold PIP2 cell lysis buffer (150
mm NaCl, 50 mm Tris-HCl, 2 mm EGTA, 0.2
mm Na3VO4, 1% Triton X-100, and 0.5% Nonidet
P-40, pH 7.5) supplemented with 1× protease inhibitor mixture (BD
Pharmingen) was added into each well. Cells were lifted from the coverslips,
vortexed at 4 °C for 30 min, and centrifuged at 16,000 × g
for 10 min at 4 °C. The supernatant was carefully removed, and its protein
concentration was determined by the BCA method using bovine serum albumin as a
standard as previously described
(6).
from samples with sulfanilamide
(0.5%, 20 min) before injection into the analyzer eliminated the signal,
indicating that other NO metabolites such as nitrosothiol, nitrosamine, or
metal nitrosyl did not contribute to the nitrite signal. To measure protein
concentrations in culture wells, coverslips containing H441 cells were washed
with ice-cold PBS, and 100 μl of cold PIP2 cell lysis buffer (150
mm NaCl, 50 mm Tris-HCl, 2 mm EGTA, 0.2
mm Na3VO4, 1% Triton X-100, and 0.5% Nonidet
P-40, pH 7.5) supplemented with 1× protease inhibitor mixture (BD
Pharmingen) was added into each well. Cells were lifted from the coverslips,
vortexed at 4 °C for 30 min, and centrifuged at 16,000 × g
for 10 min at 4 °C. The supernatant was carefully removed, and its protein
concentration was determined by the BCA method using bovine serum albumin as a
standard as previously described
(6).
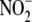 concentration was then divided by
the corresponding protein amount in each well.
concentration was then divided by
the corresponding protein amount in each well.
Western Blot of NF-κB p65 Subunit—Nuclear extracts were prepared as described before (26). The nuclear extracts were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes and blotted with a rabbit anti-p65 polyclonal antibody (Santa Cruz Biotechnology) and then striped and reblotted with a rabbit anti-histone deacetylase 1 (HDAC1) polyclonal antibody (Santa Cruz Biotechnology). The HDAC1 levels served as loading controls.
Electrophoretic Mobility Shift Assay for NF-κB—Nuclear extracts were prepared as previously described (27). H441 cells were washed with cold PBS, and the collected cells were resuspended in and incubated with hypotonic buffer (10 mm N-2-hydroxyethylpiperazine-N′-ethane sulfonic acid (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, 0.1% Nonidet P-40) on ice for 10 min. The cells were then centrifuged at 500 × g for 5 min, and the pellets were lysed in 0.1% Nonidet P-40 lysis buffer. The cell lysates were centrifuged at 14,000 rpm for 10 min, and nuclear extracts (2 μg; present in supernatant) were incubated at room temperature for 15 min in 10 μl of reaction buffer containing 10 mm Tris-Cl (pH 7.5), 1 mm MgCl2, 0.5 mm EDTA, 0.5 mm dithiothreitol, 50 mm NaCl, and 4% glycerol, with [32P]-end-labeled, double-stranded oligonucleotide probe specific for the NF-κB site, 5′-GCCATGGGGGGATCCCCGAAGTCC-3′ (Geneka Biotechnology Inc., Montreal, Quebec, Canada) and 1 μg of poly(dI-dC)·poly(dI-dC). The complexes were resolved with 5% native SDS-PAGE electrophoresis. Dried gels were exposed with Kodak Biomax MS film (Eastman Kodak Co., Rochester, NY) for 1 h at -70 °C. Samples from control non-inoculated and rRA2-infected H441 cells were all run on the same gel.
IKKγ Knockdown by siRNA Transfection—H441 cells grown to 60% confluence were transfected with 10 nm scrambled siRNA (catalog no. D-001210-01, Dharmacon, Lafayette, CO) or a 10 nm pool of 4 human specific IKKγ siRNAs (catalog no. D-003767-00) using siLentFect reagent (Bio-Rad). Twenty-four hours later, H441 cells were infected with rRA2 (m.o.i. = 1). The cells were sorted and collected by flow cytometry 2 days after infection, total RNA was extracted, and iNOS mRNA levels were measured with real-time RT-PCR as described above. To confirm the efficiency of transfection and IKKγ knockdown, extracts from transfected cells were prepared, and the level of IKKγ expression was determined by Western blot assay using IKKγ antibody (Santa Cruz Biotechnology).
Statistics—All values are expressed as the means ± 1 S.E. Data were analyzed by one-way analysis of variance followed by the Bonferroni modification of t test or the Student's t test as appropriate. Means with a p value less than 0.05 were considered significantly different from each other.
RESULTS
H441 Cells Are Infected with RSV rRA2—Significant numbers of GFP positive (i.e. H441 cells incubated with rRA2-GFP and expressing green fluorescence; GFP(+)) cells were seen after infection of H441 cells with normal but not UV-inactivated rRA2 (Fig. 1A). Cells exposed to rRA2 that did not show green fluorescence were termed as GFP negative (GFP(-)) cells. The percentage of infected cells (infection rate, GFP(+)/total cells in the field) depended on m.o.i. and length of infection; for m.o.i. = 1 the infection rates were 2.0 ± 0.7, 3.7 ± 0.4, 9.7 ± 1.1, and 14.0 ± 1.9%, whereas for m.o.i. = 5 they were 4.7 ± 1.1, 19.7 ± 1.1, 26.3 ± 1.0, and 32.3 ± 1.8% at 2, 4, 6, and 8 days after infection respectively (n = 3, Fig. 1B).
FIGURE 1.

Infection of H441 cells with GFP-RSV rRA2. A, H441 cells 6 days post-infection with either GFP-RSV rRA2 (rRA2) or UV-inactivated GFP-RSV rRA2 (UV-rRA2) at m.o.i. = 1. Cells were imaged under bright field (BF), green (GFP), or blue filters (Nuclei, Hoechst 33258 staining) with a 10× objective lens. Scale bars equal 200 μm. B, infection rates (number of GFP(+)/total cell number) after rRA2 infection with m.o.i. = 1 (solid) or 5 (striped bars). Data are the means ± 1 S.E. (n = 3 independent experiments).
rRA2 Does Not Increase Apoptosis in H441 Cells—No significant cell detachment in rRA2-infected cells was observed compared with non-inoculated cells for equal times in culture (data not shown). These data are consistent with the reported lack of cytotoxic effect of RSV in polarized cells (4, 28). On the other hand, it has been suggested that the fusion protein of RSV triggers p53-dependent apoptosis in A549 cells(29). Therefore, we utilized annexin V-PE to detect possible apoptosis caused by rRA2 infection. As shown in Fig. 2, although there is a time-dependent increase of annexin V-PE positive H441 cells cultured in vitro, no significant difference was seen among non-inoculated cells, cells infected with rRA2, wild type RSV strain A2, and UV-inactivated rRA2 (all m.o.i. = 1) at any time points. Furthermore, in another set of experiments we infected H441 cells with rRA2 for 6 days, sorted them, collected GFP(+) cells, and incubated them with annexin V-PE. The percentage of annexin V-PE cells was 12.2 ± 1.0%, not significantly different from those of the control non-inoculated cells (9.5 ± 2.7%) and wild type A2-infected cells (10.0 ± 1.6%) 6 days after infection (n = 4 from two experiments). These results showed that rRA2 does not increase the extent of apoptosis in H441 cells.
