

Abstract
Kinetic analysis of decarboxylation catalyzed by S154A, Q215A and S154A/Q215A mutant yeast orotidine 5′-monophosphate decarboxylases with orotidine 5′-monophosphate (OMP) and with a truncated nucleoside substrate (EO) activated by phosphite dianion shows: (1) The side-chain of Ser-154 stabilizes the transition state through interactions with the pyrimidine rings of OMP or EO. (2) The side-chain of Gln-215 interacts with the phosphodianion group of OMP or with phosphite dianion. (3) The interloop hydrogen bond between the side-chains of Ser-154 and Gln-215 orients the amide side-chain of Gln-215 to interact with the phosphodianion group of OMP or with phosphite dianion.
Orotidine 5′-monophosphate decarboxylase (OMPDC)1 is a remarkable enzyme because it employs no metal ions or other cofactors, but yet effects an enormous ca. 30 kcal/mol stabilization of the transition state for the decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP) (1) through an unstable vinyl carbanion intermediate (Scheme 1) (2). X-ray crystal structures of OMPDC from a variety of sources complexed with UMP and other ligands reveal intricate networks of interactions between the protein and the bound ligand (3–6), which should provide insight into the interactions that stabilize the transition state for decarboxylation of OMP. However, the extensive X-ray crystallographic data has not led to a consensus opinion about the structure of the transition state, or about the interactions responsible for its ca. 30 kcal/mol stabilization at the active site of OMPDC (1, 7, 8).
Scheme 1.

The substrate OMP can be partitioned into the “pieces” 1-(β-D-erythrofuranosyl)orotic acid (EO) and phosphite dianion, HPO32−, which bind to OMPDC at distinct sites with low affinity (Kd ≈ 0.1 M) (9). The binding site for EO is competent to catalyze the slow decarboxylation of its orotate base moiety to give 1-(β-D-erythrofuranosyl)uridine (EU) with (kcat/Km)EO = 0.021 M−1 s−1 (9). The separate binding of phosphite dianion strongly activates bound EO towards decarboxylation, with (kcat/Km)EO•HPi/(kcat/Km)EO = 80,000 (Scheme 2) (9). We conclude that stabilization of the transition state for OMPDC-catalyzed decarboxylation of EO is enhanced by binding of phosphite dianion at a second site. Tethering the two substrate pieces to give the whole substrate OMP places the orotate base and the oxydianion activator at a single molecule and results in a large entropic advantage over binding and reaction of the individual pieces (10). Similarly, both triosephosphate isomerase and glycerol 3-phosphate dehydrogenase exhibit strong activation by bound phosphite dianion of catalysis of proton transfer (11) and hydride transfer (12), respectively.
Scheme 2.

These results show that there is a switch that turns on the binding interactions between OMPDC and the nonreacting substrate phosphodianion or bound phosphite dianion upon moving from the ground-state Michaelis complex (poorly expressed interactions) to the transition state for decarboxylation of OMP. However, the operation of this switch is not understood, and there is a need for a protocol that distinguishes the catalytic rate acceleration arising from interactions between amino acid side-chains and the reacting pyrimidine ring moiety from that arising from interactions of the protein with the bound phosphate/phosphite dianion.
Figure 1 shows the X-ray crystal structure in the region of the active site of wildtype yeast OMPDC complexed with the intermediate analog 6-hydroxyuridine 5′-monophosphate (BMP) (3). Gln-215 lies in the mobile “active site loop” at the end of the seventh β-strand, while Ser-154 lies in a second mobile loop at the end of the fifth β-strand. The nitrogen of the amide side-chain of Gln-215 is equidistant (3.2 Å) from an oxygen of the phosphate group of BMP and O-2 of its pyrimidine ring. The side-chain hydroxyl of Ser-154 appears both to accept a hydrogen bond from the pyrimidine NH, and to act as a hydrogen bond donor to the oxygen of the amide side-chain of Gln-215. The latter hydrogen bond clamps the separate enzyme loops over the pyrimidine ring and the phosphodianion group of the bound ligand. We argued that this network of interactions, which involves amino acid side-chains in separate distant loops, and which spans the pyrimidine and phosphate ends of the bound ligand, is highly likely to play a significant role in catalysis of the decarboxylation of OMP.
