

Abstract
Platelet-activating factor acetylhydrolase (PLA2G7) is a potent pro- and anti-inflammatory molecule that has been implicated in multiple inflammatory disease processes, including cardiovascular disease. The goal of this study was to investigate the genetic effects of PLA2G7 on coronary artery disease (CAD) risk in two large, independent datasets with CAD. Using a haplotype tagging (ht) approach, 19 ht single nucleotide polymorphisms (SNPs) were genotyped in CATHGEN case–control samples (cases = 806 and controls = 267) and in the GENECARD Family Study (n = 1101 families, 2954 individuals). Single SNP analysis using logistic regression revealed nine SNPs with significant association in all CATHGEN subjects (P = 0.0004–0.02). CATHGEN cases were further stratified into subgroups based on age of CAD onset (AOO) and severity of disease; 599 young affecteds (YA, AOO <56) and 207 old affected (OA, AOO >56). Significant genetic effects were observed in both OA and YA (P = 0.0001–0.02). The GENECARD probands demonstrated results similar to those seen in the YA CATHGEN cases (P = 0.002–0.05). Of the 19 SNPs genotyped, 3 SNPs result in non-synonymous coding changes (I198T, A379V and R92H). Two of the coding SNPs, R92H and A379V, constitute two of the most significantly associated SNPs, even after Bonferroni correction and appear to represent independent associations (r2 = 0.09). Multiple additional polymorphisms in low linkage disequilibrium with these coding SNPs were also strongly associated. In summary, PLA2G7 represents an important, potentially functional candidate in the pathophysiology of CAD based on replicated associations using two independent datasets and multiple statistical approaches. Further functional studies involving a combination of risk alleles are warranted.
Introduction
A substantial amount of evidence has accumulated over recent years supporting the role of inflammatory processes in the development of atherosclerosis [reviewed in (1)]. Inflammation can occur at all stages of atherosclerosis and is also highly correlated with the accumulation of lipids in the arterial wall. Clinical evidence corroborates the atherogenic role of inflammation, with studies indicating a strong correlation between inflammatory risk markers and coronary events (2–4). One of the hypotheses linking inflammation to cardiovascular disease risk stems from the oxidation of low-density lipoproteins (LDLs). Research suggests that oxidative modification of LDL is necessary for the preliminary stages of atherosclerosis, including uptake of LDL particles by macrophages into the intimal atheroma (5). The inflammatory process of hydrolyzing oxidized phospholipids in LDL to generate lysophosphatidylcholine (lyso-PC) and free oxidized fatty acids is mediated by platelet-activating factor acetylhydrolase (PLA2G7), a circulating plasma enzyme bound primarily to LDL, with a small proportion bound to high-density lipoprotein (HDL).
Heritability studies indicate that 62% of the variation in PLA2G7 activity is due to genetic factors (6). In several independent studies, elevated levels of PLA2G7 have been strongly implicated as a risk factor for the development of atherosclerosis (7–10). In humans, PLA2G7 is upregulated in preadipocytes/stromal vascular cells in obese individuals compared with non-obese subjects in case–control studies (11). Moreover, in a study evaluating RNA profiles in mice aortas with varying degrees of atherosclerosis, PLA2G7 was identified as an expression candidate for early and intermediate disease progression, and consistently differentiated non-diseased and diseased aortas (12). An increase in PLA2G7 expression has also been identified in macrophages of both human and rabbit atherosclerotic lesions (13) and in an atherosclerosis-prone mouse model (14). Recently, Kolodgie et al. (15) found PLA2G7 expression to be low in early atherosclerotic disease, with highest expression in ruptured diseased lesions.
Recent studies have attempted to understand the genetic effects of single nucleotide polymorphisms (SNPs) on PLA2G7 function and atherosclerotic outcome. However, the results from these studies have only served to illustrate further the genetic complexity of PLA2G7 and CAD. For example, a rare nonsynonymous SNP (V279F), found only within Japanese, Turk, Kyrgyze and Azerbaijan populations (16,17), has been associated with reduced enzymatic levels of PLA2G7 in heterozygous individuals and complete loss of enzymatic function in homozygous individuals (17,18). The mutation was subsequently identified as an independent risk factor for the development of coronary artery disease (CAD) in Japanese men (19). Several studies have identified an additional nonsynonymous coding polymorphism (A379V) in Caucasian populations associated with plasma PLA2G7 levels (20–22). However, the genetic effect of the A379V polymorphism on the enzymatic levels and atherosclerotic outcome varies among studies, with some studies showing V379 allele to be associated with high enzyme levels (20,22,23) whereas others indicating lower levels (21). Liu et al. found significant association with A379V in subjects with premature myocardial infarctions (MIs) (21), with V379 allele associated with a decreased risk of MI. Although several studies have examined multiple-coding polymorphisms in this gene, none, to our knowledge, has taken into account the genetic architecture of the PLA2G7 gene and comprehensively investigated multiple intragenic polymorphisms within the gene. In this present study, we have performed comprehensive genotyping of PLA2G7 using haplotype tagging SNPs (htSNPs) across the entire gene region and tested these SNPs for association in two independent datasets to determine whether additional variants independent of the known coding polymorphisms are associated with CAD.
Results
SNP selection and genotyping
Single nucleotide polymorphisms were selected for genotyping using SNPselector (24) to capture genetic variation in both Caucasian and African-American sample sets. In total, 19 polymorphisms were assigned to unique linkage disequilibrium (LD) bins using the criteria r2 ≥ 0.7 and minor allele frequency (MAF) ≥ 0.05, in essence capturing the majority of haplotypic diversity of the gene (Table 1). As reported from the HapMap samples, LD structure varied between the African and Caucasian samples, thus we genotyped all population-specific tagging SNPs that had a reported MAF of ≥0.05 in both populations. Three previously reported SNPs located in the coding region of PLA2G7 resulted in non-synonymous amino acid substitutions, rs1051931 (A379V), rs1805018 (I198T) and rs1805017 (R92H). The remaining 16 SNPs were located in intronic or UTR regions of the gene (Fig. 1). The 19 SNPs were chosen from published resources based on a strict r2 criteria and were thus expected to be in low LD based on reported LD structure in Caucasians and Africans. LD was calculated separately for both Caucasian affected and unaffected individuals, with no statistical differences seen between the two groups (Fig. 1). As reported in other studies, the LD is generally lower in African Americans than in Caucasians and thus the plot of LD in the unaffected Caucasians represents the upper limit of observed LD. As expected based on the selection of tagSNP data, the majority of SNPs were not highly correlated, although a few SNPs were identified in our dataset as being highly correlated (r2 ≥ 0.7) with at least one additional SNP. However, based upon the low correlation of the majority of the genotyped SNPs and the strict criteria used to select the tagSNPs, we believe that we have captured the majority of genetic variation of PLA2G7.
Table 1.
