

Abstract
A convenient method is described for the dehydration of β-ketoesters to generate conjugated and deconjugated alkynyl esters and conjugated allenyl esters. This sequential one-pot method involves the formation of a vinyl triflate monoanion intermediate that leads to the selective formation of alkynes or allenes depending on additives and conditions used. Product outcomes appear to be a function of unique mono- and dianion mechanisms which are described.
Introduction
Highly unsaturated carbonyl compounds such as allenyl1 and alkynyl esters2 have proven increasingly useful in organic synthesis. Conjugated allenyl carbonyls are particularly promising as synthetic intermediates due to their chiral axis and electronically distinctive cumulated double bonds that can be chemoselectively manipulated. Despite their synthetic promise,3 numerous challenges remain in the preparation and application of allenyl and alkynyl carbonyls. We have previously reported procedures involving amide bases to convert conjugated alkynyl esters to the corresponding allenyl esters with alkylation at the α-position.4 However, this method was limited to α-silylation, stannylation, and methylation with other alkylating agents leading to the deconjugated alkynyl ester product depending on reaction conditions.5 We have also described methods for the isomerization of α-alkynyl esters to their corresponding allenes involving manganese coordination chemistry.6 However, all of our previously disclosed methods to prepare β-alkynyl and α-allenyl esters require conjugated alkynyl esters starting materials which are not always readily available. As an alternative, we envisioned a one-pot transformation whereby a ketone carbonyl in substrates containing α-electron withdrawing groups can be dehydrated to generate doubly unsaturated products. Indeed, simple acyclic and cyclic ketones have been converted to their alkynyl and allenyl dehydration products via vinyl triflates7 and enol phosphonates.8 Hydrocarbon alkynes have also been prepared through a decarboxylative elimination of vinyl triflates.9 However, to our knowledge, no dehydration strategy has ever been reported for the preparation of allenyl esters. In this paper we describe our development of a dehydration approach starting from cheap and readily available β-keto esters to form three different allene-alkyne isomers depending on reaction conditions.
Results and Discussion
Our initial experiments involved the treatment of β-keto esters 1 with two equivalents LiHMDS at − 78°C to form dienolates followed by trapping with Tf2O.10 Subsequent warming to room temperature leads to conjugated alkynyl esters 2 in good yields for a variety of readily available β-keto ester substrates excepting those with heteroatom substitution at the γ-position (Table 1). The choice of a metal HMDS base (i.e. lithium) was critical to the formation of 2 (and in transformations discussed below) whereas the use of LDA failed to give alkyne products. Generally 2-alkynoates are prepared through formation of bonds on either side of the triple bond either by the carboxylation of terminal alkynyl anions11 or by the less favorable alkylation of propiolates that usually require activated electrophiles.12 Both methods suffer drawbacks from either lack of availability of terminal alkynes or poor nucleophilicity of propiolate anions. Thus the present dehydration approach serves as an important alternative to existing 2-alkynoate synthesis methods.
Table 1.
Synthesis of conjugated alkynyl esters from β-ketoesters
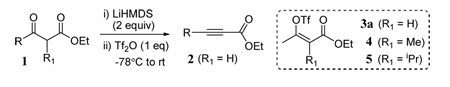 | ||||
|---|---|---|---|---|
| entry | R | R1 | product | % yielda |
| 1 | Me | H | 2a | 84b |
| 2 | Et | H | 2b | 90 |
| 3 | iPr | H | 2c | 72 |
| 4 | nPr | H | 2d | 82 |
| 5 | iBu | H | 2e | 77 |
| 6 | ClCH2 | H | 2f | 0 |
| 7 | PhS | H | 2g | 0 |
| 8 | Me | Me | 4 | 87 |
| 9 | Me | iPr | 5 | 83 |
solated yields.
Quenching at −78°C gives 3a.
It is interesting to note that these dehydration reactions did not produce any of the allene isomer. Even substrates containing α-substituents (entries 8 and 9), which were expected to eliminate the triflate via an E1cb mechanism, failed to give allene products. Instead, vinyl triflates 4 and 5 were obtained after aqueous quenching. To further study this reaction, triflate intermediate 3a was obtained by a low temperature aqueous quench of substrate 1a (R = Me, R1 = H) after the addition of base and triflic anhydride.13 Refluxing 3a in THF in the presence of Et3N gave near quantitative yield of alkynyl ester 2a (Scheme 1). However, vinyl triflates 4 and 5 did not give allene products under these conditions even with extended reaction times. The treatment of 4 and 5 with a much stronger base (excess LiHMDS) did give allene products.
Scheme 1.

