

Abstract
Recent advances have suggested a crucial role of the innate immunity in shaping adaptive immune responses. How activation of innate immunity promotes adaptive T-cell responses to pathogens in vivo is not fully understood. It has been thought that Toll-like receptor (TLR)–mediated control of adaptive T-cell responses is mainly achieved by the engagement of TLRs on antigen-presenting cells to promote their maturation and function. In this study, we showed that direct TLR2–myeloid differentiating factor 88 (MyD88) signaling in CD8 T cells was also required for their efficient clonal expansion by promoting the survival of activated T cells on vaccinia viral infection in vivo. Effector CD8 T cells that lacked direct TLR2-MyD88 signaling did not survive the contraction phase to differentiate into long-lived memory cells. Furthermore, we observed that direct TLR2 ligation on CD8 T cells promoted CD8 T-cell proliferation and survival in vitro in a manner dependent on the phosphatidylinositol 3-kinase (PI3K)–Akt pathway activation and that activation of Akt controlled memory cell formation in vivo. These results identify a critical role for intrinsic TLR2-MyD88 signaling and PI3K-Akt pathway activation in CD8 T-cell clonal expansion and memory formation in vivo and could lead to the development of new vaccine approaches.
Introduction
Toll-like receptors (TLRs) constitute a major class of pattern recognition receptors that are involved in discrimination of “self” from “nonself” by the innate immune system.1 So far, 12 TLRs have been identified in mammals, and each TLR appears to recognize a unique set of highly conserved microbial structures or products known as pathogen-associated molecular patterns.2 On recognition of pathogen-associated molecular patterns, all TLRs, except TLR3, trigger a series of signaling cascades through the common adaptor protein, myeloid differentiating factor 88 (MyD88), that binds the cytosolic domain of TLR, the Toll/interleukin-1 (IL-1) receptor domain, and the death domain of the IL-1 receptor–associated kinase, leading to activation of nuclear factor-κB (NF-κB) that is critical for induction of innate immunity to a given microbial challenge.2
The TLR-MyD88 pathway also plays an important role in adaptive T-cell immunity.3 It has been thought that this is mainly achieved by the engagement of TLRs on antigen-presenting cells (APCs), such as dendritic cells (DCs), which promote DC function, including up-regulation of costimulatory molecules,4 enhancement of antigen processing5 and presentation,6 as well as secretion of Th1 polarizing pro-inflammatory cytokines by the DCs.7,8 However, recent studies have shown that TLRs are also expressed on T cells9 and that, in vitro, various TLR ligands directly promote proliferation of CD3 monoclonal antibody-stimulated T cells10–13 as well as their survival in the absence of APCs.13,14 It remains to be defined whether direct TLR signaling on T cells is critical for adaptive T-cell responses to a bona fide infection in vivo.
To address this question, we used a model of CD8 T-cell responses to an acute infection with vaccinia virus (VV) in vivo. We have recently shown that innate immune recognition of VV is mediated by TLR2 and dependent on MyD88 and that this TLR2-MyD88 pathway is critical for adaptive CD8 T-cell immunity against VV infection in vivo.15 In this study, we sought to gain a clearer understanding of the role of direct TLR2-MyD88 signaling in CD8 T-cell responses by examining the behavior of TLR2-deficient (TLR2−/−) and MyD88-deficient (MyD88−/−) CD8 T cells in wild-type (WT) recipients in response to VV infection in vivo. We observed that both TLR2−/− and MyD88−/− CD8 T cells had a severely diminished clonal expansion in response to VV infection. However, their ability to produce interferon-γ (IFN-γ) on a per-cell basis appeared not to be affected. The reduced clonal expansion of MyD88−/− CD8 T cells was not caused by a defect in T-cell activation but by poor survival of the activated T cells. Furthermore, long-lived memory CD8 T cells could not develop in the absence of direct TLR2-MyD88 signaling. We further demonstrated that direct TLR2 stimulation on CD8 T cells enhanced their proliferation and survival in vitro, which was dependent on activation of the phosphatidylinositol 3-kinase (PI3K)–Akt pathway. In addition, the activation of Akt was required for CD8 T-cell survival and memory cell formation in vivo. Collectively, our data indicate that direct TLR2-MyD88 signaling in CD8 T cells is required for CD8 T-cell clonal expansion, survival, and formation of long-lived memory cells via activation of the PI3K-Akt pathway after VV infection in vivo and highlight a previously unappreciated role for TLR signaling in regulating adaptive T-cell responses via direct action on antigen-specific CD8 T cells.
Methods
Mice
B10.D2 and RAG-2−/− mice on the B10.D2 background were purchased from The Jackson Laboratory (Bar Harbor, ME) and Taconic Farms (Germantown, NY), respectively. TLR2−/− and MyD88−/− mice were kindly provided by Dr Shizuo Akira (Osaka University, Osaka, Japan) and have been backcrossed for 9 generations onto the B10.D2 genetic background. Clone 4 HA-TCR–transgenic mice were a gift from Dr Linda Sherman (Scripps Research Institute, La Jolla, CA) and possess a T-cell receptor (TCR) that recognizes a Kd-restricted hemagglutinin (HA) epitope (518IYSTVASSL526).16 These mice were backcrossed to the Thy1.1, B10.D2 background for at least 9 generations. We also intercrossed clone 4 HA-TCR–transgenic mice with MyD88−/− and TLR2−/− mice to generate MyD88−/− and TLR2−/− clone 4 HA-TCR mice, respectively.
All mice used for experiments were between 6 and 10 weeks of age. All experimental procedures involving the use of mice were done in accordance with protocols approved by the Animal Care and Use Committee of the Duke University Medical Center.
Adoptive transfer and vaccination
For adoptive transfer of clone 4 CD8 T cells, single-cell suspensions were prepared from pooled spleen and peripheral lymph nodes of WT, MyD88−/−, or TLR2−/− clone 4 HA-TCR–transgenic mice and subjected to 2 rounds of CD8+ selection via anti-CD8 microbeads (Miltenyi Biotec, Auburn, CA). Purified CD8 T cells were then transferred into recipient mice through tail vein injection.