SNP Information for SNPs genotyped on PLA2G7
| SNP | Location (bp) | Gene location | Minor allele | Frequency Control (n = 267) | YA (n = 599) | OA (n = 207) | MI (n = 425) | GC Proband (n = 718) |
|---|---|---|---|---|---|---|---|---|
| RS974670 | 46 775 302 | Intron | T | 0.31 | 0.30 | 0.38 | 0.33 | 0.37 |
| RS12528857 | 46 777 895 | Intron | A | 0.22 | 0.17 | 0.16 | 0.19 | 0.22 |
| RS1051931 | 46 780 902 | Exon 12 A379V | A | 0.15 | 0.20 | 0.26 | 0.19 | 0.18 |
| RS2216465 | 46 783 978 | Intron | G | 0.43 | 0.38 | 0.29 | 0.38 | 0.35 |
| RS4498351 | 46 784 742 | Intron | A | 0.43 | 0.42 | 0.44 | 0.43 | 0.47 |
| RS1805018 | 46 787 262 | Exon 8 I198T | G | 0.06 | 0.09 | 0.07 | 0.08 | 0.07 |
| RS6899519 | 46 789 859 | Intron | C | 0.34 | 0.29 | 0.21 | 0.29 | 0.27 |
| RS1362931 | 46 790 038 | Intron | T | 0.15 | 0.18 | 0.24 | 0.17 | 0.17 |
| RS1805017 | 46 792 181 | Exon 5 R92H | A | 0.34 | 0.28 | 0.21 | 0.28 | 0.27 |
| RS6929105 | 46 793 245 | Intron | T | 0.21 | 0.28 | 0.31 | 0.26 | 0.25 |
| RS12195701 | 46 795 378 | Intron | T | 0.19 | 0.16 | 0.13 | 0.16 | 0.20 |
| RS3799863 | 46 795 750 | Intron | T | 0.04 | 0.06 | 0.05 | 0.05 | 0.06 |
| RS3799862 | 46 795 890 | Intron | G | 0.21 | 0.28 | 0.30 | 0.26 | 0.24 |
| RS12528807 | 46 804 466 | Intron | C | 0.06 | 0.07 | 0.05 | 0.07 | 0.08 |
| RS9381475 | 46 807 251 | Intron | T | 0.22 | 0.20 | 0.14 | 0.19 | 0.21 |
| RS1421378 | 46 811 472 | 3′-UTR | G | 0.49 | 0.49 | 0.38 | 0.48 | 0.44 |
| RS1421379 | 46 813 953 | 3′-UTR | A | 0.20 | 0.19 | 0.14 | 0.18 | 0.20 |
| RS9349373 | 46 814 552 | 3′-UTR | A | 0.19 | 0.19 | 0.15 | 0.18 | 0.20 |
| RS1862008 | 46 818 238 | 3′-UTR | G | 0.20 | 0.19 | 0.15 | 0.18 | 0.20 |
RS numbers for each SNP is listed, followed by chromosomal location, gene location and minor allele for each SNP. Minor allele frequency is listed for control samples, YA, OA, MI subset and GENECARD probands (GC Proband), followed by the number of individuals in each group (n).
Figure 1.

PLA2G7 gene schematic and linkage disequilibrium (LD). Nineteen SNPs genotyped in PLA2G7 are depicted in the schematic and are placed according to their genetic location. Coding SNPs are denoted by a double solid arrow. Open arrow indicate intergenic regions, and solid arrows indicate SNPs located in intronic regions. LD plot was visualized using the program Haploview. R2 values for Caucasian control samples from CATHGEN are depicted.
Single marker association of PLA2G7 in the CATHGEN case-control sample sets
Using logistic regression modeling, nine polymorphisms were significantly associated with CAD outcome after adjusting for race and sex in the combined old affected (OA) and young affected (YA) samples (P = 0.005–0.03). However, after stratifying the combined group based on age of onset (AOO) (YAs and OAs), the significant association appears to be driven primarily by the OA subgroup (AOO > 55, n = 207), with 14 SNPs showing significant association (P = 0.0001–0.02) and an additional SNP trending toward association (P = 0.06). Using the conservative Bonferroni correction for all 19 SNPs (0.05/19), 9 SNPs fell below the significance threshold of 0.0026 (Table 2). The two coding polymorphisms, rs1051931 (A379V) and rs1805017 (R92H), were among the nine most significantly associated SNPs (P ≤ 0.0001), with V379 and the R92 alleles more frequent in the cases than in the controls. To verify that known cardiovascular risk factors were not contributing significantly to the identified association, we also adjusted for the following cardiovascular risk factors; history of hypertension, history of diabetes mellitus, history of dyslipidemia, body mass index and smoking history. Association remained significant after adjustment for these factors in the OA subjects (P= 0.0002–0.02), with five SNPs falling below the Bonferroni significance threshold (Table 2).
Table 2.
SNPs meeting Bonferroni threshold for significance in the CATHGEN sample set for 19 SNPs
| SNP | Young affected (n = 599) | Old affected (n = 207) | All affected (n = 806) | MI (n = 425) | GC Proband (n = 718) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| P-value* | OR | P-value* | OR | P-value* | OR | P-value* | OR | P-value* | OR | |
| Adjusted for race and sex | ||||||||||
| RS12528857 | 0.02 | 0.70 | 0.002 | 0.56 | 0.004 | 0.67 | 0.02 | 0.69 | 0.85 | 1.02 |
| RS1051931 | 0.01 | 1.50 | 0.0001 | 2.10 | 0.002 | 1.61 | 0.002 | 1.68 | 0.08 | 1.30 |
| RS2216465 | 0.05 | 0.79 | 0.001 | 0.59 | 0.008 | 0.74 | 0.01 | 0.72 | 0.04 | 0.79 |
| RS6899519 | 0.03 | 0.76 | 0.0001 | 0.51 | 0.002 | 0.68 | 0.006 | 0.69 | 0.15 | 0.86 |
| RS1362931 | 0.05 | 1.37 | 0.002 | 1.82 | 0.02 | 1.45 | 0.01 | 1.52 | 0.21 | 1.21 |
| RS1805017 | 0.01 | 0.73 | 0.0001 | 0.50 | 0.0007 | 0.66 | 0.0008 | 0.63 | 0.00 | 0.70 |
| RS6929105 | 0.002 | 1.55 | 0.0001 | 2.10 | 0.0004 | 1.62 | 0.0009 | 1.66 | 0.07 | 1.23 |
| RS3799862 | 0.00 | 1.52 | 0.0002 | 1.97 | 0.0006 | 1.58 | 0.0008 | 1.66 | 0.02 | 1.36 |
| RS9381475 | 0.16 | 0.81 | 0.0006 | 0.51 | 0.02 | 0.73 | 0.03 | 0.71 | 0.34 | 0.88 |
| Adjusted for race, sex and cardiovascular risk factors | ||||||||||
| RS12528857 | 0.10 | 0.77 | 0.01 | 0.59 | 0.02 | 0.71 | 0.09 | 0.75 | 0.63 | 1.07 |
| RS1051931 | 0.10 | 1.33 | 0.003 | 1.85 | 0.02 | 1.47 | 0.02 | 1.52 | 0.22 | 1.23 |
| RS2216465 | 0.08 | 0.79 | 0.004 | 0.61 | 0.01 | 0.73 | 0.01 | 0.71 | 0.06 | 0.79 |
| RS6899519 | 0.09 | 0.79 | 0.0003 | 0.50 | 0.007 | 0.70 | 0.01 | 0.69 | 0.21 | 0.87 |
| RS1362931 | 0.44 | 1.15 | 0.02 | 1.62 | 0.16 | 1.25 | 0.13 | 1.33 | 0.28 | 1.20 |
| RS1805017 | 0.04 | 0.75 | 0.0002 | 0.49 | 0.003 | 0.67 | 0.002 | 0.62 | 0.01 | 0.71 |
| RS6929105 | 0.11 | 1.29 | 0.0009 | 1.92 | 0.02 | 1.40 | 0.03 | 1.43 | 0.09 | 1.23 |
| RS3799862 | 0.09 | 1.30 | 0.002 | 1.81 | 0.02 | 1.40 | 0.02 | 1.47 | 0.06 | 1.33 |
| RS9381475 | 0.35 | 0.86 | 0.0007 | 0.48 | 0.05 | 0.74 | 0.04 | 0.70 | 0.62 | 0.93 |
Odds ratios (OR) and P-values were obtained using standard logistic regression and are reported for all CATHGEN case samples, as well as GENECARD (GC) probands relative to the CATHGEN old normal controls (ON). Bolded numbers indicate SNPs meeting Bonferroni significance. YA, young affected; OA, old affected; AA, all affected; MI, CATHGEN samples with MI.