Preference for vinylic E2 elimination
Our experiments with vinyl triflates 3a, 4, and 5 reveal a strong mechanistic preference for triflate elimination across the double bond even though the γ-hydrogens present in these substrates can also assume an anti-coplanar orientation. Similar observations have been made with β-bromo-α,β- unsaturated ketones.14 This strong regioselective preference for triflate elimination is helpful in deciding among mechanistic possibilities for the one-pot process leading to products 2. The likely reactive intermediate in this reaction is triflate monoanion 6 (Figure 1). Following an E1cb pathway, this intermediate would be expected to give allene product, which is not observed even with low temperature quenching. It is possible that the observed conjugated alkyne product is formed by an intramolecular abstraction of the α-proton via a 4-membered ring transition state.15 However, α-deuterio-analogs of intermediate 6 failed to yield product 7 with the anticipated γ-deuterium (Figure 1). Thus it appears that a more complex mechanism is operative here.
Figure 1.

Bimolecular proton transfer mechanism
Conjugated alkynyl esters with substitution at the γ-position can also be prepared from acetoacetates (Scheme 2). In a one-pot procedure, the dianion of ethyl acetoacetate is treated with benzyl bromide followed by the addition of an additional equivalent of LiHMDS. The addition of Tf2O at −78°C followed by warming to room temperature led to a reasonable yield of alkynoate 8. Given the general availability of arylmethyl halides,16 this method will likely prove useful especially since the synthesis of 8 and related products by alkylation of 2-butynoates has not proved feasible.17
Scheme 2.

Synthesis of γ-substituted alkynyl ester from unsubstituted acetoacetate
Our experience with the reactivity of substrates 4 and 5 prompted us to use an additional equivalent of strong base after the formation of intermediate vinyl triflate anions in an attempt to produce allenes in a one-pot procedure. Thus a series of β-keto esters were sequentially treated with LiHMDS (2 equiv), triflic anhydride (1 equiv), and a final equivalent of LiHMDS followed by quenching at low temperatures (Table 2). For substrates containing no α-substitution (entries 1–4), deconjugated alkynyl esters 9 were obtained in reasonable yields especially with the addition of HMPA prior to quenching. In a previous work we observed a similar preference for deconjugated products using HMPA.5 Presumably this agent acts to destabilize lithium complexes of the monoanion and HMDS, which have a tendency to quench the anion through intracomplex protonation.18 Deconjugated alkynoate 9a (entry 1) was also produced in good yields despite its well-known propensity for isomerization to allene form 10a.
Table 2.
Synthesis of 3-alkynoates 9 and allenyl esters 10.
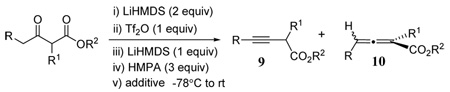 | ||||||
|---|---|---|---|---|---|---|
| entry | R | R1 | R2 | additive | Product (% yield)a | |
| 1 | H | H | Et | None | 9a (65) | 10a (0b) |
| 2 | Me | H | Et | None | 9b (82) | 10b (0) |
| 3 | Et | H | Et | None | 9c (69) | 10c (0) |
| 4 | nPr | H | Et | None | 9d (76) | 10d (0) |
| 5 | H | Me | Et | None | 9e (53) | 10e (23) |
| 6 | H | nBu | Et | None | 9f (36) | 10f (36) |
| 7 | H | iPr | Et | None | 9g (20) | 10g (43) |
| 8 | H | nBu | tBu | None | 9h (20) | 10h (53) |
| 9 | H | H | Et | ZnCl2 | 9a (0) | 10a (68) |
| 10 | Et | H | Et | ZnCl2 | 9c (72) | 10c (0) |
| 11 | H | Me | Et | ZnCl2 | 9e (0) | 10e (68) |
| 12 | H | nBu | Et | ZnCl2 | 9f (0) | 10f (78) |
| 13 | H | iPr | Et | ZnCl2 | 9g (0) | 10g (73) |
| 14 | H | H | Ph | ZnCl2 | 9i (0) | 10i (27c) |
| 15 | H | H | tBu | ZnCl2 | 9j (0) | 10j (76) |
| 16 | H | nBu | tBu | ZnCl2 | 9h (0) | 10h (74) |
All yields are based on isolated products. For entries 5–8, isomers were isolated as inseparable mixtures and product ratios estimated by NMR.
Care must be taken to use neutral silica gel during flash chromatographic isolation to avoid isomerization to allene.
Numerous decomposition byproducts most likely due to ketene formation from phenoxide elimination.
It is important to note that this method (Table 2) did not lead to any conjugated alkynyl ester products. This product outcome can be rationalized in terms of the preference for the amide base to deprotonate intermediate 6 (formed after step 2) exclusively at the γ-position (Scheme 3). Overwhelming spectroscopic, crystallographic, and computational investigations establish that lithium cations generally interact with enolates at the oxygen center pointing to the predominance of oxyanion resonance forms.19 Therefore, as a first approximation, a comparison of the two oxyanion resonance forms resulting from selective deprotonation can be made to the exclusion of other resonance contributors. Deprotonation of monoanion 6 at the γ-position gives oxyanion 11 in which the alkene and alkyne bonds are in conjugation (alkynenolate). Whereas deprotonation at the α-position produces oxyanion 12 containing a cumulated system (cumulenolate) with diminished conjugation of the π-bonds. Although conjugated alkynyl ester products are more thermodynamically stable, their formation appears to require the intermediacy of the less favorable anion 12. Indeed, in related ketone systems, a semiempirical computational study revealed that alkynenolates were over 5 kcal/mol more stable than their cumulenolate isomers.20
Scheme 3.