For transfer of endogenous polyclonal T cells, spleen and peripheral lymph nodes from WT, MyD88−/−, or TLR2−/− mice were pooled and made into single-cell suspensions, which then underwent 2 rounds of CD8+ selection using anti-CD8 microbeads. Flow-through from the CD8+ selection of WT cells was collected and subjected to CD4+ selection using anti-CD4 microbeads (Miltenyi Biotec). Cells were injected into RAG-2−/− recipients via the tail vein.
Recombinant vaccinia virus encoding HA (rVV-HA) and adenovirus-encoding HA (Ad-HA) has been previously described.17 In all experiments, mice were infected with either rVV-HA or Ad-HA intraperitoneally.
Antibodies and flow cytometry
Monoclonal antibodies used for staining were CyChrome-conjugated anti-CD8; fluorescein isothiocyanate–conjugated anti-CD62L, anti-CD69, anti-CD4, and anti–IFN-γ; phycoerythrin-conjugated anti-Thy1.1, anti-Thy1.2, anti-CD44, anti-CD25, and annexin V; allophycocyanin-conjugated anti–IFN-γ and streptavidin; and biotinylated anti-Thy1.1 (all from BD Biosciences, San Jose, CA). Collection of flow cytometric data was carried out using a FACSCanto (BD Biosciences), and events were analyzed using CellQuest or FACS DIVA software (BD Biosciences).
Intracellular cytokine staining
The intracellular staining was performed as previously described.15,17 Briefly, for clone 4 CD8 T cells, cells were incubated with 2 μg/mL of the Kd-HA518-526 peptide in the presence of 5 μg/mL brefeldin A for 6 hours at 37°C. For polyclonal CD8 T cells, cells were stimulated with 2 μg/mL of the VV-specific peptide F2L (26SPYAAGYDL34)18 for 6 hours at 37°C in the presence of 5 μg/mL brefeldin A. After washing, cells were stained with surface markers and permeabilized to detect intracellular IFN-γ using the Cytofix/Cytoperm Kit (BD Biosciences).
In vitro stimulation of T cells
Carboxyfluorescein succinimidyl ester (CFSE)–labeled WT CD8 T cells were placed into culture in 96-well plates in 200 μL total volume at a concentration of 2 × 105 cells/well in RPMI 1640 medium supplemented with 10% fetal bovine serum and recombinant murine IL-2 (100 U/mL). Cells were stimulated with 1 μg/mL plate-bound anti-CD3 antibody alone, or plate-bound anti-CD3 antibody coupled with 10 μg/mL plate-bound anti-CD28, 2 μg/mL Pam3Cys with or without addition of 10 μM of the PI3K inhibitor LY294002, 100 ng/mL lipopolysaccharide (all from Sigma-Aldrich, St Louis, MO), or 5 μg/mL cytosine-phosphate-guanosine (CpG) oligodeoxynucleotide (5′-TCCATGACGTTCCTGATGCT-3′; Integrated DNA Technologies, Coralville, IA). After 18 hours of stimulation, T cells were removed from stimulation, washed once, and placed back into culture in T-cell media supplemented with 100 U/mL IL-2 in the absence of further stimulation for a total of 4 days. For Western blot analysis, cells were removed from stimulation at 6, 12, and 18 hours, washed once, and total cell lysates were obtained for analysis.
RNA isolation and semiquantitative RT-PCR
Total RNA was isolated from CD8 T cells using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. One-step semiquantitative reverse-transcription polymerase chain reaction was performed using 5-fold serial dilution of template RNA and primers specific for TLR2 (forward 5′-TTGCTCCTGCGAACTCCTAT-3′, reverse 5′-CAATGGGAATCCTGCTCACT-3′), TLR3 (forward 5′-CCCCCTTTGAACTCCTCTTC-3′, reverse 5′-TTTCGGCTTCTTTTGATGCT-3′), TLR4 (forward 5′-GCTTTCACCTCTGCCTTCAC-3′, reverse 5′-CGAGGCTTTTCCATCCAATA-3′), TLR7(forward 5′-GGTATGCCGCCAAATCTAAA-3′, reverse 5′-TTGCAAAGAAAGCGATTGTG-3′), TLR8 (forward 5′-CAAACAACAGCACCCAAATG-3′, reverse 5′-GGGGGCACATAGAAAAGGTT-3′), TLR9 (forward 5′-GCAAGCTCAACCTGTCCTTC-3′, reverse 5′-TAGAAGCAGGGGTGCTCAGT-3′), and β-actin (forward 5′-AGCCATGTACGTAGCCATCC-3′, reverse 5′-CTCTCAGCTGTGGTGGTGAA-3′).
Western blot analysis
Western blot analysis was conducted as previously described.19 Briefly, samples were transferred to a nitrocellulose membrane after separation on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (Bio-Rad, Hercules, CA). Membranes were blotted overnight at 4°C with anti–phospho-Akt (Ser473; Cell Signaling Technology, Danvers, MA), washed 3 times, probed with an Alexa Fluor 680–conjugated anti–mouse Ig secondary antibody (Invitrogen, Carlsbad, CA), followed by visualization using the Odyssey infrared imaging system (LI-COR). Membranes were then stripped and probed with an anti–total Akt antibody (Cell Signaling Technology) to serve as a loading control.
Retrovirus preparation and infection
Preparation of retrovirus was performed as previously described.19 The retroviral constructs pMSCV encoding green fluorescent protein (GFP), and pMSCV-dn-Akt (D462*) encoding GFP, and dominant-negative Akt retroviral vectors were gifts from Z. Songyang (Baylor College of Medicine, Houston, TX). Retroviral transduction was performed as previously described.20 Briefly, freshly prepared retroviral supernatants were added to the HA-specific CD8 T cells in 96-well culture plates 36 hours after activation with 40 ng/mL phorbol-12-myristate-13-acetate and 500 ng/mL ionomycin in the presence of 8 μg/mL polybrene and murine IL-2 (100 U/mL). Cells were then centrifuged at 1300g for 2 hours at 25°C. Media containing the retroviral supernatant were removed after an additional 6 hours at 37°C, and the transduction procedure was repeated the following day. Two days after the second transduction, GFP+CD8+ T cells were sorted by fluorescence-activated cell sorter (FACS) using a high-speed cell sorter FACSVantage (BD Biosciences) and 3 × 103 cells were injected intravenously into naive B10.D2 mice.