Figure 1.
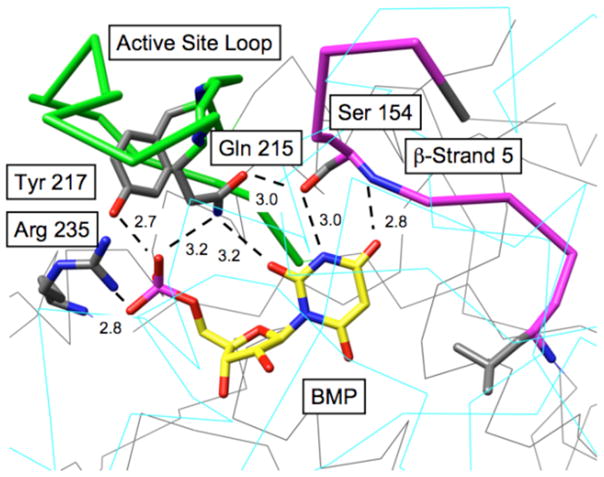
X-ray crystal structure (1DQX) of yeast OMPDC complexed with the intermediate analog BMP (3).
We report here the effects of the single S154A and Q215A (13) mutations, and of the S154A/Q215A double mutation, on the kinetic parameters for decarboxylation of OMP and of EO, and for the phosphite-activated decarboxylation of EO (Table 1). Initial velocities, vo (M s−1), for decarboxylation of OMP were determined spectrophotometrically, while those for the reactions of EO were determined by HPLC. Values of kcat and Km for decarboxylation of OMP, second-order rate constants (kcat/Km)EO for decarboxylation of EO in the absence of HPO32−, and third-order rate constants (kcat/Km)EO•HPi/Kd for activation of the decarboxylation of EO by phosphite dianion were determined as described in the Supporting Information. The data provide no evidence for saturation of the mutant enzymes by either substrate piece, so that Kd > 0.1 M for the EO and HPO32− pieces (9).
Table 1.
Effects of the S154A, Q215A and S154A/Q215A Mutations on the Kinetic Parameters for Decarboxylation of the Whole Substrate OMP and of the Substrate Pieces EO and Phosphite Dianion Catalyzed by Yeast OMPDC
| OMPa | EOb | EO + HPO32−c | ||||||
|---|---|---|---|---|---|---|---|---|
| Yeast OMPDC |
kcat s−1 |
Km μM |
kcat/Km M−1 s−1 |
ΔΔ G‡OMP kcal/mol |
(kcat/Km)EO M−1 s−1 |
ΔΔG‡EO kcal/mol |
(kcat/Km)EO•HPi/Kd M−2 s−1 |
ΔΔG‡EO•HPi kcal/mol |
| Wildtype | 15d | 1.6d | 9.4 × 106 | 0.021e | 1.2 × 104e | |||
| S154A | 0.082 | 130 | 630 | 5.7 | 8.7 × 10−5 | 3.2 | 0.25 | 6.4 |
| Q215A | 21 | 50 | 4.2 × 105 | 1.8 | 0.011 | 0.4 | 240 | 2.3 |
| S154A/Q215A | 0.042 | 110 | 380 | 6.0 | 1.1 × 10−4 | 3.1 | 0.14 | 6.7 |
Reactions in the presence of 10 mM MOPS at pH 7.1, 25 °C and I = 0.105 (NaCl). Standard deviations obtained from the nonlinear least squares fit of the initial velocity data to the Michaelis-Menten equation were: kcat≤ 4%; Km≤ 10%. Values of kcat = 14 s−1 and Km = 22 μM were determined for OMPDC from Escherichia Coli.
Reactions in the presence of 50 mM MOPS at pH 7.1, 25 °C and I = 0.15 (NaCl). Standard deviations in the values of (kcat/Km)EO are estimated to be ≤ 10%.
Reactions in the presence of 10 mM MOPS and phosphite dianion at pH 7.0, 25° C and I = 0.15 (NaCl). Standard deviations in the values of (kcat/Km)EO•HPi/Kd are estimated to be ≤ 10%.