FIGURE 2.

Infection of H441 cells with rRA2 does not increase apoptosis. A, representative flow cytometry records of H441 cells not incubated with annexin V-PE serving as background. Shown is annexin V-PE binding to control (non-inoculated) H441 cells treated with 10 μg/ml actinomycin D for 48 h (B), non-inoculated cells (C) or cells infected with rRA2 for 6 days (D). Numbers of cells in P2 and P3 indicate annexin V-PE-negative and -positive cells, respectively. E, flow cytometry analysis of the percentage of annexin V-PE-positive cells in non-inoculated H441 cells (solid), cells infected with rRA2 (stripes at 45 degrees), infected with wild type RSV strain A2 (horizontal stripes), and UV-inactivated rRA2 (cross-hatched pattern) at different times after infection. Floating and adherent cells were pooled together; bars are the means ± 1 S.E.; data are from at least 4 samples from two independent experiments (all at m.o.i. = 1).
Infection of H441 Cells with rRA2 Decreases Their Whole-cell Na+ Currents—When patched in the current clamp mode, resting membrane potentials of control non-inoculated cells was -34.1 ± 2.1 mV (n = 7), whereas those of the GFP(+) and GFP(-) cells were -32.0 ± 1.5 mV (n = 6) and -34.6 ± 2.0 mV (n = 5), respectively (mean ± 1 S.E., n = number of cells patched). Mean membrane capacitances were 15.2 ± 2.1 (n = 14), 19.6 ± 2.8 (n = 14), and 20.3 ± 3.2 pF (n = 7) for control non-inoculated, GFP(-), and GFP(+) cells, respectively; none of these values differed significantly. It should be noted that membrane capacitances of H441 cells that were part of a group of cells were not different from those of the stand-alone cells (15.2 ± 2.1 pF, n = 14, versus 14.7 ± 1.2 pF, n = 6; p > 0.05). These findings indicate that H441 cells, cultured as described above, lacked effective gap junctions.
H441 cells patched in the whole-cell voltage clamp mode exhibited near linear current-voltage relationships. A large fraction of the inward Na+ currents was inhibited by amiloride (10 μm), added in the bath solution (Fig. 3, A-C). GFP(+) cells had significantly lower basal and amiloride-sensitive currents as compared with control non-inoculated cells. Mean values of inward Na+ currents of GFP(-) cells were also smaller than those of the control non-inoculated cells, although the differences were not statistically significant (Fig. 3, D-F). The basal and amiloride-sensitive current densities at a step pulse of -60 mV for control non-inoculated, GFP(-), and GFP(+) cells were -13.3 ± 1.1 and -9.0 ± 1.3 pA/pF (n = 7), -11.4 ± 1.8 and -6.4 ± 2.0 pA/pF (n = 7), and -7.6 ± 1.5 pA/pF and -0.9 ± 0.4 pA/pF (n = 8), respectively. We have chosen to present current densities for each cell as the ratios of the current divided by the membrane capacitance to correct for differences in cell surface area that may affect whole-cell currents.
FIGURE 3.

Infection of rRA2 decreases whole-cell Na+ currents in GFP(+) H441 cells. Characteristic whole-cell current traces in a control non-inoculated H441 cell before (A) and after (B) perfusion with 10 μm amiloride. The amiloride-sensitive currents (C) were calculated by digitally subtracting the current remaining after perfusion of amiloride from that before amiloride perfusion. D and E, whole-cell basal (D) and amiloride-sensitive (E) Na+ current I-V relationships of non-inoculated cells (squares), cells exposed to rRA2 but showing no green fluorescence (GFP(-), triangles) and exposed to rRA2 and showing green fluorescence (GFP(+), circles). Cells were held at -40 mV and the currents (in pA) were normalized for surface area by dividing them by capacitance (in pF), then plotted to their corresponding driving potentials as described under “Materials and Methods.” Values are the means ± S.E., n = 6-8 per group. F, comparison of whole-cell basal (solid) and amiloride-sensitive (striped bar) current densities at -60 mV of control non-inoculated cells, GFP(-), and GFP(+) cells. Values are the means ± S.E., n = 6 to 8 for each group. Measurements were obtained 3-6 days post-infection (p < 0.05 (*) and p < 0.01 (**) are from the corresponding value of non-inoculated cells, n = 6-8 for each group, m.o.i. = 1).
Infection of H441 Cells with rRA2 Decreases Single Channel ENaC Activity—In the next series of experiments we recorded single channel currents from control non-inoculated H441 cells and GFP(+)-infected cells using the cell attached patch configuration by applying 60-mV pulses into the pipette. Because resting membrane potentials (Vm; measured during whole-cell recordings) were about -34 mV for control (non-inoculated) cells and -32 mV for GFP(+) cells, the patch potentials (Vpatch = Vm - Vpipette) were about -94 to -92 mV, respectively, when 60-mV pulses were applied. Characteristic single channel current traces recorded from a control non-inoculated and a GFP(+) cell along with their corresponding amplitude histograms are shown in Fig. 4, A-F. The average amplitudes for the level 1 opening were 0.38 ± 0.02 (n = 6) and 0.37 ± 0.03 pA (n = 5) for control non-inoculated cells and GFP(+) cells, respectively, corresponding in both cases to a single channel conductance of about 4 picosiemens. NPo values in control non-inoculated cells were 0.26 ± 0.03 for level 1 and 0.48 ± 0.10 for all levels (n = 6), whereas in GFP(+) cells the corresponding values were 0.07 ± 0.04 and 0.10 ± 0.07, respectively (n = 5); both values of the GFP(+) cells were significantly lower compared with those of the control non-inoculated cells (Fig. 4G, p < 0.01). Single channels with conductance of 8, 15, or 25 picosiemens were occasionally observed in patches of GFP(+) but not in control non-inoculated cells; however, the number of these events were too few for proper analysis.
FIGURE 4.

Infection with rRA2 decreases Na+ single channel NPo in GFP(+) H441 cells. A, characteristic single channel Na+ current traces and amplitude histogram of a non-inoculated H441 cell patched in the cell-attached mode. Only a small portion of the entire record (∼10 min of recording) is shown. B, expanded view of the indicated portion of the record. The closed and open states of a channel are shown. The patch potential was approximately -100 mV (see “Materials and Methods”). Channel conductance was about 4 picosiemens, characteristic of ENaC. C, all events histogram for a 10-min recording showing the number of events (y axis) versus current in pA (x axis). D-F, characteristic single channel Na+ current traces and amplitude histogram of a GFP(+) cell 4 days after infection. Notice markedly decreased number of events in the all events histogram. G, NPo values of non-inoculated and GFP(+) cells. Values are the means ± S.E. (**, p < 0.01, n = 6 for control non-inoculated cells and 5 for GFP(+) cells, m.o.i. = 1).