Statistical analysis
Results were expressed as mean plus or minus SD. Differences between groups were examined for statistical significance using the Student t test.
Results
Defective generation of virus-specific effectors on VV infection in the absence of TLR2-MyD88 signaling in CD8 T cells
To investigate the role of direct TLR signaling in the CD8 T-cell response, we first examined the expansion of endogenous CD8 T cells on VV infection in vivo. We purified CD8 T cells from WT, MyD88−/−, and TLR2−/− mice and transferred them into RAG-2–deficient (RAG-2−/−) mice. These WT, MyD88−/−, and TLR2−/− CD8 T cells displayed a similar naive phenotype of CD44lowCD62LhighCD25lowCD69low before transfer (Figure S1A, available on the Blood website; see the Supplemental Materials link at the top of the online article). Because CD4 T cells have been shown to play a critical role in CD8 T-cell immunity,21 we also cotransferred WT CD4 T cells along with the CD8 T cells into RAG-2−/− recipients. Seven days later, all transferred CD8 T cells maintained a phenotype of CD25lowCD69low but showed up-regulation of CD44 and down-regulation of CD62L (Figure S1B), suggesting that these T cells were partially activated during homeostatic proliferation. In addition, all of them demonstrated the ability to produce IFN-γ on stimulation with phorbol-12-myristate-13-acetate/ionomycin (Figure S1C). Furthermore, the splenic absolute numbers of CD8 T cells were comparable among mice transferred with WT, MyD88−/−, or TLR2−/− CD8 T cells (Figure S1D), suggesting that homeostatic proliferation and homing of WT, MyD88−/−, and TLR2−/− CD8 T cells were similar. Similar results were obtained when cells were analyzed 14 days after transfer (data not shown), suggesting that sufficient homeostatic expansion of CD8 T cells was achieved after 7 days in RAG-2−/− mice. Mice were then infected with VV, and splenocytes were analyzed 7 days later for the percentage and absolute number of VV-specific CD8 T cells by IFN-γ intracellular staining on restimulation with VV-specific immunodominant epitope peptide.18 Mice transferred with WT CD8 T cells had a significant (P < .001) expansion of virus-specific CD8 T cells on VV infection compared with the uninfected naive mice (Figure 1). In contrast, in mice transferred with MyD88−/− or TLR2−/− CD8 T cells, the expansion of virus-specific CD8 T cells was significantly (P < .001) reduced (Figure 1). These results indicate that direct TLR2-MyD88 signaling is required for the generation of virus-specific CD8 effectors on VV infection in vivo.
Figure 1.

Endogenous MyD88−/− and TLR2−/− CD8 T cells exhibit defective virus-specific effector response to VV infection in vivo. A total of 106 purified WT, MyD88−/−, or TLR2−/− polyclonal CD8 T cells were transferred along with 2 × 106 WT polyclonal CD4 T cells into RAG-2−/− hosts. Seven days after transfer, mice were infected intraperitoneally with 5 × 106 pfu VV or left uninfected (naive) as control. Seven days after infection, splenocytes were stained with anti-CD4 and anti-CD8 antibodies and subjected to intracellular staining with an anti–IFN-γ antibody after a 6-hours stimulation with the VV-specific peptide F2L (26SPYAAGYDL34). (A) The percentage of IFN-γ+ CD8+ T cells among total splenocytes for each respective population is indicated. (B) The absolute cell number per spleen of IFN-γ+CD8+ T cells for both naive and infected groups are shown with SDs indicated (n = 6 per group). For all groups: naive versus VV, P < .001. For VV-infected hosts: MyD88−/− or TLR2−/− versus WT, P < .001. Data shown are representative of 3 independent experiments.
Clonal expansion is severely compromised in MyD88−/− and TLR2−/−-transgenic T cells on VV infection in vivo
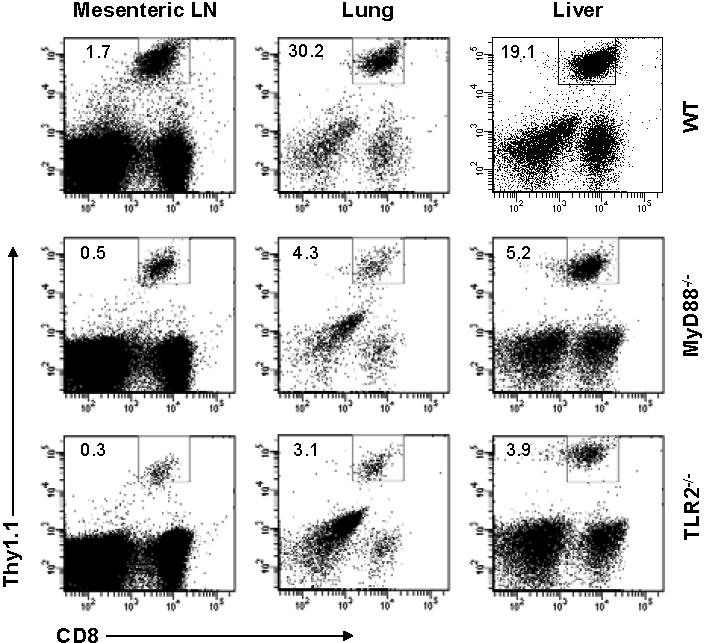
What then contributed to the defective generation of VV-specific CD8 effectors in the absence of direct TLR2-MyD88 signaling on VV infection in vivo? To address this question, we used a model of influenza HA-specific CD8 T-cell response to infection with rVV-HA. We first intercrossed the clone 4 HA-TCR–transgenic mice that express a TCR recognizing a Kd-restricted HA epitope (518IYSTVASSL526)17,19,22,23 with MyD88−/− and TLR2−/− mice to generate MyD88−/− and TLR2−/− clone 4 CD8 T cells, respectively. We then transferred 104 of purified naive WT, MyD88−/−, or TLR2−/− clone 4 CD8 T cells (Thy1.1+) into WT recipients (Thy1.2+) that were subsequently infected with rVV-HA. To assess relative contribution of TLR-induced maturation of APC function to CD8 T-cell priming, we also transferred WT clone 4 CD8 T cells into MyD88−/− hosts followed by rVV-HA infection. Seven days after infection, splenocytes were analyzed for clonal expansion and effector differentiation of the clone 4 CD8 T cells. Massive clonal expansion and effector differentiation, as measured by the production of IFN-γ, were detected in WT mice that received WT clone 4 CD8 T cells (Figure 2A,B). By contrast, the extent of clonal expansion was significantly (P < .001) diminished when MyD88−/− and TLR2−/− clone 4 CD8 T cells were transferred into WT mice (Figure 