Nine SNPs were associated in the YA subgroup (AOO < 55, n = 599), although association was not as strong as that seen in the OA subgroup (P = 0.002–0.05). None of the associated SNPs in the YA subgroup met the Bonferroni threshold value of significance. However, the directions of association identified by the odds ratios in the YA group were consistent for all SNPs identified in the OA group (Fig. 2).
Figure 2.
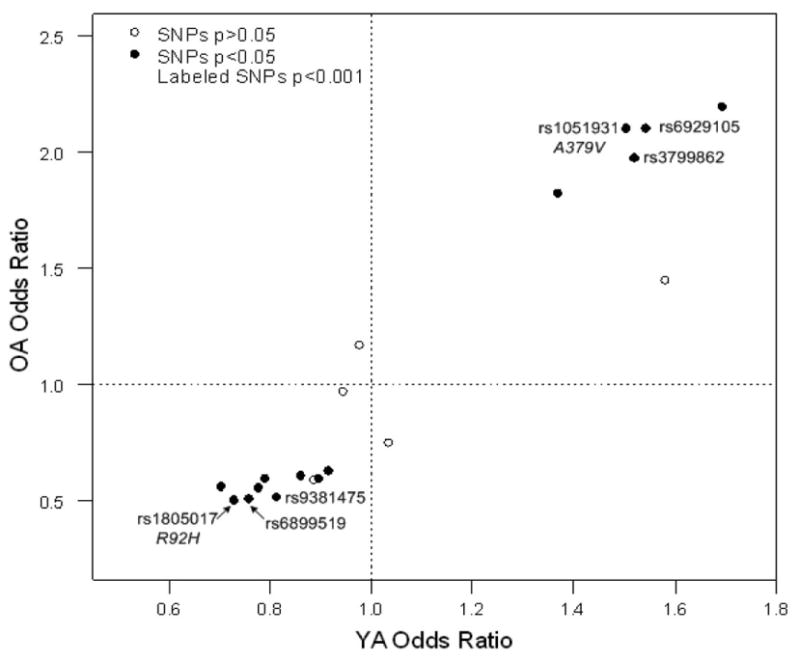
PLA2G7 genotype odds ratio plot for old affected (OA) and young affected (YA) individuals. Solid circles represent SNPs with P-values of <0.05 in the OA, open circles represent P-values of >.0.05 in OA. Circles with a SNP name on graph indicate SNPs with P < 0.001 in OA. Odds ratios are adjusted for race and sex.
To validate the findings of Liu et al. (20), subjects were stratified into subgroups based upon documented history of MI (n = 425). Interestingly, the results of this analysis closely mimicked that of the OA sample set in regards to the associated SNPs in the MI subgroup (Table 2). Ten SNPs were identified as being associated (P < 0.05) with an additional two SNPs trending toward association (0.05 < P < 0.1). Rs1805017 (R92H), rs6929105 and rs3799862 were the most significantly associated SNPs (P = 0.0008–0.0009), with the odds ratios remaining consistent with those found in both young affecteds and old affecteds. Additionally, rs1051931 (A379V) remained consistently associated in the MI subgroup (P = 0.002), with the V379 allele identified more often in individuals presenting with an MI than controls, consistent with seen in the OA and YA sample set. Four SNPs, rs1051931 (A379V), rs1805017 (R92H), rs6929105 and rs3799862, met the Bonferroni threshold for significance. The results remained consistent after adjustment for known CAD risk factors (Table 2).
As significant differences were seen between the YA and OA subgroups, we analyzed the SNPs using AOO as an outcome to more accurately determine whether the association was due to older AOO or disease severity. No significant association was identified with AOO, suggesting that the SNPs may be detecting an association with disease severity (data not shown).
Owing to the potential for population stratification, data were also analyzed by stratifying the sample set based on race (Caucasian and African American only). The race-stratified analyses demonstrated consistent OR for most SNPs although fewer SNPs were significant for the African American due to the reduction in sample size (n = 17 African-American OA subset; data not shown). Thus, the associations identified with polymorphisms in PLA2G7 do not appear to be ethnicity specific, although these results should be verified in a larger minority cohort.
We evaluated several models of multiple marker association to examine whether the observed single SNPs associations are independent or driven by the low LD observed in our dataset. The assessment of independent effects through backward stepwise logistic regression modeling revealed four SNPs (rs974670, rs1051931 (A379V), rs9381475 and rs1421378) independently associated with CAD case status after adjustment for known cardiovascular risk variables (P = 0.01, 0.0008, 0.01 and 0.02, respectively). We also examined evidence for multiplicative interactions between these SNPs. No interaction terms were significant when added to the SNP main effects models suggesting that statistical interactions, should they exist at all, are too modest to be detected with these sample sizes. Finally, we examined haplotype analyses as specific cases of a genetic interaction model. As the LD between these SNPs is modest (r2 < 0.45), specific combinations of SNP alleles may represent an underlying risk associated with a distinct haplotype background. We used Haplo.Stats to construct all pairwise (Supplementary Material, Table S1) and four-way haplotypes from the four SNPs (Supplementary Material, Table S2) from the backward stepwise regression. Haplotype results suggest that two SNPs, A379V and rs9381475, are consistently contributing to the significant haplotype P-values and thus appear to be driving the association. These multi-SNP models do not appear to strengthen the associations identified in the single SNP analysis and do not further refine the multiple-observed single SNP associations, thus suggesting that the effects of each SNP, particularly the four SNPs identified in the backward stepwise logistic regression, are independent.
Single SNP association analysis in the GENECARD proband dataset
We constructed a third case set comprised of US GENECARD (GC) probands (AOO men ≤51 and women ≤56) to compare with our CATHGEN controls. All probands had a documented history of CAD, as defined in Meterials and methods section. Modest association was detected with our GC probands. Three SNPs were identified as being significantly associated (P < 0.05), whereas two additional SNPs trended toward association (0.05 < P < 0.1). Although none of the SNPs met the strict criteria for significance using the Bonferroni correction, the three significant and two trending SNPs in GC probands were consistent with the SNPs that met Bonferroni correction in the OA CATHGEN sample set (Table 2). The direction of effect identified by the OR remained consistent between all associated SNPs and that seen in the CATHGEN case set.
Single SNP analysis using family-based association analysis in GENECARD families
Three SNPs (rs9381475, rs1421379 and rs1862008) showed significant association in GENECARD 1 (G1) families using association in the presence of linkage (APL) analysis (Table 3). However, of those three SNPs, only one SNP (rs9381475) replicated the association seen in the CATHGEN Bonferroni corrected analysis. This association was not replicated in the second set of GENECARD families (G2) or in all GENECARD families combined. It should be noted that the PLA2G7 gene is not located in a previously identified region of linkage in the GENECARD study, suggesting association results may have a modest effect size with or without substantial genetic heterogeneity.