Infection of H441 Cells with rRA2 Decreases ENaC mRNA Levels—Both whole-cell and single channel recordings indicate that ENaC activity was reduced after rRA2 infection. Results of our RT-PCR experiment showed that GFP(+)-infected cells had significantly lower levels of α and β ENaC mRNAs compared with control non-inoculated cells; GFP(-) cells also showed lower β ENaC mRNAs levels (p < 0.01 and p < 0.05, n = 4 independent experiments; Fig. 5). γ ENaC mRNA levels also tended to be lower in both GFP(+) and GFP(-) cells as compared with non-inoculated cells (Fig. 5); however, these values were not statistically significant. Because of the low yield of GFP(+) cells obtained by cell sorting, Western blotting studies of ENaC proteins were not feasible.
FIGURE 5.

Infection of H441 cells with rRA2 decreases ENaC mRNA levels. H441 cells were infected with rRA2 at m.o.i. = 1. 3-6 days later cells were sorted according to green fluorescence levels and α, β, and γ ENaC as well as HPRT mRNAs were measured with real-time RT-PCR as described in “Materials and Methods”. Bars are the mean values ± S.E. of ENaC mRNA levels in non-inoculated (solid), GFP(-) (diagonal stripes), and GFP(+) (cross-hatched stripes) cells, calculated as described under “Materials and Methods.” ENaC mRNA values of each group were normalized by dividing their values by the corresponding values of the non-inoculated cells. Values are the means ± 1 S.E., n = 4 independent experiments (p < 0.01 (**) and p < 0.05 (*) are as compared with the corresponding non-inoculated value).
Inhibition of ENaC Activity by RSV Is Mediated Partly by UTP—To address the involvement of UTP in the inhibition of Na+ currents by rRA2, we pretreated H441 cells with A77-1726 (20 μm; shown to decrease UTP levels in RSV infected mice) (30) for at least 12 h, then infected them with rRA2. A77-1726 was present until the whole-cell patch clamp recording 3 days later. A77-1726-treated GFP(+) cells exhibited a larger basal Na+ current which was inhibited by amiloride (Fig. 6A). A77-1726 had no effect on basal and amiloride-sensitive currents of non-inoculated H441 cells; however, both the basal and amiloride-sensitive currents in A77-1726-treated GFP(+) cells were significantly higher than those of the DMSO (vehicle)-treated GFP(+) cells (-10.0 ± 1.3 and -4.9 ± 1.4 (n = 5) versus -7.5 ± 1.5 and -1.2 ± 0.4 pA/pF (n = 6); p < 0.05 and p < 0.01, Fig. 6B). On the other hand, real-time RT-PCR measurements showed that the observed decrease of α and β ENaC mRNA levels in GFP(+) cells were not restored by incubation with A77-1726 (Fig. 6, C and D).
FIGURE 6.

Inhibition of Na+ currents in GFP(+) cells by rRA2 is partially mediated by UTP. A, characteristic time course recordings of whole-cell Na+ currents in a GFP(+) H441 cell pretreated with either A77-1726 (20 μm, solid circles) or vehicle (0.1% DMSO, open circles) for 12 h, infected with rRA2 (m.o.i. = 1), and patched 3 days later. The cells were held at -40 mV, and currents were elicited by a step pulse to -60 mV every 0.5s. The cells were perfused with amiloride (amil., 10 μm) as indicated. Pretreatment with A77-1726 partially restored amiloride sensitivity. Notice the lack of amiloride (amil.)-sensitivity in cells treated with vehicle (open circles). B, mean values ± S.E. of basal (solid) and amiloride-sensitive (striped bars) current densities at -60 mV for non-inoculated H441 cells incubated with A77-1726 (20 μm, n = 5) for 3 days and GFP(+) cells incubated with either DMSO (n = 5) or A77-1726 (20 μm, n = 6) 3 days after rRA2 infection. (p < 0.05 (*) and p < 0.01 (**) are from their corresponding non-inoculated values). C and D, α (C) and β (D) ENaC mRNA levels in non-inoculated (solid bars), GFP(-)(diagonal bars), and GFP(+)(cross-hatched bars) cells treated by DMSO or A77-1726 (20 μm). ENaC mRNA levels of each cell group were measured by real-time RT-PCR from cells sorted and collected by flow cytometry 3-5 days after rRA2 infection and then divided by those of the non-inoculated cells (m.o.i. = 1; **, p < 0.01 compared with non-inoculated cells; data are from four independent experiments).
rRA2 Infection Increases NO Production through Up-regulation of iNOS—Because our results indicated that inhibition of UTP does not restore amiloride-sensitive currents of rAR2-infected cells to control non-inoculated levels, we investigated the contribution of NO to this process. Immunostaining of H441 cells with a polyclonal antibody against iNOS followed by a secondary fluorescent antibody showed higher levels of fluorescence (Fig. 7, A and B) in infected (both GFP(+) and GFP(-)) cells as compared with control non-inoculated cells. Mean fluorescent intensity of rRA2-infected cells was 38% higher as compared with control non-inoculated cells 4 days after infection (p < 0.05, n = 3, as determined in 3 different experiments, Fig. 7C). Meanwhile, fluorescence levels of UV-inactivated rRA2-infected cells were similar to non-inoculated cells. Moreover, as seen in Fig. 7B, GFP(+) H441 cells (arrows) had significantly higher levels of red fluorescence, indicative of higher levels of iNOS protein as compared with GFP(-) cells and non-inoculated cells. Consistent with the up-regulation of iNOS protein levels, nitrite levels in the culture medium of H441 cells infected with rRA2 for 48 h were 67% higher as compared with the corresponding levels of non-inoculated cells (Fig. 8); on the other hand, nitrite levels in the medium of H441 cells infected with UV-inactivated RSV remained at control levels (Fig. 8). As positive control, non-inoculated H441 cells treated with LPS (1 μg/ml) and human IFN-γ (200 ng/ml) for 48 h showed a 72% increase of nitrite concentration compared with untreated cells.
FIGURE 7.

Infection of H441 cells with rRA2 up-regulates iNOS levels. A, upper row, characteristic 10× images of non-inoculated H441 cells immunostained with normal rabbit IgG (IgG), non-inoculated cells (noninoc. iNOS), or cells infected with UV-inactivated rRA2 for 4 days (UV-rRA2 iNOS) and immunostained with a rabbit polyclonal anti-iNOS antibody. Lower row, characteristic 10× images of H441 cells infected with rRA2 for 4 days (rRA2; iNOS, left and middle panels) or incubated with LPS (1 μg/ml) and human IFN-γ (200 ng/ml) for 48 h (LPS+IFN-γ) and immunostained with a rabbit polyclonal anti-iNOS antibody. The middle panel is a merged composite iNOS staining and green filter image showing green fluorescence of GFP(+) cells. The secondary antibody was goat anti-rabbit IgG conjugated to AlexaFluor 594. The scale bar equals to 200 μm. B, characteristic 40× images of non-inoculated cells (non-inoc.) and cells infected with rRA2 for 3 days (rRA2; middle and right panels) immunostained with a rabbit polyclonal antibody to iNOS then labeled by a goat anti-rabbit IgG conjugated to AlexaFluor 594. The middle panel was imaged with a green filter to show GFP(+) cells (arrows). The right panel is a composite merged image obtained with green and red filters to show both GFP(+) cells and iNOS stains. The scale bar equals to 50 μm. C, iNOS fluorescence intensity for the indicated groups. Mean fluorescence intensities of corresponding cell groups were divided by those of the non-inoculated cell groups. Date are the means ± S.E. and are from 3 independent experiments (*, p < 0.05 as compared with the non-inoculated value; m.o.i. = 1).