2A,B). However, the effector differentiation of MyD88−/− and TLR2−/− clone 4 CD8 T cells appeared to be intact as their production of IFN-γ on a per-cell basis (as measured by mean fluorescence intensity [MFI]) was similar to that of WT clone 4 CD8 T cells (Figure 2C). The diminished clonal expansion was not the result of preferential sequestration of MyD88−/− or TLR2−/− clone 4 CD8 T cells in other compartments as a similar degree of reduction in clonal expansion was found in several other lymphoid and extralymphoid tissues (Figure S2). WT clone 4 CD8 T cells also displayed a significant (P < .05) reduction in clonal expansion in MyD88−/− recipients, but to a lesser degree compared with that of MyD88−/− clone 4 T cells in WT recipients (Figure 2). This is consistent with the notion that TLR-induced APC maturation is important for promoting adaptive T-cell responses.3
Figure 2.

Clonal expansion is severely compromised in MyD88−/− and TLR2−/− clone 4 CD8 T cells in response to viral infection in vivo. A total of 104 naive WT, MyD88−/−, or TLR2−/− clone 4 CD8 T cells (Thy1.1+) were transferred into WT (WT→WT, MyD88−/− →WT, or TLR2−/−→WT) or MyD88−/− (WT→MyD88−/−) recipient mice (Thy1.2+) that were either left uninfected (naive) or infected with 5 × 105 pfu rVV-HA intraperitoneally. Seven days after infection, splenocytes were analyzed for the expansion of clonotypic T cells and their function by IFN-γ intracellular staining. (A) The percentages of clonotypic T cells among total lymphocytes (top) and IFN-γ–producing clonotypic T cells among total CD8+ T cells (bottom) in rVV-HA infected mice are indicated. (B) The absolute cell number per spleen of CD8+Thy1.1+ clonotypic T cells in both uninfected and rVV-HA infected hosts with SDs are indicated (n = 4 per group). For all groups: naive versus rVV-HA, P < .001. For rVV-infected hosts: WT→MyD88−/− versus WT→WT, P < .05; MyD88−/−→WT or TLR2−/−→WT versus WT→WT, P < .001. (C) The mean fluorescence intensity (MFI) of IFN-γ–producing clonotypic cells is indicated. WT→MyD88−/−, MyD88−/−→WT, or TLR2−/−→WT versus WT→WT, P > .05. Data shown are representative of 3 independent experiments.
Recent studies have demonstrated the need to closely mimic the endogenous CD8 T-cell response when transgenic T cells are used for adoptive transfer experiments.24,25 Therefore, we repeated the experiment using 500 WT or MyD88−/− clone 4 CD8 T cells. After transfer of 500 clone 4 T cells, mice were infected with 5 × 105 plaque-forming units (pfu) rVV-HA, and both transgenic and endogenous HA-specific CD8 T-cell responses were assessed 7 days later. We found that endogenous HA-specific CD8 T-cell responses were similar among all groups, including those receiving no transgenic T cells (Figure S3), suggesting that transfer of 500 clone 4 T cells did not suppress the endogenous HA-specific CD8 T-cell response. Under this condition, we demonstrated once again that the clonal expansion and effector function of MyD88−/− clone 4 T cells were significantly (P < .001) reduced compared with the WT controls (Figure S3).
Collectively, these results indicate that TLR2-MyD88 signaling in clone 4 CD8 T cells is critical for their clonal expansion in response to VV infection in vivo.
Direct MyD88 signaling is critical for the survival of activated CD8 T cells in vivo
We next investigated what was responsible for the defective clonal expansion of CD8 T cells in the absence of MyD88 signaling. A diminished clonal expansion could be the result of insufficient initial activation/proliferation or increased death of the activated T cells. We first investigated whether initial T-cell activation/proliferation was affected by lack of direct MyD88 signaling. To test this, we transferred 106 naive WT or MyD88−/− clone 4 CD8 T cells into WT mice followed by infection with rVV-HA. Higher clone 4 T-cell numbers were used here because transfer of 104 cells was below the limit of detection at early time points. At 24 hours after infection, both WT and MyD88−/− clone 4 CD8 T cells displayed a similarly activated phenotype of CD44high, CD62Llow, CD25high, and CD69high compared with the naive CD8 T-cell phenotype of CD44low, CD62Lhigh, CD25low, and CD69low (Figure 3A). Three days after infection, both WT and MyD88−/− clone 4 CD8 T cells underwent several rounds of division by CFSE dilution (Figure 3B). These data indicate that activation/proliferation of CD8 T cells in response to viral infection in vivo is not affected by a defect in intrinsic MyD88 signaling in CD8 T cells. We then asked whether CD8 T cells activated in the absence of direct MyD88 signaling were more susceptible to cell death. We used annexin V staining to assess CD8 T cells undergoing early apoptosis 7 days after infection. Indeed, MyD88−/− clone 4 CD8 T cells displayed a significant increase in annexin V+ cells compared with WT clone 4 CD8 T cells (Figure 3C). Taken together, these results suggest that the diminished clonal expansion of CD8 T cells that lack direct MyD88 signaling is not caused by a reduction in T-cell activation but by poor survival of activated CD8 T cells.