Table 3.
Family-based association analysis in the GENECARD family study
| SNP | Minor Allele | MAF | Genecard 1 APL_PVAL | Genecard 2 APL_PVAL | Genecard 1 and 2 APL_PVAL |
|---|---|---|---|---|---|
| RS974670 | T | 0.38 | 0.964 | 0.398 | 0.520 |
| RS12528857 | A | 0.22 | 0.160 | 0.658 | 0.108 |
| RS1051931 | A | 0.20 | 0.595 | 0.376 | 0.407 |
| RS2216465 | G | 0.33 | 0.704 | 0.560 | 0.536 |
| RS4498351 | A | 0.39 | 0.650 | 0.290 | 0.167 |
| RS1805018 | G | 0.05 | 0.752 | 0.948 | 0.419 |
| RS6899519 | C | 0.23 | 0.346 | 0.515 | 0.958 |
| RS1362931 | T | 0.19 | 0.630 | 0.974 | 0.341 |
| RS1805017 | A | 0.24 | 0.780 | 0.750 | 0.812 |
| RS6929105 | T | 0.25 | 0.731 | 0.928 | 0.990 |
| RS12195701 | T | 0.20 | 0.174 | 0.812 | 0.159 |
| RS3799863 | T | 0.05 | 0.767 | 0.656 | 0.276 |
| RS3799862 | G | 0.34 | 0.575 | 0.769 | 0.841 |
| RS12528807 | C | 0.08 | 0.078 | 0.682 | 0.626 |
| RS9381475 | T | 0.20 | 0.002 | 0.094 | 0.530 |
| RS1421378 | G | 0.42 | 0.454 | 0.683 | 0.816 |
| RS1421379 | A | 0.20 | 0.014 | 0.808 | 0.392 |
| RS9349373 | A | 0.13 | 0.055 | 0.361 | 0.219 |
| RS1862008 | G | 0.19 | 0.004 | 0.066 | 0.502 |
For each SNP, APL P-values are reported for GENECARD 1, GENECARD 2 and GENECARD 1 and 2 combined. Minor allele and minor allele frequency (MAF) is reported for the combined GENECARD dataset. Bolded SNPs indicate P ≤ 0.05.
Aorta differential expression and genotyping results
Aorta expression from the Affymetrix U95 chip using the PLA2G7 gene tag, 37068_at, was compared between aorta case and aorta control RNA samples (n = 104) using logistic regression analysis. PLA2G7 expression was not differentially expressed between diseased and non-diseased aortas in either the Sudan IV staining or raised lesion models (P > 0.05). Although none of the SNP genotypes significantly contributed to differential PLA2G7 expression levels, rs1805018 (I198T) trended toward significant association (P = 0.07, Supplementary Material, Table S3). Genotypes were also analyzed for the association with disease in the aorta (diseased versus non-disease), with no statistical significance detected (data not shown).
Discussion
This current genetic study of PLA2G7 represents, to our knowledge, the first comprehensive htSNP analysis of PLA2G7 to accurately capture the polymorphic variation of the gene in relation to cardiovascular disease phenotypes. The strengths of the study include an extensively phenotyped case–control cohort with documented cardiovascular disease, a large sample size which allows for sufficient power for stratification based on age of cardiovascular disease onset or clinical presentation, and a family-based cohort with documented early age of CAD onset. We have captured the genetic diversity of PLA2G7 using haplotype tagSNPs, have assessed the genetic contribution of known putatively functional coding polymorphisms and have used multiple statistical analyses to define further the dependent and independent SNP associations. Multiple individual polymorphisms showed significant associations, which continued to be significant after detailed evaluation of the extent of interaction among polymorphisms. We have identified several single SNP and haplotype associations that survive conservative Bonferroni comparisons, with a subset also surviving Bonferroni correction in different sample sets. Additionally, we were able to replicate our findings in multiple independent datasets, thus identifying a true genetic association (25).
Our results strongly support the findings of multiple genetic studies in which PLA2G7 coding polymorphisms were associated with cardiovascular disease phenotypes (20,21,23,26). The majority of previous studies have consistently identified genetic association of A379V with cardiovascular disease, which is supported by our findings. However, there remains substantial discordance between the published studies regarding the direction of the genetic effect. Given the identification of multiple, additional significantly associated SNPs, it is not surprising that the SNP effects are not consistent from one sample to the next. One possible explanation for the variability found in the previous studies may be explained by the CAD classification criteria used in our sample set. Individuals in the CATHGEN study were stratified by age and disease severity as assessed by coronary angiography. Individuals in the OA subgroup not only had an increased AOO compared with the YAs, but also had significantly more disease, (CADi ≥ 72, indicating >95% stenosis in at least three vessels). No other PLA2G7 genetic study, to our knowledge, has separated case subjects into subsets based on age or severity of disease. On the basis of our findings, SNPs in PLA2G7 appear to be associated with increasing disease severity, as indicated by the strength of association identified in our OA subgroup and lack of association with AOO as an outcome. Additionally, the results in the MI subgroup nearly mimicked that seen in the OA subgroup, again indicating a disease severity effect distinct from age. Thus, the discrepancy identified in previous studies may be confounded by the lack of stratification of severely diseased subgroups.
Three known nonsynonymous polymorphisms were genotyped in our study. Although all three polymorphisms were associated, the R92H and A379V variants were more significantly associated with CAD than the I198T polymorphism. Several studies have investigated the effect of these variants on the enzyme activity. In one such study, Ninio et al. (20), identified an apparent increase in plasma PLA2G7 activity with the V379 allele, although it was noted that the genotype explained <1% of the variability of plasma PLA2G7 activity after adjustment for main confounders. A separate study by Kruse et al. (22), investigating the effects of all three coding polymorphisms on PAF activity in asthmatics, found that the V379and T198 variants showed an increase in Km values and Vmax values, whereas no differences were noted for either allele of R92H. However, H92, T198 and V379 variants all resulted in decreased substrate affinity of PAF, thus potentially prolonging the activities of this potent inflammatory protein. These studies support our findings for the risk amino acids V379and T198 in our sample, but are inconclusive with regards to our findings of the risk R92. Although we were unable to test the effects of the coding SNPs and plasma PLA2G7 enzymatic activity, we were able to obtain aorta tissue samples. Our results using mRNA expression studies in diseased and non-diseased aorta samples do not indicate differential gene expression in diseased arteries nor do they indicate any SNP influence on disease burden in the aorta. However, it should be noted that the number of RNA samples available from the aortas was small, thus giving us limited power. Recently, Kolodgie et al. (15) reported low or absent PLA2G7 expression in early lesions of human coronary arteries, contrasted by high PLA2G7 expression in thin-cap fibroatheromas and ruptured plaques. Our aorta samples are more likely to reflect early atherosclerosis phenotypes with very limited contribution of severe disease represented by extensive atheroma or ruptured plaques. Multiple studies have indicated an association between genotype and PLA2G7 activity; however, no study to our knowledge has analyzed the effect of these polymorphisms on PLA2G7 expression in the aorta. On the basis of these results, polymorphisms in PLA2G7 do not appear to modulate PLA2G7 expression in the aorta, although a larger dataset is warranted to confirm this finding.