FIGURE 8.

Infection of H441 cells with rRA2 increases nitrite (NO-2) concentrations in the medium. Nitrite concentrations in cell culture medium of non-inoculated (solid) cells or cells infected with rRA2 (diagonal lines) or UV-inactivated rRA2 (dense cross-hatching) for 48 h. A fourth group of H441 cells were incubated with LPS (1 μg/ml) and human interferon-γ (IFN, 200 ng/ml) for 48 h (vertical lines). Nitrite concentrations were measured by chemiluminescence and adjusted to protein concentrations as described under “Materials and Methods”; data are normalized by dividing the nitrite concentrations of the various groups by the values of the non-inoculated cell groups. Values are the means ± 1 S.E. (n = 6 from two independent experiments; *, p < 0.05 compared with the non-inoculated mean value).
Because of the limited number of GFP(+) cells, measurements of nitrite were conducted in mixed populations containing both GFP(+) and GFP(-) cells. rRA2 Infection Activates NF-κB—In a number of inflammatory conditions, iNOS up-regulation has been linked to activation of NF-κB (31). NF-κB has also been shown to play a critical role in the up-regulation of host immune response to RSV (32). Our data show significant increases in nuclear translocation of the NF-κB p65 subunit in H441 cells at 24- and 48-h post-rRA2 infection as compared with non-inoculated cells (Fig. 9, A and B). NF-κB activity was also significantly increased after rRA2 infection as shown by EMSA (Fig. 9C). In contrast, only little increase in nuclear translocation of p65 was observed in cells exposed to UV-inactivated rRA2.
FIGURE 9.

Infection of H441 cells with rRA2 up-regulates NF-κB activity. Western blots of p65 subunits of non-inoculated H441 cells (non-inoc.) or infected with either rRA2 or UV-inactivated rRA2 (m.o.i. = 1) for either 24 (A) or 48 h (B). Nuclear extracts were separated by SDS-PAGE and immunoblotted with antibodies to NF-κB p65 subunit. Each lane represents a different experiment. C, EMSA gel image of non-inoculated cells or cells infected with rRA2 for either 24 or 48 h (m.o.i. = 1). Infection with rRA2 increased NF-κB activity at either time point. Results of a typical experiment which were repeated two times.
Knockdown of NF-κB IKKγ Subunit Reverses the Up-regulation of iNOS—In the next series of experiments, we investigated the relationship between activation of NF-κB and up-regulation of iNOS in rRA2-infected cells. H441 cells were transfected with IKKγ siRNA for 24 h and then infected with rRA2 as described under “Materials and Methods.” Forty-eight hours later cells were sorted by flow cytometry, equal numbers of non-inoculated and GFP(-)- and GFP(+)-infected cells were collected, and iNOS mRNA was measured by real-time RT-PCR. IKKγ knockdown by the specific siRNA was demonstrated by Western blotting with an anti-IKKγ antibody (Fig. 10A). As shown in Fig. 10B, GFP(+) cells transfected with scrambled siRNA showed a 2.5-fold increase of iNOS mRNA compared with control non-inoculated cells, consistent with the immunofluorescent findings of iNOS up-regulation shown in Fig. 7. On the other hand, the iNOS mRNA levels of GFP(+) cells after transfection with IKKγ siRNA were not different from iNOS mRNA levels of either non-inoculated or GFP(-) cells. These data suggest that rRA2 infection activates NF-κB, leading to increase in iNOS expression and NO production.
FIGURE 10.

Knockdown of IKKγ reverses the up-regulation of iNOS in rRA2 infected H441 cells. A, H441 cells were transfected with either scrambled or specific IKKγ siRNAs as described under “Materials and Methods.” Twenty-four hours later cell extracts were immunostained with anti-IKKγ and anti-actin antibodies. Data are the result of a typical experiment which was repeated two times with identical results. B, H441 cells were transfected with either scrambled or specific IKKγ siRNAs for 24 h then infected with rRA2 (m.o.i. = 1). Cells were sorted, the same numbers of non-inoculated, GFP(-) and GFP(+) cells were collected by flow cytometry 48 h later, and iNOS mRNA was measured with real-time RT-PCR as described under “Materials and Methods.” iNOS mRNA levels of each cell group were normalized by dividing their values by the values of the non-inoculated groups. Values are the means ± 1 S.E., n = 4 independent experiments. Solid bars, non-inoculated; diagonal stripes, GFP(-); dense cross-hatching, GFP(+) cells (**, p < 0.01 as compared with the non-inoculated values).
Inhibition of iNOS Partially Reverses the Amiloride-sensitive Currents—To investigate whether the up-regulation of iNOS and NO production contributed to the observed decrease of ENaC activity after RSV infection, we pretreated H441 cells with 1 μm 1400W, a specific iNOS inhibitor, or its vehicle (0.1% DMSO) for 12 h, then infected the cells with rRA2 and recorded basal and amiloride-sensitive Na+ currents from GFP(+) cells 72 h later. As shown in Fig. 11A, the nitrite concentration in the culture medium from 1400W-treated infected cells was 48 ± 12% lower than that of those treated with DMSO (p < 0.01, n = 6 from 3 independent experiments), indicating that 1400W decreases NO production by inhibiting iNOS. Additionally, when recorded in the whole-cell mode, basal and amiloride-sensitive current densities at -60-mV step pulse were -7.2 ± 1.0 and -2.8 ± 0.6 pA/pF for 1400W-treated GFP(+) cells (n = 5), whereas those of the DMSO-treated GFP(+) cells were -6.1 ± 1.6 and -1.3 ± 0.4 pA/pF, respectively (n = 4). The amiloride-sensitive currents of the 1400W-treated GFP(+) cells were twice as high as compared with vehicle-treated GFP(+) cells (p < 0.05, Fig. 11B). These data indicated that the increase of NO by up-regulation of iNOS after rRA2 infection contributes at least in part to the observed ENaC activity inhibition. On the other hand, the decrease of α and β ENaC mRNA levels of GFP(+) cells was not restored by incubation with 1400W (Fig. 11, C and D), similar to the results obtained with A77-1726 preincubation (Fig. 6, C and D).
FIGURE 11.