Figure 3.

MyD88 signaling in CD8 T cells is critical for their survival after activation and proliferation in vivo. (A,B) A total of 106 WT or MyD88−/− clone 4 CD8 T cells (Thy1.1+) were transferred into B10.D2 mice, which were infected with 5 × 106 pfu rVV-HA intraperitoneally or left uninfected (naive). (A) At 24 hours after infection, splenocytes were analyzed for T-cell activation phenotypically by staining with anti-CD44 and anti-CD62L (left panels) or anti-CD25 and anti-CD69 antibodies (right panels). FACS plots are gated on CD8+Thy1.1+ clonotypic cells, and percentages shown are of those cells possessing an activated phenotype of CD44highCD62Llow or CD25highCD69high. (B) Three days after infection, proliferation of the clonotypic cells was analyzed by the CFSE dilution assay. Events were gated on CD8+Thy1.1+ cells. (C) A total of 104 WT or MyD88−/− clone 4 CD8 T cells were transferred into B10.D2 mice that were either left uninfected (naive) or infected with 5 × 105 pfu rVV-HA (rVV-HA). Seven days after infection, splenocytes were harvested and the extent of clonotypic cells undergoing apoptosis was analyzed by annexin V staining. Events were gated on CD8+Thy1.1+ cells, and percentages shown are of those cells that are annexin V+. Representative data of 3 independent experiments with 3 mice per group for each experiment are shown.
Direct TLR2-MyD88 signaling is required for the formation of memory CD8 T cells
We next determined the ability of effector CD8 T cells that were primed in the absence of direct TLR2-MyD88 signaling to develop into long-term memory cells. After the peak of clonal expansion at day 7, WT clone 4 effector CD8 T cells in the WT recipients underwent marked contraction and those that survived differentiated into long-lasting memory cells between days 14 and 28 (Figure 4A). This is consistent with previous observations in other models of bacterial or viral infections.26–28 Similarly, WT clone 4 effector CD8 T cells generated in the MyD88−/− hosts were also capable of differentiating into memory cells with a reduction in memory size that was proportional to the size of effectors (Figure 2). In contrast, the small number of MyD88−/− or TLR2−/− clone 4 effector CD8 T cells that were present by day 7 in the WT recipients could not survive the contraction phase to develop into memory cells (Figure 4A,B), indicating that direct MyD88 signaling is critical for the generation of long-lived memory cells. These data demonstrate that TLR2-MyD88 signaling within virus-specific CD8 T cells is required for efficient clonal expansion and formation of long-lived memory cells.
Figure 4.

A critical role for direct TLR2-MyD88 signaling in the formation of memory CD8 T cells. A total of 104 naive WT, MyD88−/−, or TLR2−/− clone 4 CD8 T cells (Thy1.1+) were transferred into WT (WT→WT, MyD88−/− →WT, or TLR2−/−→WT) or MyD88−/− (WT→MyD88−/−) recipient mice (Thy1.2+) that were infected with 5 × 105 pfu rVV-HA intraperitoneally. (A) Splenocytes were harvested at days 0, 7, 14, 28, or 42 after infection to determine the absolute number of the CD8+Thy1.1+ population per spleen, shown with SDs indicated (n = 4 per group). (B) At 42 days after infection, splenocytes were stained with anti-CD8 and anti-Thy1.1. The percentages of clonotypic T cells among total lymphocytes (top) and IFN-γ–producing clonotypic T cells among total CD8+ T cells (bottom) are indicated. Data shown are representative of 3 independent experiments.
Intrinsic TLR2 signaling increases cellular proliferation and survival in vitro
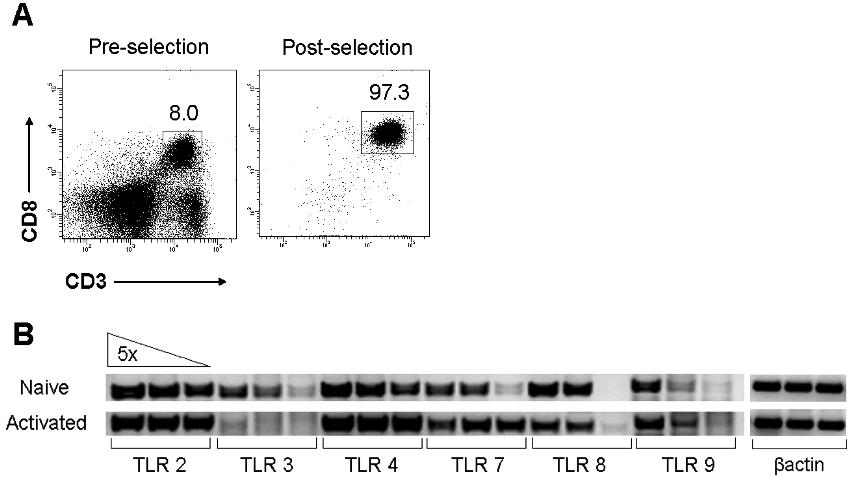
To further elucidate the mechanism by which direct signaling through the TLR2-MyD88 pathway functions to promote the survival of activated CD8 T cells, we turned to an in vitro culture system. Naive polyclonal CD8 T cells were purified by 2 rounds of positive selection using CD8 microbeads with a purity of more than 97% (Figure S4A), which is similar to that achieved by FACS sorting (data not shown). We first showed that naive and activated polyclonal CD8 T cells express multiple TLRs, including TLR2 (Figure S4B). Naive WT polyclonal CD8 T cells were stimulated with plate-bound anti-CD3 alone or in combination with plate-bound anti-CD28 or the soluble TLR2 ligand, Pam3Cys. Cells were removed from stimulation after 18 hours, and cellular division and survival as measured by CFSE dilution and annexin V staining, respectively, was measured at day 4. Stimulation of WT CD8 T cells by anti-CD3 alone induced minimal increases in cell division and survival over naive, unstimulated controls (Figure 5). In contrast, those cells stimulated with both anti-CD3 and Pam3Cys proliferated vigorously after 4 days (Figure 5A). This increase in proliferation was accompanied by an increase in survival, with an approximate 10-fold increase over naive cells in the percentage of cells surviving as evidenced by annexin V− staining (Figure 5B). This augmentation was dependent on MyD88, as CD8 T cells deficient for MyD88 showed complete abrogation of the increased proliferation and survival gained by Pam3Cys stimulation (Figure 5). Consistent with a role for CD28 costimulation in promoting CD8 T-cell proliferation and survival, cells stimulated with both anti-CD3 and anti-CD28 plate-bound antibodies also showed an increase in proliferation and survival, but in an MyD88-independent manner (Figure 5). However, CD28 costimulation only moderately enhanced the survival of CD8 T cells stimulated with anti-CD3 and Pam3Cys (Figure S5).