On the basis of our findings, and those of published genetic and functional studies of PLA2G7, we postulate that individuals carrying a combination of risk alleles, including the risk variants R92 and V379, have not only have an increase in the activity of the enzyme of PLA2G7, which could lead to an increase in the amount of inflammatory molecules such as lyso-PC and free fatty acids, but also an increase in PAF activity due to lower substrate affinity, thus prolonging the inflammatory actions of this potent inflammatory molecule. Taken together, these pro-inflammatory actions could lead to the increased risk of developing CAD. As PLA2G7 levels tend to increase in individuals with a more severe CAD phenotype, it is not surprising that we find the strongest association in individuals with a more severe phenotype. Additional functional studies are warranted to confirm the effect of the multiple risk alleles on the enzymatic activity. In silico analysis failed to identify that any of the genotyped SNPs were located in putative transcription binding sites, enhancer elements, splicing junctions, intron/exon boundaries, triplex repeats or additional known functional motifs. Additionally, none of the SNPs, including the three coding SNPs, were located in highly conserved regions by multiple species sequence comparison (http://genome.ucsc.edu). Thus, the presence of multiple, independent, associated SNPs suggests the relationship between genetic variation, PLA2G7 activity, and ultimately CAD outcome is very complex.
In addition to candidate gene association studies, PLA2G7 has been linked to cardiovascular disease by localization to linkage peaks identified by three genome-wide screens investigating the genetic causes to carotid artery intima media thickness (27), elevated HDL cholesterol levels in premature atherosclerosis (28) and HDL levels (29), respectively. The Wellcome Trust Case Control Consortium (WTCCC) genome-wide association study (GWAS) for CAD included three SNPs in PLA2G7 that did not show association with CAD (P > 0.05) (30). The two PLA2G7 SNPs in common between the GWAS and our own study were not strongly associated in either our CATHGEN or GENECARD sample set and are not in LD (r2 < 0.15) with any of the highly associated SNPs presented here. This illustrates a potential limitation of the GWAS data that may reveal important functional candidates (31,32), whereas other potentially important genes may be missed. Regardless of the method used to identify genetic association, by GWAS or focused candidate analysis (e.g. mouse studies or expression studies), candidate genes require focused and complete genetic analysis. An alternative approach to GWAS to identifying susceptible candidate genes is to use a genomic convergence approach, which gathers multiple lines of evidence, including replication in phenotypically similar but independent populations, linkage, gene expression studies, proteomics, etc. to assess the strength of the identified association. This approach has been used and successfully applied to a variety of genetic association studies (33,34).
In sum, we have identified multiple SNPs within the PLA2G7 gene that are associated with CAD in two independent CAD cohorts. These results are consistent across ethnic and phenotypic designations. Given the multitude of studies indicating PLA2G7 levels as an independent risk factor in the development of CAD, it is imperative that researchers discover the causative nature of variation among individuals. Multiple studies suggest measurement of PLA2G7 levels to be included as additional risk predictors in the diagnosis of CAD. Moreover, with the recent surge of interest in identifying a set of risk polymorphisms to be included in CAD-risk profiling, identifying a set of true risk susceptibility markers is mandatory for clinical success. Previous genetic studies of PLA2G7 have focused on three coding variants, but all have largely disregarded the remaining genetic variability of this highly potent CAD inflammatory marker. Thus, the importance of the present study recognizes the need to investigate all variable regions of PLA2G7 and determine whether multiple variants independent of the previously reported coding SNPs infer significant CAD risk. We have concluded that risk alleles in PLA2G7 may have an additive effect and that the presence of multiple risk alleles appears to confer strongest genetic risk. On basis of significant associations identified with multiple independent SNPs, it is highly plausible that multiple SNPs may act in concert to contribute to a dysfunctional protein product. Further investigation is warranted to identify the biological mechanisms by which this protein is contributing to CAD and determine the true functional variants associated with CAD for potential use in CAD genetic risk profiling and drug development.
Materials and Methods
Subject selection
CAD case–control (CATHGEN)
CATHGEN participants were recruited through the cardiac catheterization laboratories at Duke University Hospital (Durham, NC, USA) with approval from the Duke Institutional Review Board. All participants undergoing catheterization were offered participation in the study and signed informed consent. Medical history and clinical data were collected and stored in the Duke Databank for Cardiovascular Disease maintained at the Duke Clinical Research Institute (35).
Table 4 describes the clinical characteristics of the CATHGEN and GENECARD participants. Controls and cases were chosen on the basis of extent of CAD as measured by the CAD index (CADi). CADi is a numerical summary of the coronary angiographic data that incorporates the extent and anatomical distribution of coronary disease and is based upon outcomes at follow-up (36). In our cohort, CADi has been shown to be a better predictor of clinical outcome than more traditional anatomical characterizations of the extent of CAD (37). Affected status was determined by the presence of significant CAD defined as a CADi of ≥32 (38), the equivalent of requiring at least a 95% stenosis in at least one major epicardial vessel. For patients >55 years of age, a higher CADi threshold (CADi ≥ 74; the equivalent of three vessel disease with at least a 95% lesion in the proximal left anterior descending coronary artery) was used to adjust for the higher baseline extent of CAD in this group. Medical records were reviewed to determine the AOO of CAD, i.e. the age at first documented surgical or percutaneous coronary revascularization procedure, MI or cardiac catheterization meeting the above-defined CADi thresholds. The CATHGEN cases were stratified into two groups for the analysis: a YA group (AOO ≤ 55 years), which provides a consistent comparison for the GENECARD family study and an OA group (AOO > 55 years). Additionally, individuals from each stratified group (YA and OA) were further stratified based on prior myocardial event. Myocardial event was defined as history of MI, as based on patient report and review of the medical records. An MI is recorded only when the patient has documentation of typical evolutionary ECG or serum myocardial enzyme changes with a consistent clinical history. An MI was also recorded when it was a result of or related to the index catheterization or documented MI at follow-up. Controls were defined as ≥60 years of age and having no history of MI or CAD (in the past, related to index enrollment catheterization, or on follow-up), and no history of heart transplant, peripheral or cerebrovascular disease, surgical or percutaneous coronary revascularization and no history of cardiomyopathy (no history of depressed ejection fraction (EF), and EF > 50% as identified on left ventriculogram if done during cardiac catheterization or on echocardiogram done within 1 month of index catheterization). Although these controls may not be representative of population-based controls (i.e. they presented for cardiac catheterization), the angiographic data are key for establishing a true control sample free of coronary atherosclerosis.
Table 4.