Data from the literature (16).
Data from the literature (9).
The second-order rate constants kcat/Km for turnover of OMP by OMPDC provide a measure of the barrier for conversion of the enzyme and OMP to the transition state for decarboxylation of OMP (Table 1). The third-order rate constants (kcat/Km)EO•HPi/Kd for the catalyzed reactions of the substrate pieces provide a measure of the barrier for conversion of the enzyme and the two substrate pieces EO and HPO32− to the transition state for decarboxylation of EO (Table 1). The data in Table 1 were used to calculate the effect of each single mutation and of the double mutation on the stability of the transition state for decarboxylation of OMP (ΔΔG‡OMP) and for decarboxylation of the substrate pieces EO and HPO32− (ΔΔG‡EO and ΔΔG‡EO•HPi). The large effect of the S154A mutation on kcat/Km for OMP (ΔΔG‡OMP = 5.7 kcal/mol) highlights the critical importance of the CH2OH side-chain of Ser-154 in catalysis of decarboxylation at a distant site. For each mutation, ΔΔG‡OMP and ΔΔG‡EO•HPi are similar, which shows that the absence of a covalent connection to the phosphodianion group has only a small effect on the interaction of the resulting substrate pieces with the side-chains of Ser-154 and Gln-215 at OMPDC.
The 1.8 kcal/mol effect of the Q215A mutation on kcat/Km for OMP results mainly from a change in Km for the binding of OMP (2.0 kcal/mol effect), which shows that the interactions between the amide side-chain of Gln-215 and OMP are fully expressed at the Michaelis complex.2 The Q215A mutation has little effect on (kcat/Km)EO for decarboxylation of EO (ΔΔG‡EO = 0.4 kcal/mol), which suggests that the interaction of the amide side-chain with O-2 of the pyrimidine ring is weak. However, the value of ΔΔG‡EO•HPi = 2.3 kcal/mol is similar to ΔΔG‡OMP = 1.8 kcal/mol for OMP. Therefore, the two-part substrate [EO + HPO32−] pinpoints the otherwise unclear role of the amide side-chain of Gln-215 as stabilization of the Michaelis complex by its interaction with the phosphodianion group of OMP at the binary complex E•OMP and, by analogy, with bound phosphite dianion at the ternary complex E•EO•HPO32−.
Scheme 3 shows a cycle (14, 15) for the mutations at OMPDC that was constructed using the data in Table 1. Around 20% of the ca. 30 kcal/mol total transition state stabilization for OMPDC is due to interactions of the bound transition state with the side-chains of Ser-154 and Gln-215 (ΔΔG‡OMP = 6.0 kcal/mol for the S154A/Q215A double mutant). However, the effect of the individual mutations depends upon the order in which they are made. For example, the 6 kcal/mol total interaction can be partitioned into a ca. 2 kcal/mol ground-state interaction between the amide side-chain of Gln-215 and the phosphodianion group of OMP (Q215A mutation of the wildtype enzyme), and a ca. 4 kcal/mol interaction between the CH2OH side-chain of Ser-154 and the pyrimidine ring of OMP (S154A mutation of the Q215A mutant enzyme). The opposite order of these mutations leads to expression of nearly the entire 6 kcal/mol effect of the S154A/Q215A double mutation in ΔΔG‡OMP for the S154A single mutation, with only a small 0.3 kcal/mol effect of the second Q215A mutation.
Scheme 3.

These data provide strong evidence for cooperativity in the interactions of the side-chains of Ser-154 and Gln-215 with the bound transition state for decarboxylation, as a result of the requirement for a hydrogen bond between the CH2OH group of Ser-154 and the amide side-chain of Gln-215 (Figure 1) that properly orients the amide to interact with the phosphodianion group of OMP. The S154A mutation of the wildtype enzyme therefore leads to loss of the transition state stabilization contributed by the side-chains of both Ser-154 and Gln-215. There is an additional hydrogen bond between the backbone amide of Ser-154 and O-4 of the pyrimidine ring which “anchors” the CH2OH group of Ser-154 (Figure 1). Therefore, the transition state stabilization arising from the hydrogen bond between this side-chain and the pyrimidine NH is maintained at the Q215A mutant enzyme.