Figure 5.

Intrinsic TLR2-MyD88 signaling increases cellular proliferation and survival after in vitro stimulation and is dependent on the PI3K pathway. (A,B) Polyclonal CFSE-labeled WT or MyD88−/− CD8 T cells were stimulated in vitro with plate-bound anti-CD3 antibody alone (αCD3) or anti-CD3 antibody coupled with plate-bound anti-CD28 (αCD3 αCD28), Pam3Cys (αCD3 Pam3Cys), Pam3Cys and the PI3K inhibitor LY294002 (αCD3 Pam3Cys LY294002), or left unstimulated (naive) as a control. After 18 hours of stimulation, T cells were removed from stimulation and placed back into culture in the absence of further stimulation for a total of 4 days. At this time, both the CFSE profile (A) as well as the survival of CD8+ cells by annexin V staining (B) were determined by flow cytometry. Percentage of annexin V− cells among total CD8+ T cells is indicated. Representative data from 4 independent experiments are shown.
The additive effect we observed with Pam3Cys was also seen to some extent with the addition of other various TLR ligands, most notably with the addition of CpG DNA (Figure S6). This is in line with a recent report demonstrating that the addition of CpG to CD4+ T cells in culture serves both a costimulatory and survival function.12 Collectively, these data demonstrate that direct TLR signaling promotes CD8 T-cell proliferation and survival in vitro.
Stimulation of TLR2 on CD8 T cells increases activation of the PI3K-Akt pathway
We next sought to determine which signaling pathway downstream of TLR engagement was responsible for mediating the observed increase in CD8 T-cell survival. The serine-threonine Akt has emerged as an important regulator of cell survival.29–33 In mature CD4 T cells, TCR stimulation as well as CD28, IL-2R, and OX-40 signaling have led to Akt activation.34–37 Furthermore, a recent study showed that stimulation of CD4 T cells with CpG DNA induced an association of PI3K with MyD88, leading to phosphorylation of PI3K downstream targets, Akt and glycogen synthetase kinase-3 (GSK-3).12 We therefore hypothesized that direct TLR2 stimulation in CD8 T-cell survival and memory formation was mediated by the activation of the PI3K-Akt pathway. To test this, we first examined whether addition of the PI3K inhibitor LY294002 to CD8 T-cell cultures abolished CD8 T-cell proliferation and survival gained by TLR2 stimulation. Indeed, the TLR2-stimulated enhancement of CD8 T-cell proliferation and survival was dependent on activation of PI3K as addition of LY294002 blocked all increase in cellular proliferation and survival brought about by the addition of Pam3Cys (Figure 5). However, different from CD4 T cells, CD28-mediated enhancement of survival was independent of PI3K as addition of LY294002 did not diminish cell survival (Figure S5).
We next tested whether TLR2 ligation on CD8 T cells promoted activation of Akt, a downstream target of PI3K, as determined by Western blot analysis (Figure 6). Our data demonstrate that addition of Pam3Cys increased the phosphorylation of Akt. Collectively, these observations suggest that CD8 T-cell survival and proliferation brought about by direct TLR2 ligation are dependent on the activation of the PI3K-Akt pathway.
Figure 6.

Addition of Pam3Cys increases activation of the PI3K-Akt pathway in polyclonal CD8 T cells. Polyclonal CD8 T cells were stimulated in vitro with plate-bound anti-CD3 antibody alone (αCD3 only), coupled with either Pam3Cys (αCD3 + Pam3Cys) or plate-bound anti-CD28 (αCD3 + αCD28), or left unstimulated (naive) as a control. After 6, 12, and 18 hours of stimulation, CD8 T cells were removed from culture and total cell lysates were collected for Western blot analysis of phosphorylated (pAkt) as well as total Akt, which served as a loading control. Data shown are a representative blot of 5 independent experiments.
Activation of Akt is required for CD8 memory formation in vivo
Our observations that MyD88−/− CD8 T cells failed to form memory cells in vivo (Figure 4) and direct TLR2 ligation on CD8 T cells increased activation of the PI3K-Akt pathway and T-cell survival (Figures 5,6) suggested the importance of the PI3K-Akt pathway activation in CD8 memory T-cell formation in vivo. To address this question, WT clone 4 CD8 T cells (Thy1.1+) were first transduced with a retroviral construct containing either GFP alone (empty) as control or GFP and a dominant negative form of Akt (dn-Akt). The transduced GFP+ clone 4 CD8 T cells were sorted, and 3 × 103 cells were transferred into naive B10.D2 (Thy1.2+) recipients that were subsequently infected with rVV-HA. At 42 days after infection, memory cell formation and functionality were assessed by challenging the mice with recombinant Ad-HA. Seven days after challenge, expansion and effector function, as assayed by intracellular staining for IFN-γ, of memory cells were determined. We found that the percentage of clone 4 HA-specific CD8 T cells was significantly reduced in mice transferred with dn-Akt–transduced T cells compared with those that received the control T cells (Figure 7). However, the endogenous HA-specific CD8 T-cell responses were comparable between 2 groups. These data demonstrate that activation of Akt in CD8 T cells is critical for CD8 memory T-cell formation in response to VV infection in vivo.
Figure 7.