1400W partially reverses the inhibition of amiloride-sensitive Na+ currents by rRA2 infection in GFP(+) cells. A, nitrite concentration of rRA2 infected (m.o.i. = 1) cells as measured by chemiluminescence 48 h after rRA2 infection. Cells were treated either by vehicle (0.1% DMSO; solid bars) or 1400W (1 μm; diagonal lines). Nitrite concentrations were normalized to those of the DMSO-treated cells. Values are the means ± 1 S.E., n = 6 from 3 independent experiments (**, p < 0.01, m.o.i. = 1). B, mean values ± 1 S.E. of amiloride-sensitive currents of GFP(+) cells at -60 mV incubated with either DMSO (solid bars, n = 4) or 1400W (diagonal lines, n = 5) 3 days after rRA2 infection. C and D, α and β ENaC mRNA levels in non-inoculated (solid bars), GFP(-)(diagonal lines), and GFP(+)(cross-hatched lines) cells (m.o.i. = 1) treated with DMSO or 1400W (1 μm). ENaC mRNA levels were measured by real-time RT-PCR from cells sorted and collected by flow cytometry 3-5 days after rRA2 infection and normalized by dividing them by those of the non-inoculated cell groups. Values are the means ± 1 S.E.; data are from 4 independent experiments (**, p < 0.01 compared with non-inoculated cells).
Combined Effects of UTP and NO Inhibition on Whole-cell Na+ Currents of RSV-infected H441 Cells—In the next series of experiments, we preincubated H441 cells with both 20 μm A77-1726 and 1 μm 1400W for 12 h, infected them with rRA2 (m.o.i. = 1), and recorded whole-cell currents 72 h later. As shown in Fig. 12, basal and amiloride-sensitive currents of GFP(+) cells preincubated with both A77-1726 and 1400W were -11.3 ± 1.3 and -8.2 ± 1.4 pA/pF (n = 7) versus -6.9 ± 1.0 and -1.2 ± 0.2 pA/pF, respectively, for DMSO controls (n = 10). Both basal and amiloride-sensitive currents of GFP(+) cells treated with A77-1726 and 1400W were restored to the same levels of those of the non-inoculated cells (-11.7 ± 0.9 and -7.5 ± 1.0 pA/pF, respectively, p > 0.05, n = 7).
FIGURE 12.

Incubation of H441 cells with both A77-1726 and 1400W completely reverses the inhibition of amiloride-sensitive Na+ currents by rRA2 infection. H441 cells were preincubated with either vehicle (0.1% DMSO) or a combination of A77-1726 (20 μm) and 1400W (1 μm) for 12 h, then infected with rRA2 (m.o.i. = 1). DMSO or A77-1726 and 1400W were presented until whole-cell patch clamp recording 72 h after infection. Values are the means ± 1 S.E. Plotted are current densities at -60mV. GFP(+) cells incubated with DMSO exhibited significantly lower basal (solid bars) and amiloride-sensitive Na+ currents (diagonal lines) as compared with those of the non-inoculated cells (*, p < 0.05; **, p < 0.01, n = 7). Treatment with A77-1726 and 1400W restored the basal and amiloride-sensitive currents of GFP(+) cells to near control levels (n = 7).
DISCUSSION
Herein, for the first time, we present direct evidence that RSV infection inhibits amiloride-sensitive Na+ transport across human airway cells both at the whole-cell and single channel levels. Infection of H441 cells with RSV resulted in a 41% inhibition of the basal whole-cell Na+ currents and 88% of the amiloride-sensitive currents. Furthermore, RSV infection decreased ENaC mRNA levels of infected cells. These findings are consistent with our in vivo data, showing that RSV infection decreased Na+-dependent alveolar fluid clearance and abolished the amiloride sensitivity of AFC (7, 30). Infection with RSV did not decrease α, β, or γ ENaC/glyceraldehyde-3-phosphate dehydrogenase mRNA values in lung tissues of these mice (6). However, only a small fraction of epithelial cells become infected with RSV, so studies in mixed cell populations may mask its effects on infected cells. To our knowledge, this is the first demonstration that an actively replicating virus alters active Na+ transport in human cells at the single cell level.
Clara cells constitute up to 80% of the epithelial cell population of the distal and terminal airways and are the important sites of RSV infection. They play an important role in the regulation of epithelial electrolyte/fluid secretion in terminal airways (33) and secrete Clara cell 10-kDa protein (CC10) which is thought to play important roles in protecting against oxidative stress and inflammatory response in the respiratory tract (34). H441 cells express highly selective amiloride-sensitive Na+ conductances and cAMP-activated Cl- currents after hormone treatment and have been used as a model for epithelial sodium channel investigation (35, 36). Recently we demonstrated that infection of confluent monolayers of H441 cells with wild type RSV strain A2 decreases their amiloride-sensitive short circuit currents and α ENaC levels by activating UTP (36). Furthermore, Na+ transport through H441 cells was shown to be inhibited by UTP (36). However, our previous studies did not investigate the effects of reactive oxygen-nitrogen intermediates in the RSV induced down-regulation of ENaC.
We used a form of the RSV virus in which the gene expressing the small hydrophobic (SH) protein was replaced by the gene for green fluorescent protein (15). The precise function of the SH protein, one of three RSV transmembrane glycoproteins, has not been elucidated (37). Genetically engineered RSV from which SH has been deleted grows well in cell culture and is modestly attenuated in animals, and the SH protein appears to be dispensable for virus replication in humans (38, 39). When expressed in bacteria, it increases membrane permeability to low molecular weight compounds, and it has been suggested that it functions as a viroporin (40). More recently, the SH protein has been shown to decrease apoptosis at least in part by inhibiting tumor necrosis factor α (41). Because the RSV fusion protein triggered p53-dependent apoptosis in A549 cells (29), there is concern that infection of cells with RSV viruses lacking the SH protein may cause significant degrees of apoptosis. However, in our studies rRA2-infected cells did not exhibit higher levels of annexin V-PE binding as compared with either non-inoculated or wild type RSV-infected cells (Fig. 2). In addition, the inhibition effects of rRA2 on ENaC function were completely reversible by the combined inhibition of iNOS and de novo UTP synthesis, which were unlikely to occur in apoptotic cells. These two sets of data, therefore, strongly argue against the possibility that the observed rRA2 down-regulation of ENaC in H441 cells was due to the induction of apoptosis due to the lack of SH protein. GFP expression was critical in our studies, as this allowed the identification of individual infected cells for biophysical and biochemical studies. Transfection of H441 cells with plasmids expressing GFP alone did not alter either basal or amiloride-sensitive currents in H441 cells (data not shown).
Another potential concern is that most of our experiments were conducted using non-purified rRA2 viruses, which may contain LPS and other cytokines known to up-regulate iNOS. The labile nature of this virus and possible loss of titer has led a number of investigators to use non-purified RSV in a variety of experiments (see for example Refs. 42 and 43). The fact that UV-inactivated rRA2 did not up-regulate iNOS or down-regulate ENaC indicates that contaminants (if present) were not responsible for the noted responses. Nevertheless, to address this potential concern we infected H441 cells with sucrose-gradient purified rRA2 and found that these purified viruses inhibited whole-cell ENaC currents in H441 cells to the same extent as the not-purified rRA2 (whole-cell basal and amiloride-sensitive current densities of GFP(+) cells infected by purified rRA2 being -8.0 ± 1.4 and -0.8 ± 0.3 pA/pF, respectively (n = 6), versus -7.6 ± 1.5 and -0.9 ± 0.4 pA/pF of GFP(+) cells infected by not-purified virus (n = 8)). Thus we feel that in our experiment ENaC down-regulation was the result of events initiated by viral infection per se.