Clinical characteristics of CATHGEN and GENECARD proband participants
| Category | Characteristic | CATHGEN | GENECARD | |||||
|---|---|---|---|---|---|---|---|---|
| Young affected (n = 599) | Old affected (n = 207) | Unaffected controls (n = 266) | Probands (n = 757) | Genecard 1 (n = 1127) | Genecard 2 (n = 1757) | Total Genecard set (n = 2862) | ||
| Mean age of onset (SD) | 46.3b | 65.7 | NA | 43.6a | NA | NA | NA | |
| Race | Caucasian (%) | 70.51b | 89.86a | 74.69 | 85.49a | 92.9 | 88.4 | 90.2 |
| African American (%) | 22.37b | 8.7a | 20 | 7.65a | 2.7 | 4.6 | 3.8 | |
| American Indian (%) | 5.76b | 1.45a | 4.08 | 2.77a | 3.8 | 4.6 | 4.3 | |
| Other (unknown) (%) | 1.36b | 0a | 1.22 | 4.09a | 0.6 | 2.4 | 1.7 | |
| Gender | Male (%) | 78.63a | 72.46a | 39.47 | 68.73a | 66.0 | 61.0 | 62.8 |
| Family history of CAD (%) | 54.09a,b | 42.03a | 22.18 | 100a | 89.2 | 72.5 | 78.9 | |
| Body mass index, kg/m2 (SD) | 30.8 (6.5)a,b | 28.4 (6.5) | 29 (7.7) | 31.6 (7.6) | 29.5 (5.7) | 30.0 (7.6) | 29.8 (7.0) | |
| Ever smoked (%) | 68.61a,b | 52.17a | 39.10 | 26.65a | 74.7 | 71.6 | 72.8 | |
| History of diabetes (%) | 33.06a | 31.4a | 17.29 | 24.9a | 20.1 | 19.6 | 19.8 | |
| History of hypertension (%) | 69.62b | 77.29a | 63.53 | 63.3a | 54.2 | 53.7 | 53.9 | |
| History of coronary artery bypass graft | 36.23%b | 51.21% | NA | 48.88%a | 38.9% | 32.9% | 35.4% | |
| History of myocardial infarction | 313b | 81 | NA | 480a | 674 | 1023 | 1683 | |
| Mean systolic blood pressure, mmHg (SD) | 139.7 (23.9)a,b | 150.1 (26.1) | 149.5 (23.7) | 126.7 (19.3)a | 146.1 (24.7) | 137.9 (23.0) | 138.2 (23.1) | |
| Mean diastolic blood pressure, mmHg (SD) | 78.1 (14.3) | 77.2 (12.7) | 76.6 (13.4) | 76.1 (12.1) | 81.3 (11.0) | 81.4 (12.3) | 81.4 (12.2) | |
| Total cholesterol, mg/dl (SD) | 194.7 (62.8)b | 175.6 (39.9)a | 193.4 (49.9) | 219.3 (56.4)a | 188.9 (116.9) | 208.2 (81.4) | 199.1 (100.7) | |
| Low-density lipoprotein, mg/dl (SD) | 111.9 (42.7) | 104 (33.3) | 108.7 (36.4) | 142.1 (73.9)a | 87.2 (97.4) | 113.4 (76.7) | 101.2 (88.5) | |
| High-density lipoprotein, mg/dl (SD) | 40.2 (12)aa,b | 44.2 (14.5)a | 52 (18) | 43.5 (50.9) | 25.2 (35.6) | 37.2 (37.0) | 31.6 (37.0) | |
| Triglycerides, mg/dl (SD) | 227.2 (268.8)a,b | 157.4 (88.3) | 159.6 (137.6) | 240.1 (310.3) | 170.5 (194.5) | 215.7 (278.6) | 193.6 (241.5) | |
Clinical characteristics are listed for each subgroup of the CATHGEN sample set (young affected, old affected and unaffected controls) as well as for the GENECARD probands and the entire GENECARD family cohort.
Significant difference (P < 0.05) between GENECARD probands and Cathgen young affected cases or CATHGEN cases and controls.
Significant difference (P < 0.05) between CATHGEN YA and OA samples.
Genetics of Early Onset Cardiovascular Disease
The Genetics of Early Onset Cardiovascular Disease (GENECARD) study is a multi-center collaborative effort among investigators at Duke University (Durham, NC, USA), Vanderbilt University (Nashville, TN, USA), and five additional international sites. Details of the design of the study are described in detail elsewhere (39). Briefly, families were recruited throughout a 4-year period, from March 1998 to March 2002. All study subjects signed a consent form approved by the responsible institutional review board or local ethics committee. Qualified participants were required to have at least one of the following: MI or unstable angina (acute coronary syndrome), coronary catheterization demonstrating significant disease (at least a 50% stenosis in one major epicardial coronary vessel), interventional coronary revascularization procedure (percutaneous transluminal coronary intervention or coronary artery bypass grafting) or a functional test documenting reversible myocardial ischemia with cardiac imaging. The qualifying event or procedure must have occurred at or before age 51 years in men and 56 years in women. For families to be eligible for the inclusion in the study, they were required to include at least two siblings, each of whom met the above-described diagnostic criteria and were available for sampling and data collection. For family-based association studies, unaffected siblings were also collected, defined as having no clinical evidence of CAD and age > 55 years for men or > 60 years for women. Owing to the timing of the collection of the participants, GENECARD families were originally split into two groups for linkage and association analysis. GENECARD 1 (G1) contained 420 families (individuals = 1167) and GENECARD 2 (G2) contained 373 families (individuals = 1757). Separating these families into two distinct groups provided a consistent validation set to test associated SNPs as all families were recruited according to the same criteria. Thus analyses are reported for both groups (G1 and G2) separately, and also as a combined cohort for maximum statistical power.
Aorta sample collection
A collection of aorta samples (n = 104) were collected for use in microarray expression studies. Aortas were harvested from donor hearts and prepared as described previously (40). In addition to preparation for mRNA studies, DNA from 205 samples was also extracted from aorta tissue in the Center for Human Genetics (CHG) DNA Bank to be used in genetic association studies. Disease burden was measured in the aorta using procedures developed by the Pathobiological Determinants of Atherosclerosis in the Young study (PDAY) (41). Procedures consisted of measuring a proportion of the aorta with Sudan IV staining (early atherosclerotic lesions) and a proportion of the aorta with raised lesions (severe disease). In addition to the disease burden measures, normalized expression levels on the Affymetrix U95A microarray chip using PLA2G7 tag 37068_at were used as phenotypes for genetic analysis. As harvested tissue was obtained from deceased heart donors, the clinical data associated with these aortas are very limited and consists of age, sex and race.
SNP selection and genotyping methods
A minimal set of htSNPs using MAF ≥5% and r2 ≥ 0.7 were selected using the SNPselector program (42) to cover the predicted LD structure of the PLA2G7 gene in both Caucasian and African-American populations. SNPselector incorporates available information from HapMap (www.hapmap.org), The SNP Consortium (http://snp.cshl.org), Japanese SNP database (http://snp.ims.u-tokyo.ac.jp/index.html) and the Affymetrix 120K SNP (http://www.affymetrix.com/index.affx) to generate the most likely LD bins and determines the optimal tagSNP for each bin. Genomic DNA for the GENECARD and CATHGEN samples was extracted from whole blood and purified using the PureGene system (Gentra Systems, Minneapolis, MN, USA). SNPs were genotyped using the ABI 7900HT Taqman SNP genotyping system (Applied Biosystems, Foster City, CA, USA), which incorporates a standard PCR-based, dual fluor, allelic discrimination assay in a 384-well plate format with a dual laser scanner. Allelic discrimination assays were purchased through Applied Biosystems. Quality control samples, composed of 12 reference genotype controls, two Center d'Etude du Polymorphism Humain (CEPH) pedigree individuals and one no-template sample, were included in each quadrant of each 384-well plate. The quality control samples were used to provide duplicated samples within one quadrant, across quadrants within one plate, and across plates. The results of the CEPH and quality control samples were compared to identify possible sample plating errors and genotype inconsistencies. Genotype call rates were >95% for all SNPs. SNPs that did not meet this criterion or had quality control errors were not used in the analysis. No departures from Hardy Weinberg Equilibrium were observed.