Activation of and signaling through Akt is required for CD8 memory cell formation in vivo. (A,B) Naive WT clone 4 CD8+ T cells were transduced with retroviral constructs encoding GFP only (empty) as a control or GFP coupled with a dominant-negative form of Akt (dnAkt). The transduced cells were sorted, and 3 × 103 GFP+CD8+ (Thy1.1+) T cells were transferred into B10.D2 recipients (Thy1.2+), which were then vaccinated with 5 × 105 pfu rVV-HA intraperitoneally. At 42 days later, mice were boosted with 2 × 109 pfu Ad-HA intraperitoneally. Seven days after infection, splenocytes were analyzed for the expansion of clonotypic T cells and their function by IFN-γ intracellular staining. (A) The percentages of clonotypic T cells among total lymphocytes (top) and IFN-γ–producing clonotypic T cells among total CD8+ T cells (bottom) are indicated. (B) The absolute cell numbers per spleen of CD8+Thy1.1+ clonotypic T cells in both empty vector and dnAkt groups with SDs are indicated (n = 4 per group). Empty versus dnAkt, P < .001. Data shown are representative of 3 independent experiments.
Discussion
Recent advances have suggested a crucial role of the innate immune system in shaping adaptive immunity.3 How activation of innate immunity promotes adaptive T-cell responses to pathogenic infection in vivo is not fully understood. It has been thought that TLR-mediated control of adaptive T-cell immunity is mainly achieved by the engagement of TLRs on APCs to promote their maturation and function.4–8 In this study, we show that, in addition to TLR-induced maturation of APC function, direct TLR signaling in CD8 T cells is required for their clonal expansion in response to VV infection in vivo. This dependency on direct TLR2-MyD88 signaling for clonal expansion is achieved by promoting the survival of effector CD8 T cells because those primed in the absence of direct TLR signaling display poor survival. However, in vitro ligation of TLR2 on CD8 T cells promotes both the proliferation and survival of CD8 T cells. This is in line with the previous observation.13 The reason why direct TLR2 signaling on VV infection in vivo only promotes CD8 T-cell survival, but not proliferation, is not clear. Because CD28 costimulation also enhances T-cell proliferation and survival, we speculate that CD8 T-cell proliferation may not be affected in the absence of TLR2 signaling in vivo because it occurs early during DC–T-cell interactions, which also provide CD28 costimulation. However, when activated CD8 T cells leave secondary lymphoid organs, their survival may solely depend on direct TLR signaling as CD28 costimulation provided by DCs is no longer available.
In addition to TLR2, CD8 T cells also express other TLRs that use MyD88 as the adaptor. Furthermore, MyD88 also mediates IL-1 receptor (IL-1R) and IL-18R signaling.38 Although we cannot completely rule out the contribution of other TLRs or IL-1/IL-18 signaling on CD8 T cells, we think that the observed deficiency in MyD88−/− CD8 T-cell responses to VV infection is mainly the result of a lack of TLR2 signaling for 2 reasons: (1) so far, only TLR2 has been shown to recognize VV in vivo15; and (2) both polyclonal and clone 4 TLR2−/− CD8 T cells behaved similarly to their MyD88−/− counterparts in response to VV infection in vivo.
The course of CD8 T-cell response after an acute infection or vaccination consists of 3 distinct phases: clonal expansion and development of effector functions, subsequent contraction of the majority of effector T cells, and generation of long-lived memory cells from the surviving cells.26–28 The signals that regulate the survival of effector T cells and promote the generation of memory cells remain largely unknown. Previous studies have suggested that effector CD8 T cells that up-regulate the expression of IL-7Rα39 or CD8αα homodimer40 possess the ability to escape death during contraction and develop into long-lived memory cells. However, other studies have demonstrated that IL-7Rα expression might not necessarily identify memory CD8 T-cell precursors41,42 and that memory CD8 T cells can develop in the absence of CD8αα.43 Here we provide evidence that, although TLR signaling through both CD8 T cells and APCs is required for efficient clonal expansion, it is direct TLR2-MyD88 signaling that is critical for effector CD8 T cells to survive the contraction phase to develop into memory cells.
It is not clear why the majority of effector CD8 T cells that have received direct TLR signals still undergo massive contraction. One possibility could be that TLR ligands provided by the pathogen become limited at the onset of contraction resulting from clearance of the pathogen. Another possibility could be that the effector T cells may become progressively refractory to TLR signals, especially after the withdrawal of antigenic exposure and other survival signals, such as type I IFNs.44 It has been shown that repeated exposure of APCs to lipopolysaccharide can induce endotoxin tolerance either through down-regulation of TLR4 expression or reduction of TLR4-MyD88 complex formation.45,46 Whether similar TLR pathway down-regulation occurs in activated effector CD8 T cells requires further investigation.
How does direct TLR2-MyD88 signaling in CD8 T cells promote their survival? Previous studies in vitro have shown that the TLR9 ligand CpG DNA enhances survival of activated CD4 T cells in an MyD88-dependent manner, and this increased survival is associated with NF-κB activation and up-regulation of the survival molecule, Bcl-XL.14 This suggests that direct TLR ligation on T cells may confer survival signals via a pathway that activates both the PI3K signaling pathway and NF-κB.35,47 Consistent with this, a recent report shows that stimulation of CD4 T cells with anti-CD3 monoclonal antibody in the presence of CpG DNA induces an association of PI3K with MyD88, leading to phosphorylation of PI3K downstream targets, Akt and GSK-3.12 Similarly, we have demonstrated here that Akt activation as well as proliferation and survival of activated CD8 T cells are enhanced on direct stimulation of TLR2 on CD8 T cells. This observed increase in CD8 T-cell proliferation and survival was entirely dependent on activation of the PI3K-Akt pathway. Furthermore, Akt activation was critically required for CD8 memory T-cell formation in response to viral infection in vivo.