Previously we have shown that the deleterious effects of RSV-A2 on Na+-dependent AFC and oxygen saturation of BALB/c mice were both prevented and reversed by systemic or intranasal administration of inhibitors of the de novo pathway of pyrimidine synthesis (such as leflunomide or A77-1726, its active metabolite). Furthermore, the protective effects of leflunomide and A77-1726 were obviated to a large extent by concomitant administration of uridine which stimulates UDP and UTP synthesis via the salvage pathway (7, 30). Results presented in this study demonstrate H441 cells incubated with A77-1726 before infection partially restored amiloride-sensitive whole-cell currents. These results definitively establish the involvement of UTP on ENaC down-regulation in RSV infection. Our previous studies showed that RSV infection increased both UTP and ATP levels in the BAL of BALB/c mice and intranasal administration of A77 1 day post-infection reduced both UTP and ATP to base-line levels (30). These findings are consistent with the observation that de novo purine and pyrimidine synthesis pathways are usually concordantly regulated. However, the RSV-induced decrease of AFC was reversed by intratracheal instillation of agents that degrade UTP but not ATP (6, 7).
The molecular mechanisms by which RSV infection up-regulates UTP production and how elevated UTP mediates the inhibition of Na+ channel currents have not been elucidated. P2Y receptors (the main targets of UTP) are G protein-coupled and act via the inositol phosphate pathway to stimulate Ca2+ release from intracellular stores but can also act via multiple secondary signal transduction pathways, including protein kinase C (44). Activation of protein kinase C has been shown to reduce ENaC activity and modify its subunit composition (45, 46). Our previous data indicate that infection of mice with RSV results in activation of protein kinase Cζ, as indicated by its phosphorylation and translocation from the cytoplasm to the plasma membrane (47).
iNOS expression in murine cells is thought to be induced by bacterial LPS and a number of pro-inflammatory cytokines, such as tumor necrosis factor-α, interleukin-1β, interleukin 6, and interferon-γ (48); however, activation of iNOS in the majority of human cells requires a complex cytokine combination that includes tumor necrosis factor-α, interleukin-1β, and interferon-γ (49). NF-κB plays a pivotal role in regulating iNOS expression (50). Our data clearly show that replicating, but not UV-inactivated, rRA2 activates NF-κB which in turn up-regulates iNOS and increases NO steady state levels, whereas knockdown of IKKγ (which is essential to canonical NF-κB activation) prevents iNOS up-regulation in GFP(+)-infected cells. Furthermore, direct inhibition of iNOS by incubation of H441 cells with the specific iNOS inhibitor 1400W partially alleviates the rRA2 inhibition of amiloride-sensitive currents. Our findings are in agreement with previous reports showing that CD14 and TLR4 receptors in monocytes are essential for RSV activation of NF-κB and inflammatory cytokines (32, 51). It should be noted that we only investigated the canonical NF-κB pathway and cannot exclude the possibility that the translocation of p65 is a result of activation of non-canonical pathway as reported recently by Liu et al. (26). Nevertheless, our data provide the first evidence that NO generated by iNOS plays an important role in down-regulation of amiloride-sensitive Na+ channels in individual human respiratory epithelial cells infected with RSV.
Several reports have described that infection of lung epithelial cells with respiratory virus damages ion channels. Exposure of murine tracheal tissue to intact virus or even viral antigens from pneumotropic influenza A virus (52), a murine para-influenza virus (Sendai virus) (53), or RSV (54) resulted in rapid inhibition of active Na+ transport. Influenza A virus also reduced ENaC activity in rat alveolar type II cells (55). However, it should be noted that all of these reports described acute effects, usually within 60 min of infection. More importantly, they utilized inactive viruses, indicating that the acute effects of exposure to these pathogens on active Na+ transport are likely mediated solely by binding and attachment of viral glycoproteins (such as hemagglutinin) with cellular receptors. In contrast, we concentrated on long term effects to better mimic events during human infections. In our studies the fact that UV-inactivated rRA2 did not activate NF-κB and failed to up-regulate iNOS and increase NO steady state levels clearly indicates virus replication is necessary for the inhibition of Na+ currents. This is consistent with our in vivo results that the inhibitory effects of RSV on AFC during prolonged infection require active viral replication (6). We also observed small decreases in amiloride-sensitive currents and ENaC mRNA levels in GFP(-) cells, consistent with paracrine effects of UTP and NO on ENaC. Therefore, the chronic effects of RSV on respiratory epithelium in infected subjects, as shown by AFC and our current observation of inhibition of Na+ channel activity, appear to be mediated by a different mechanism involving RSV infection and replication within respiratory epithelial cells.
It is well documented that RSV infection results in profound global responses in gene expression both in vivo and in vitro (56-58). In particular, Zhang et al. (57) reported that more than 1200 genes were affected by RSV infection in A549 or small airway epithelial cells, with the numbers of up-regulated and down-regulated genes nearly equal at early time points after infection (6 and 12 h) and down-regulated genes predominating at later times (36 h). Our results showing that both α and β ENaC mRNAs were significantly decreased in GFP(+)-infected cells are, therefore, consistent with these reports. However, the underlying mechanism of this down-regulation is still unclear. Although preincubation with both A77-1726 and 1400W totally restored the amiloride-sensitive currents in GFP(+)-infected cells, neither restored the decreased ENaC mRNA levels in GFP(+) cells; therefore, our data suggested that both UTP and NO inhibit ENaC activity at post-transcriptional levels. Reactive oxygen-nitrogen intermediates have been shown to down-regulate amiloride-sensitive Na+ channels and vectorial Na+ transport through cGMP-dependent and -independent mechanisms involving post-transcriptional ENaC regulation (11, 12, 22). It is important to point out that it is unlikely that these effects are limited to ENaC alone, as reactive oxygen-nitrogen intermediates have been implicated in the regulation of the activities of large number of ion channels, including CFTR (59-62), calcium-activated K+ channels (63), and type 1 ryanodine receptors (64, 65).
In summary, our data presented herein represent the first evidence that RSV infection inhibits Na+ channel activity in whole-cell and single channel levels in human respiratory epithelial cells. These findings should be distinguished from those of the prior in vitro studies, which have only investigated the effects of short term exposure of epithelial cells to viral antigens on ion transport. Because these latter studies did not permit sufficient time for viral replication and gene expression within epithelial cells, their relevance to the pathogenesis of respiratory disease in an infected subject over a period of several days of viral replication remains unclear. Moreover, our findings are the first report that NO is involved in the inhibition of Na+ transport after RSV infection through up-regulation of iNOS by activation of NF-κB pathway. This in turn suggests that counter measures against NO as a potential therapy for RSV-induced respiratory fluid imbalance deserve further investigation.