The 6.0 kcal/mol effect of the double S154A/Q215A mutation on the stability of the transition state for decarboxylation of OMP may also be partitioned into a 2.5 kcal/mol effect on the stability of the Michaelis complex (Km effect) and a 3.5 kcal/mol effect that is expressed specifically at the transition state for decarboxylation of bound OMP (kcat effect).
The small effect of 0.5 kcal/mol on the stability of the Michaelis complex upon making the S154 mutation at the Q215A mutant enzyme provides strong evidence that the effect of the double mutation on Km results mainly from loss of the stabilizing interactions between the amide side-chain of Gln-215 and the substrate phosphodianion group. The 500-fold decrease in kcat due to the S154 mutation at the Q215A mutant enzyme likely represents strengthening of the hydrogen bond between the CH2OH side-chain of Ser-154 and the substrate pyrimidine NH specifically at the carbanion-like transition state for formation of the C-6 vinyl carbanion intermediate (2). This might be the result of reorganization of the substrate at the binding pocket that accompanies the loss of CO2 from the substrate.
In summary, application of our two-part substrate analysis to single and double mutant enzymes provides a detailed description of the stabilizing interactions between two amino acid side-chains at the active site of OMPDC and of their interactions with the transition state for enzyme-catalyzed decarboxylation of OMP. We expect that this protocol will be useful in defining the role in catalysis of the many other interactions between OMPDC its bound ligands.
Supplementary Material
Details of the kinetic protocols for decarboxylation of OMP and EO; Michaelis-Menten plots of vo/[E] (s−1) vs [OMP], plots of vo/[E] (s−1) vs [EO], and plots of the observed second-order rate constants (kcat/Km)app (M−1 s−1) for turnover of EO vs [HPO32−]; details of cloning and mutagenesis of yeast OMPDCs (18 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by Grants GM39754 to JPR and GM65155 to JAG from the National Institutes of Health.
Footnotes
Abbreviations: OMPDC, orotidine 5′-monophosphate decarboxylase; OMP, orotidine 5′-monophosphate; UMP, uridine 5′-monophosphate; EO, 1-(β-D-erythrofuranosyl)orotic acid; EU, 1-(β-D-erythrofuranosyl)uridine; BMP, 6-hydroxyuridine 5′-monophosphate.
The 30-fold effect of the Q215A mutation on Km for OMP reported here is larger than the ca. 4-fold effect from earlier work conducted at lower ionic strength [ref. 13].
References
- 1.Miller BG, Wolfenden R. Ann Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 2.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci USA. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu N, Mo Y, Gao J, Pai EF. Proc Natl Acad Sci USA. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Natl Acad Sci USA. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris P, Poulsen JCN, Jensen KF, Larsen S. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 7.Callahan BP, Miller BG. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Gao J, Byun KL, Kluger R. Top Curr Chem. 2004;238:113–136. [Google Scholar]
- 9.Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 10.Jencks WP. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amyes TL, Richard JP. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsang WY, Amyes TL, Richard JP. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller BG, Snider MJ, Wolfenden R, Short SA. J Biol Chem. 2001;276:15174–15176. doi: 10.1074/jbc.M011429200. [DOI] [PubMed] [Google Scholar]
- 14.First EA, Fersht AR. Biochemistry. 1995;34:5030–5043. doi: 10.1021/bi00015a014. [DOI] [PubMed] [Google Scholar]
- 15.Horovitz A, Fersht AR. J Mol Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 16.Porter DJT, Short SA. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of the kinetic protocols for decarboxylation of OMP and EO; Michaelis-Menten plots of vo/[E] (s−1) vs [OMP], plots of vo/[E] (s−1) vs [EO], and plots of the observed second-order rate constants (kcat/Km)app (M−1 s−1) for turnover of EO vs [HPO32−]; details of cloning and mutagenesis of yeast OMPDCs (18 pages). This material is available free of charge via the Internet at http://pubs.acs.org.