Statistical analysis
Linkage disequilibrium between pairs of SNPs was assessed using the Graphical Overview of Linkage Disequilibrium (GOLD) package (43) and were visualized using the Haploview program (44). Genotypic and allelic association in CATHGEN and GENECARD probands from the USA was examined using multivariable logistic regression modeling adjusted for race and sex, and also for race, sex, and known CAD risk factors (history of hypertension, history of diabetes mellitus, history of dyslipidemia, body mass index and smoking history) as covariates. These adjustments could hypothetically allow us to control for competing genetic pathways that are independent risk factors for CAD, thereby allowing us to detect a separate CAD genetic effect. SAS 9.1 (SAS Institute, Cary, NC, USA) was used for statistical analysis. The Haplo.Stats package was used to estimate and test for association of haplotypes in CATHGEN. Haplo.Stats expands on the likelihood approach to account for haplotype ambiguity in case-control studies by using a generalized linear model to test for haplotype association including adjustment for covariates (45). Haplo.Stats computes score statistics for the components of the genetic vectors, such as individual haplotypes. We applied backward stepwise logistic regression as implemented in SAS version 9 software (SAS, Inc. Cary, NC, USA) to reduce the number of SNPs in our model and to identify SNPs with the strongest effects, after taking into account CAD risk factors and other SNPs in the model. Family-based association in GENECARD samples was tested using the APL test (46). The APL software appropriately accounts for the non-independence of affected siblings and calculates a robust estimate of the variance.
Supplementary Material
Supplementary Material is available at HMG Online.
Acknowledgments
We thank the subjects in the GENECARD and CATHGEN studies for their participation. Wewould also like to acknowledge the essential contributions of the following individuals to making this publication possible: Elaine Dowdy, the GENECARD Investigators Network, the CATHGEN Steering Committee Members (Chris Granger, Mike Sketch, Mark Donahue, Geoff Ginsburg and Kristin Newby), Charlotte Nelson, Paul Hofmann and Judy Stafford at the Duke Clinical Research Institute, Ben Lambertson, Liyong Wang, Jessica Connelly, and the support staff at the Center for Human Genetics.
Funding: This work was supported by NIH grants HL073389 (Hauser) and HL73042-04 (Goldschmidt, Kraus). NIH grants HL073389 and HL73042-04.
Footnotes
Conflict of Interest statement. None declared.
Publisher's Disclaimer: This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels—The Pravastatin Inflammation/CRP Evaluation (PRINCE): a randomized trial and cohort study. J Am Med Assoc. 2001;286:64–70. doi: 10.1001/jama.286.1.64. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. Circulation. 1999;100:230–235. doi: 10.1161/01.cir.100.3.230. [DOI] [PubMed] [Google Scholar]
- 4.Ridker PM, Rifai N, Lowenthal SP. Rapid reduction in C-reactive protein with cerivastatin among 785 patients with primary hypercholesterolemia. Circulation. 2001;103:1191–1193. doi: 10.1161/01.cir.103.9.1191. [DOI] [PubMed] [Google Scholar]
- 5.Steinberg D. Lewis A. Conner memorial lecture—oxidative modification of LDL and atherogenesis. Circulation. 1997;95:1062–1071. doi: 10.1161/01.cir.95.4.1062. [DOI] [PubMed] [Google Scholar]
- 6.Guerra R, Zhao BR, Mooser V, Stafforini D, Johnston JM, Cohen JC. Determinants of plasma platelet-activating factor acetylhydrolase: heritability and relationship to plasma lipoproteins. J Lip Res. 1997;38:2281–2288. [PubMed] [Google Scholar]
- 7.Packard CJ, O'Reilly DSJ, Caslake MJ, McMahon AD, Ford I, Cooney J, Macphee CH, Suckling KE, Krishna M, Wilkinson FE, et al. Lipoprotein-associated phospholipase A(2) as an independent predictor of coronary heart disease. N Engl J Med. 2000;343:1148–1155. doi: 10.1056/NEJM200010193431603. [DOI] [PubMed] [Google Scholar]
- 8.Tjoelker LW, Eberhardt C, Unger J, Letrong H, Zimmerman GA, McIntyre TM, Stafforini DM, Prescott SM, Gray PW. Plasma platelet-activating-factor acetylhydrolase is a secreted phospholipase A(2) with a catalytic triad. J Biol Chem. 1995;270:25481–25487. doi: 10.1074/jbc.270.43.25481. [DOI] [PubMed] [Google Scholar]
- 9.Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR, Heiss G, Sharrett AR. Lipoprotein-associated phospholipase A(2), high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2004;109:837–842. doi: 10.1161/01.CIR.0000116763.91992.F1. [DOI] [PubMed] [Google Scholar]
- 10.Winkler K, Winkelmann BR, Scharnagl H, Hoffmann MM, Grawitz AB, Nauck M, Bohm BO, Marz W. Platelet-activating factor acetylhydrolase activity indicates angiographic coronary artery disease independently of systemic inflammation and other risk factors—the Ludwigshafen risk and cardiovascular health study. Circulation. 2005;111:980–987. doi: 10.1161/01.CIR.0000156457.35971.C8. [DOI] [PubMed] [Google Scholar]
- 11.Nair S, Lee YH, Rousseau E, Cam M, Tataranni PA, Baier LJ, Bogardus C, Permana PA. Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia. 2005;48:1784–1788. doi: 10.1007/s00125-005-1868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karra R, Vemullapalli S, Dong CM, Herderickt EE, Song XH, Slosek K, Nevins JR, West M, Goldschmidt-Clermont PJ, Seo D. Molecular evidence for arterial repair in atherosclerosis. Proc Natl Acad Sci. 2005;102:16789–16794. doi: 10.1073/pnas.0507718102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hakkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Yla-Herttuala S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–2917. doi: 10.1161/01.atv.19.12.2909. [DOI] [PubMed] [Google Scholar]
- 14.Singh U, Zhong SM, Xiong MM, Li TB, Sniderman A, Teng BB. Increased plasma non-esterified fatty acids and platelet-activating factor acetylhydrolase are associated with susceptibility to atherosclerosis in mice. Clin Sci. 2004;106:421–432. doi: 10.1042/CS20030375. [DOI] [PubMed] [Google Scholar]
- 15.Kolodgie F, Burke A, Skorija K, Ladich E, Kutys R, Makuria AT, Virmani R. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–2529. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 16.Balta G, Gurgey A, Kudayarov DK, Tunc B, Altay C. Evidence for the existence of the PAF acetylhydrolase mutation (Va1279Phe) in non-Japanese populations: a preliminary study in Turkey, Azerbaijan, and Kyrgyzstan. Thromb Res. 2001;101:231–234. doi: 10.1016/s0049-3848(00)00394-7. [DOI] [PubMed] [Google Scholar]
- 17.Stafforini DM, Satoh K, Atkinson DL, Tjoelker LW, Eberhardt C, Yoshida H, Imaizumi T, Takamatsu S, Zimmerman GA, McIntyre TM, et al. Platelet-activating factor acetylhydrolase deficiency. A missense mutation near the active site of an anti-inflammatory phospholipase. J Clin Invest. 1996;97:2784–2791. doi: 10.1172/JCI118733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishihara M, Iwasaki T, Nagano M, Ishii J, Takano M, Kujiraoka T, Tsuji M, Hattori H, Emi M. Functional impairment of two novel mutations detected in lipoprotein-associated phospholipase A2 (Lp-PLA2) deficiency patients. J Hum Gen. 2004;49:302–307. doi: 10.1007/s10038-004-0151-6. [DOI] [PubMed] [Google Scholar]