Acknowledgments
We thank Dr. Gail Wertz for the generous gift of plasmids containing RSV cDNAs. Our sincere thanks to Dr. Fu-ping Chen for virus preparation, Marion Spell for assistance with the flow cytometry study, Drs. Jaroslaw Zmijewski and Albert Tousson for technical help with the imaging studies, Devipriya Subramaniyam for assistance with EMSA, and Teri Potter for editing this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-31197 and HL-51173 and Grants U01ES015676 and 1U54NS063739 (NIEHS, Public Health Service). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: RSV, respiratory syncytial virus; IKK, IκB kinase; AFC, alveolar fluid clearance; ENaC, epithelial Na+ channels; RT, reverse transcription; iNOS, inducible nitric-oxide synthase; PBS, phosphate-buffered saline; siRNA, small interfering RNA; pF, picofarads; GFP, green fluorescent protein; m.o.i., multiplicity of infection; HPRT, hypoxanthine phosphoribosyltransferase; LPS, lipopolysaccharide; IFN-γ, interferon γ; PE, phycoerythrin; SH, small hydrophobic.
References
- 1.Sullender, W. M. (2000) Clin. Microbiol. Rev. 13 1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smyth, R. L., and Openshaw, P. J. (2006) Lancet 368 312-322 [DOI] [PubMed] [Google Scholar]
- 3.Dowell, S. F., Anderson, L. J., Gary, H. E., Jr., Erdman, D. D., Plouffe, J. F., File, T. M., Jr., Marston, B. J., and Breiman, R. F. (1996) J. Infect. Dis. 174 456-462 [DOI] [PubMed] [Google Scholar]
- 4.Zhang, L., Peeples, M. E., Boucher, R. C., Collins, P. L., and Pickles, R. J. (2002) J. Virol. 76 5654-5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammer, J., Numa, A., and Newth, C. J. (1997) Pediatr. Pulmonol. 23 176-183 [DOI] [PubMed] [Google Scholar]
- 6.Davis, I. C., Sullender, W. M., Hickman-Davis, J. M., Lindsey, J. R., and Matalon, S. (2004) Am. J. Physiol. Lung Cell. Mol. Physiol. 286 112-120 [DOI] [PubMed] [Google Scholar]
- 7.Davis, I. C., Lazarowski, E. R., Hickman-Davis, J. M., Fortenberry, J. A., Chen, F. P., Zhao, X., Sorscher, E., Graves, L. M., Sullender, W. M., and Matalon, S. (2006) Am. J. Respir. Crit. Care Med. 173 673-682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matalon, S., and O'Brodovich, H. (1999) Annu. Rev. Physiol. 61 627-661 [DOI] [PubMed] [Google Scholar]
- 9.Matalon, S., Lazrak, A., Jain, L., and Eaton, D. C. (2002) J. Appl. Physiol. 93 1852-1859 [DOI] [PubMed] [Google Scholar]
- 10.Tsutsumi, H., Takeuchi, R., Ohsaki, M., Seki, K., and Chiba, S. (1999) J. Leukocyte Biol. 66 99-104 [PubMed] [Google Scholar]
- 11.Guo, Y., Duvall, M. D., Crow, J. P., and Matalon, S. (1998) Am. J. Physiol. 274 L369-L377 [DOI] [PubMed] [Google Scholar]
- 12.Jain, L., Chen, X. J., Brown, L. A., and Eaton, D. C. (1998) Am. J. Physiol. 274 L475-L484 [DOI] [PubMed] [Google Scholar]
- 13.Lazrak, A., and Matalon, S. (2003) Am. J. Physiol. Lung Cell. Mol. Physiol. 285 443-450 [DOI] [PubMed] [Google Scholar]
- 14.Lazrak, A., Samanta, A., Venetsanou, K., Barbry, P., and Matalon, S. (2000) Am. J. Physiol. Cell Physiol. 279 762-770 [DOI] [PubMed] [Google Scholar]
- 15.Cartee, T. L., Megaw, A. G., Oomens, A. G., and Wertz, G. W. (2003) J. Virol. 77 7352-7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pattnaik, A. K., Ball, L. A., LeGrone, A. W., and Wertz, G. W. (1992) Cell 69 1011-1020 [DOI] [PubMed] [Google Scholar]
- 17.Yu, Q., Hardy, R. W., and Wertz, G. W. (1995) J. Virol. 69 2412-2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardy, R. W., and Wertz, G. W. (1998) J. Virol. 72 520-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy, R. W., Harmon, S. B., and Wertz, G. W. (1999) J. Virol. 73 170-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullender, W. M., Anderson, K., and Wertz, G. W. (1990) Virology 178 195-203 [DOI] [PubMed] [Google Scholar]
- 21.Mbiguino, A., and Menezes, J. (1991) J. Virol. Methods 31 161-170 [DOI] [PubMed] [Google Scholar]
- 22.Lazrak, A., Samanta, A., and Matalon, S. (2000) Am. J. Physiol. Lung Cell. Mol. Physiol. 278 848-857 [DOI] [PubMed] [Google Scholar]
- 23.Liu, G., Tsuruta, Y., Gao, Z., Park, Y. J., and Abraham, E. (2007) J. Immunol. 179 4125-4134 [DOI] [PubMed] [Google Scholar]
- 24.Nifli, A. P., Kampa, M., Alexaki, V. I., Notas, G., and Castanas, E. (2005) J. Dairy Res. 72 Spec. no 44-50 [DOI] [PubMed]
- 25.Feelisch, M., Rassaf, T., Mnaimneh, S., Singh, N., Bryan, N. S., Jourd'heuil, D., and Kelm, M. (2002) FASEB J. 16 1775-1785 [DOI] [PubMed] [Google Scholar]
- 26.Liu, G., Park, Y. J., and Abraham, E. (2008) FASEB J. 22 2285-2296 [DOI] [PubMed] [Google Scholar]
- 27.Tsuruta, Y., Park, Y. J., Siegal, G. P., Liu, G., and Abraham, E. (2007) J. Immunol. 179 7079-7086 [DOI] [PubMed] [Google Scholar]
- 28.Roberts, S. R., Compans, R. W., and Wertz, G. W. (1995) J. Virol. 69 2667-2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckardt-Michel, J., Lorek, M., Baxmann, D., Grunwald, T., Keil, G. M., and Zimmer, G. (2008) J. Virol. 82 3236-3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis, I. C., Lazarowski, E. R., Chen, F. P., Hickman-Davis, J. M., Sullender, W. M., and Matalon, S. (2007) Am. J. Respir. Cell Mol. Biol. 37 379-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie, Q. W., Kashiwabara, Y., and Nathan, C. (1994) J. Biol. Chem. 269 4705-4708 [PubMed] [Google Scholar]
- 32.Haeberle, H. A., Takizawa, R., Casola, A., Brasier, A. R., Dieterich, H. J., Van, R. N., Gatalica, Z., and Garofalo, R. P. (2002) J. Infect. Dis. 186 1199-1206 [DOI] [PubMed] [Google Scholar]