The mechanism by which Akt promotes survival downstream of TLR2-MyD88 signaling in CD8 T cells is unknown. Akt has many downstream substrates that have been shown to regulate survival of T cells under various conditions. It is possible that, similar to studies in activated CD4 T cells, Akt activation may regulate CD8 T-cell survival via direct regulation of prosurvival members of the Bcl-2 family.14 Indeed, previous studies from our laboratory have demonstrated that activation of Akt is responsible for the up-regulation of Bcl-XL after CD8 T-cell activation in vitro.19 Another possibility is that Akt could inactivate its downstream target, FOXO, leading to inhibition of proapoptotic molecules, such as Bim.48,49 Akt could also activate GSK-3, resulting in increased glycogen synthesis and cell survival.50,51 Future studies will be needed to address the mechanism by which Akt promotes activated CD8 T-cell survival.
In conclusion, we have shown that, in addition to TLR-mediated promotion of DC function, direct TLR2-MyD88 signaling in CD8 T cells is crucial for their clonal expansion in response to viral infection in vivo. This is achieved by promoting the survival of effector CD8 T cells via a mechanism dependent on activation of the PI3K-Akt pathway. Furthermore, effector CD8 T cells do not survive the contraction phase to differentiate into long-lived memory cells in the absence of intrinsic TLR signaling. These results identify a critical role for direct activation of TLRs in CD8 T-cell clonal expansion and memory formation after viral infection in vivo and may shed light on the design of effective vaccines.
Supplementary Material
Acknowledgments
We thank S. Akira for providing MyD88−/− and TLR2−/− mice and Z. Songyang for providing the pMSCV and pMSCV-dnAkt constructs.
This work was supported by the National Institutes of Health (grants CA111807 and CA047741) and a grant from the Alliance for Cancer Gene Therapy (Y.Y.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.Q., J.M., and X.H. performed research and collected and analyzed data; and Y.Y. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yiping Yang, Departments of Medicine and Immunology, Duke University Medical Center, Box 103005, Durham, NC 27710; e-mail: yang0029@mc.duke.edu.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 4.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 5.Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T-cell response. Immunity. 2006;25:655–664. doi: 10.1016/j.immuni.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 6.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 7.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 9.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 10.Bendigs S, Salzer U, Lipford GB, Wagner H, Heeg K. CpG-oligodeoxynucleotides co-stimulate primary T cells in the absence of antigen-presenting cells. Eur J Immunol. 1999;29:1209–1218. doi: 10.1002/(SICI)1521-4141(199904)29:04<1209::AID-IMMU1209>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 11.Caron G, Duluc D, Fremaux I, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- 12.Gelman AE, LaRosa DF, Zhang J, et al. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cottalorda A, Verschelde C, Marcais A, et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. 2006;36:1684–1693. doi: 10.1002/eji.200636181. [DOI] [PubMed] [Google Scholar]
- 14.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-β. Blood. 2007;109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan DJ, Liblau R, Scott B, et al. CD8(+) T cell-mediated spontaneous diabetes in neonatal mice. J Immunol. 1996;157:978–983. [PubMed] [Google Scholar]
- 17.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5:508–515. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 18.Tscharke DC, Woo WP, Sakala IG, et al. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J Virol. 2006;80:6318–6323. doi: 10.1128/JVI.00427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quigley M, Huang X, Yang Y. Extent of stimulation controls the formation of memory CD8 T cells. J Immunol. 2007;179:5768–5777. doi: 10.4049/jimmunol.179.9.5768. [DOI] [PubMed] [Google Scholar]
- 20.Koonpaew S, Shen S, Flowers L, Zhang W. LAT-mediated signaling in CD4+CD25+ regulatory T cell development. J Exp Med. 2006;203:119–129. doi: 10.1084/jem.20050903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 22.Novy P, Quigley M, Huang X, Yang Y. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. J Immunol. 2007;179:8243–8251. doi: 10.4049/jimmunol.179.12.8243. [DOI] [PubMed] [Google Scholar]
- 23.Quigley M, Huang X, Yang Y. STAT1 signaling in CD8 T cells is required for their clonal expansion and memory formation following viral infection in vivo. J Immunol. 2008;180:2158–2164. doi: 10.4049/jimmunol.180.4.2158. [DOI] [PubMed] [Google Scholar]
- 24.Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T-cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 27.Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 28.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 29.Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 30.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 31.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 32.Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7:1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 33.Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones RG, Parsons M, Bonnard M, et al. Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivo. J Exp Med. 2000;191:1721–1734. doi: 10.1084/jem.191.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2:37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- 36.Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 1999;11:281–288. doi: 10.1016/s1074-7613(00)80103-x. [DOI] [PubMed] [Google Scholar]
- 37.Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol. 2004;5:150–158. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 38.Adachi O, Kawai T, Takeda K, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 39.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 40.Madakamutil LT, Christen U, et al. CD8alphaalpha-mediated survival and differentiation of CD8 memory T cell precursors. Science. 2004;304:590–593. doi: 10.1126/science.1092316. [DOI] [PubMed] [Google Scholar]
- 41.Lacombe MH, Hardy MP, Rooney J, Labrecque N. IL-7 receptor expression levels do not identify CD8+ memory T lymphocyte precursors following peptide immunization. J Immunol. 2005;175:4400–4407. doi: 10.4049/jimmunol.175.7.4400. [DOI] [PubMed] [Google Scholar]
- 42.Klonowski KD, Williams KJ, Marzo AL, Lefrancois L. Cutting edge: IL-7-independent regulation of IL-7 receptor alpha expression and memory CD8 T cell development. J Immunol. 2006;177:4247–4251. doi: 10.4049/jimmunol.177.7.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandele A, Kaech SM. Cutting edge: memory CD8 T cell maturation occurs independently of CD8alphaalpha. J Immunol. 2005;175:5619–5623. doi: 10.4049/jimmunol.175.9.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nomura F, Akashi S, Sakao Y, et al. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol. 2000;164:3476–3479. doi: 10.4049/jimmunol.164.7.3476. [DOI] [PubMed] [Google Scholar]
- 46.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol. 2002;169:5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 47.Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci U S A. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stahl M, Dijkers PF, Kops GJ, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 49.Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 2004;104:784–787. doi: 10.1182/blood-2003-09-3071. [DOI] [PubMed] [Google Scholar]
- 50.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 51.Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. doi: 10.1034/j.1600-065x.2003.00008.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}