- 19.Unno N, Sakaguchi T, Nakamura T, Yamamoto N, Sugatani J, Miwa M, Konno H. A single nucleotide polymorphism in the plasma PAF acetylhydrolase gene and risk of atherosclerosis in Japanese patients with peripheral artery occlusive disease. J Surg Res. 2006;134:36–43. doi: 10.1016/j.jss.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 20.Ninio E, Tregouet D, Carrier JL, Stengel D, Bickel C, Perret C, Rupprecht HJ, Cambien F, Blankenberg S, Tiret L. Platelet-activating factor-acetylhydrolase and PAF-receptor gene haplotypes in relation to future cardiovascular event in patients with coronary artery disease. Hum Mol Genet. 2004;13:1341–1351. doi: 10.1093/hmg/ddh145. [DOI] [PubMed] [Google Scholar]
- 21.Liu PY, Li YH, Wu HL, Chao TH, Tsai LM, Lin LJ, Shi GY, Chen JH. Platelet-activating factor-acetylhydrolase A379V (exon 11) gene polymorphism is an independent and functional risk factor for premature myocardial infarction. J Thromb Haemost. 2006;4:1023–1028. doi: 10.1111/j.1538-7836.2006.01895.x. [DOI] [PubMed] [Google Scholar]
- 22.Kruse S, Mao XQ, Heinzmann A, Blattmann S, Roberts MH, Braun S, Gao PS, Forster J, Kuehr J, Hopkin JM, et al. The Ile198Thr and Ala379Val variants of plasmatic PAF-acetylhydrolase impair catalytical activities and are associated with atopy and asthma. Am J Hum Genet. 2000;66:1522–1530. doi: 10.1086/302901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wootton PTE, Stephens JW, Hurel SJ, Durand H, Cooper J, Ninio E, Humphries SE, Talmud PJ. Lp-PLA2 activity and PLA2G7 A379V genotype in patients with diabetes mellitus. Atherosclerosis. 2006;189:149–156. doi: 10.1016/j.atherosclerosis.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Gregory SG, Hauser ER, Stenger JE, Pericak-Vance MA, Vance JM, Zuchner S, Hauser MA. SNPselector: a web tool for selecting SNPs for genetic association studies. Bioinformatics. 2005;21:4181–4186. doi: 10.1093/bioinformatics/bti682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moonesinghe R, Khoury MJ, Janssens ACJW. Most published research findings are false- but a little replication goes a long way. Plos Med. 2007;4:218–221. doi: 10.1371/journal.pmed.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abuzeid AM, Hawe E, Humphries SE, Talmud PJ. Association between the Ala379Val variant of the lipoprotein associated phospholipase A2 and risk of myocardial infarction in the north and south of Europe. Atherosclerosis. 2003;168:283–288. doi: 10.1016/s0021-9150(03)00086-8. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Yang H, Quinones MJ, Bulnes-Enriquez I, Jimenez X, De La RR, Modilevsky T, Yu K, Li Y, Taylor KD, et al. A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke. 2005;36:540–545. doi: 10.1161/01.STR.0000155746.65185.4e. [DOI] [PubMed] [Google Scholar]
- 28.Canizales-Quinteros S, Aguilar-Salinas CA, Reyes-Rodriguez E, Riba L, Rodriguez-Torres M, Ramirez-Jimenez S, Huertas-Vazquez A, Fragoso-Ontiveros V, Zentella-Dehesa A, Ventura-Gallegos JL, et al. Locus on chromosome 6p linked to elevated HDL cholesterol serum levels and to protection against premature atherosclerosis in a kindred with familial hypercholesterolemia. Circ Res. 2003;92:569–576. doi: 10.1161/01.RES.0000064174.69165.66. [DOI] [PubMed] [Google Scholar]
- 29.Francke S, Manraj M, Lacquemant C, Lecoeur C, Lepretre F, Passa P, Hebe A, Corset L, Yan SL, Lahmidi S, et al. A genome-wide scan for coronary heart disease suggests in Indo- Mauritians a susceptibility locus on chromosome 16p13 and replicates linkage with the metabolic syndrome on 3q27. Hum Mol Genet. 2001;10:2751–2765. doi: 10.1093/hmg/10.24.2751. [DOI] [PubMed] [Google Scholar]
- 30.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, et al. Genomewide association analysis of coronary artery disease. N Eng J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 32.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hauser MA, Li YJ, Takeuchi S, Walters R, Noureddine M, Maready M, Darden T, Hulette C, Martin E, Hauser E, et al. Genomic convergence: identifying candidate genes for Parkinson's disease by combining serial analysis of gene expression and genetic linkage. Hum Mol Genet. 2003;12:671–677. [PubMed] [Google Scholar]
- 34.Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop AM, Callier SJ, Ban M, Goris A, Barcellos LF, et al. Allelic and functional association of the interleukin 7 receptor gene a chain (IL7Ra) with multiple sclerosis. Nat Gen. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 35.Fortin DF, Califf RM, Pryor DB, Mark DB. The way of the future redux. Am J Cardiol. 1995;76:1177–1182. doi: 10.1016/s0002-9149(99)80331-2. [DOI] [PubMed] [Google Scholar]
- 36.Smith LR, Harrell FE, Rankin JS, Califf RM, Pryor DB, Muhlbaier LH, Lee KL, Mark DB, Jones RH, Oldham HN, et al. Determinants of early versus late cardiac death in patients undergoing coronary-artery bypass graft-surgery. Circulation. 1991;84:245–253. [PubMed] [Google Scholar]
- 37.Kong DF, Shaw LK, Harrell FE, Muhlbaier LH, Lee KL, Califf RM, Jones RH. Predicting survival from the coronary arteriogram: an experience-based statistical index of coronary artery disease severity. J Am Coll Cardiol. 2002;39(Suppl A):327A. [Google Scholar]
- 38.Felker GM, Shaw LK, O'Connor CM. A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol. 2002;39:210–218. doi: 10.1016/s0735-1097(01)01738-7. [DOI] [PubMed] [Google Scholar]
- 39.Hauser ER, Mooser V, Crossman DC, Haines JL, Jones CH, Winkelmann BR, Schmidt S, Scott WK, Roses AD, Pericak-Vance MA, et al. Design of the genetics of early onset cardiovascular disease (GENECARD) study. Am Heart J. 2003;145:602–613. doi: 10.1067/mhj.2003.13. [DOI] [PubMed] [Google Scholar]
- 40.Seo D, Wang T, Dressman H, Herderick EE, Iversen ES, Dong C, Vata K, Milano CA, Rigat F, Pittman J, et al. Gene Expression Phenotypes of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1922–1927. doi: 10.1161/01.ATV.0000141358.65242.1f. [DOI] [PubMed] [Google Scholar]
- 41.Cornhill JF, Herderick EE, Vince DG. The clinical morphology of human atherosclerotic lesions. Lessons from the PDAY Study Pathobiological determinants of atherosclerosis in youth. Wien Klin Wochenschr. 1995;107:540–543. [PubMed] [Google Scholar]
- 42.Xu H, Gregory SG, Hauser ER, Stenger JE, Pericak-Vance MA, Vance JM, Zuchner S, Hauser MA. SNPselector: a web tool for selecting SNPs for genetic association studies. BioInformatics. 2005;21:4181–4186. doi: 10.1093/bioinformatics/bti682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abecasis GR, Cookson WO. GOLD—graphical overview of linkage disequilibrium. BioInformatics. 2000;16:182–183. doi: 10.1093/bioinformatics/16.2.182. [DOI] [PubMed] [Google Scholar]
- 44.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. BioInformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 45.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin ER, Bass MP, Hauser ER, Kaplan NL. Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet. 2003;73:1016–1026. doi: 10.1086/378779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material is available at HMG Online.