- 33.Kulaksiz, H., Schmid, A., Honscheid, M., Ramaswamy, A., and Cetin, Y. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 6796-6801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shijubo, N., Itoh, Y., Yamaguchi, T., and Abe, S. (2000) Ann. N. Y. Acad. Sci. 923 268-279 [DOI] [PubMed] [Google Scholar]
- 35.Lazrak, A., Thome, U., Myles, C., Ware, J., Chen, L., Venglarik, C. J., and Matalon, S. (2002) Am. J. Physiol. Lung Cell. Mol. Physiol. 282 650-658 [DOI] [PubMed] [Google Scholar]
- 36.Chen, L., Song, W., Davis, I. C., Shrestha, K., Schwiebert, E., Sullender, W. M., and Matalon, S. (2008) Am. J. Respir. Cell Mol. Biol., in press [DOI] [PMC free article] [PubMed]
- 37.Collins, P. L., and Graham, B. S. (2008) J. Virol. 82 2040-2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitehead, S. S., Bukreyev, A., Teng, M. N., Firestone, C. Y., St, C. M., Elkins, W. R., Collins, P. L., and Murphy, B. R. (1999) J. Virol. 73 3438-3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karron, R. A., Wright, P. F., Belshe, R. B., Thumar, B., Casey, R., Newman, F., Polack, F. P., Randolph, V. B., Deatly, A., Hackell, J., Gruber, W., Murphy, B. R., and Collins, P. L. (2005) J. Infect. Dis. 191 1093-1104 [DOI] [PubMed] [Google Scholar]
- 40.Kochva, U., Leonov, H., and Arkin, I. T. (2003) Protein Sci. 12 2668-2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fuentes, S., Tran, K. C., Luthra, P., Teng, M. N., and He, B. (2007) J. Virol. 81 8361-8366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindell, D. M., Lane, T. E., and Lukacs, N. W. (2008) Eur. J. Immunol. 38 2168-2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pribul, P. K., Harker, J., Wang, B., Wang, H., Tregoning, J. S., Schwarze, J., and Openshaw, P. J. (2008) J. Virol. 82 4441-4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burnstock, G., and Williams, M. (2000) J. Pharmacol. Exp. Ther. 295 862-869 [PubMed] [Google Scholar]
- 45.Ling, B. N., and Eaton, D. C. (1989) Am. J. Physiol. 256 F1094-F1103 [DOI] [PubMed] [Google Scholar]
- 46.Stockand, J. D., Bao, H. F., Schenck, J., Malik, B., Middleton, P., Schlanger, L. E., and Eaton, D. C. (2000) J. Biol. Chem. 275 25760-25765 [DOI] [PubMed] [Google Scholar]
- 47.Davis, I. C., Xu, A., Gao, Z., Hickman-Davis, J. M., Factor, P., Sullender, W. M., and Matalon, S. (2007) Am. J. Physiol. Lung Cell Mol. Physiol. 293 L281-L289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hickman-Davis, J., Gibbs-Erwin, J., Lindsey, J. R., and Matalon, S. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4953-4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo, Z., Shao, L., Du, Q., Park, K. S., and Geller, D. A. (2007) FASEB J. 21 535-542 [DOI] [PubMed] [Google Scholar]
- 50.Baeuerle, P. A., and Baltimore, D. (1996) Cell 87 13-20 [DOI] [PubMed] [Google Scholar]
- 51.Kurt-Jones, E. A., Popova, L., Kwinn, L., Haynes, L. M., Jones, L. P., Tripp, R. A., Walsh, E. E., Freeman, M. W., Golenbock, D. T., Anderson, L. J., and Finberg, R. W. (2000) Nat. Immunol. 1 398-401 [DOI] [PubMed] [Google Scholar]
- 52.Kunzelmann, K., Beesley, A. H., King, N. J., Karupiah, G., Young, J. A., and Cook, D. I. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 10282-10287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunzelmann, K., Konig, J., Sun, J., Markovich, D., King, N. J., Karupiah, G., Young, J. A., and Cook, D. I. (2004) J. Biol. Chem. 279 48760-48766 [DOI] [PubMed] [Google Scholar]
- 54.Kunzelmann, K., Sun, J., Meanger, J., King, N. J., and Cook, D. I. (2007) J. Virol. 81 3714-3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen, X. J., Seth, S., Yue, G., Kamat, P., Compans, R. W., Guidot, D., Brown, L. A., Eaton, D. C., and Jain, L. (2004) Am. J. Physiol. Lung Cell Mol. Physiol. 287 366-373 [DOI] [PubMed] [Google Scholar]
- 56.Zhang, Y., Luxon, B. A., Casola, A., Garofalo, R. P., Jamaluddin, M., and Brasier, A. R. (2001) J. Virol. 75 9044-9058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, Y., Jamaluddin, M., Wang, S., Tian, B., Garofalo, R. P., Casola, A., and Brasier, A. R. (2003) J. Virol. 77 5933-5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Janssen, R., Pennings, J., Hodemaekers, H., Buisman, A., van, O. M., de, R. L., Ozturk, K., Dormans, J., Kimman, T., and Hoebee, B. (2007) J. Virol. 81 5958-5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bebok, Z., Varga, K., Hicks, J. K., Venglarik, C. J., Kovacs, T., Chen, L., Hardiman, K. M., Collawn, J. F., Sorscher, E. J., and Matalon, S. (2002) J. Biol. Chem. 277 43041-43049 [DOI] [PubMed] [Google Scholar]
- 60.Chen, L., Bosworth, C. A., Pico, T., Collawn, J. F., Varga, K., Gao, Z., Clancy, J. P., Fortenberry, J. A., Lancaster, J. R., Jr., and Matalon, S. (2008) Am. J. Respir. Cell Mol. Biol. 39 150-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen, L., Patel, R. P., Teng, X., Bosworth, C. A., Lancaster, J. R., Jr., and Matalon, S. (2006) J. Biol. Chem. 281 9190-9199 [DOI] [PubMed] [Google Scholar]
- 62.Zaman, K., Carraro, S., Doherty, J., Henderson, E. M., Lendermon, E., Liu, L., Verghese, G., Zigler, M., Ross, M., Park, E., Palmer, L. A., Doctor, A., Stamler, J. S., and Gaston, B. (2006) Mol. Pharmacol. 70 1435-1442 [DOI] [PubMed] [Google Scholar]
- 63.Nara, M., Dhulipala, P. D., Ji, G. J., Kamasani, U. R., Wang, Y. X., Matalon, S., and Kotlikoff, M. I. (2000) Am. J. Physiol. Cell Physiol. 279 1938-1945 [DOI] [PubMed] [Google Scholar]
- 64.Eu, J. P., Xu, L., Stamler, J. S., and Meissner, G. (1999) Biochem. Pharmacol. 57 1079-1084 [DOI] [PubMed] [Google Scholar]
- 65.Campbell, D. L., Stamler, J. S., and Strauss, H. C. (1996) J. Gen. Physiol. 108 277-293 [DOI] [PMC free article] [PubMed] [Google Scholar]