Rationale for disfavored formation of 2-alkynyl ester
Dehydration of α-substituted β-ketoesters tends to favor the formation of allenes 10 especially when ZnCl2 is added prior to quenching (Table 2, entries 11–13). This was true whether the reactions were quenched at −78°C or at warmer temperatures. Although these allene products are thermodynamically favored, their formation can be better rationalized using a kinetic argument. Thus anionic intermediate 13 is expected to prefer a geometry in which the ester alkoxy group (OR2) is trans to the larger α-substituent (Figure 2) based on our previous work involving trapping experiments with intermediates similar to 134 and on other reports.21 Protonation of anion 13 at the α-position leading to transition state TS14 (Figure 2) would augment destabilizing steric interactions between the ester alkoxy and ethynyl groups as the α-carbon undergoes angle compression due to sp3 hybridization. However, γ-protonation maintains sp2 geometry at the α-carbon. This rationale appears to explain the product ratios observed for substrates where R = H (entries 5 – 8). As the size of the α-substituent (R1) increases, it is more likely that the ester alkoxy group will prefer the trans to this group in intermediate 13 thus increasing the propensity for γ-protonation. Indeed, a bulky tert-butyl ester group gave a significantly higher ratio of allene to alkyne relative to the ethyl ester (compare entries 6 and 8). It is unclear how zinc chloride acts to enhance allene formation in this reaction. In general, zinc enolates are thought to be present in solution as mainly the C-metalated species.22 Perhaps the transmetalation of intermediates such as 13 is governed by similar energetic considerations as just described for protonation.
Figure 2.

Deconjugated alkynyl ester disfavored in substrates containing α-substitution
Since large α-groups appear to favor allene products in our dehydration of β-keto esters, we sought to introduce an α-trialkyl silyl group as a control element. Thus two equivalents of LiHMDS were added to vinyl triflate intermediate 6 (Table 3) most likely generating an ynenolate dianion 15 (R = Et). We have previously observed that these unusually stable dianions can be readily prepared from conjugated alkynyl esters.4 Subsequent addition of TMSCl to 15 presumably gave bis-silyl intermediate 17, which upon aqueous quench led to a 2:1 mixture of allenylsilane 18a and propargyl product 9c most likely the result of silyl group hydrolysis during aqueous workup (entry 1). Very recently, intermediates similar to 17 derived from allenyl esters have been converted to carbinol allenoates with excellent selectivity via Mukaiyama aldol-type reactions.3
Table 3.
Synthesis of 3-alkynyl and allenyl esters via enyne dianions
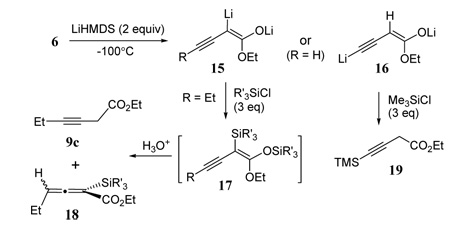 | ||||
|---|---|---|---|---|
| entry | R | R’3Si | Product (% yield)a | |
| 1 | Et | TMS | 9c (25) | 18a (55) |
| 2 | Et | TIPS | - | 18b (78) |
| 3 | H | TMS | 19 (87) | - |
Isolated yields.
The same reaction sequence with 15 (R = Et) using TIPSCl led to the exclusive formation of allenyl ester 18b. This finding with the much larger TIPS group in the α-position parallels the trend of substrates with α-substitution from the monoanion reactions (Table 2 and Figure 2). The synthesis of product 18b from 6 is also significant since no other substrate with γ-substitution discussed thus far has been converted exclusively to an allenyl ester product. Deprotonation of 6 where (R = H) led to intermediate 16 which upon silylation gave deconjugated product 19.
Conclusion
Conditions have been developed to selectively convert a series of β-ketoesters to conjugated allenyl, alkynyl, or β-deconjugated alkynyl esters. Following a neutral, monoanion, or dianion mechanistic rationale, our method involves a one-pot stepwise addition of LiHMDS, triflic anhydride, and quenching under varying conditions to bring about dehydration. Our investigation has also uncovered an efficient synthesis of ketene acetal intermediate 17 that preferentially adds proton at the γ-position to form allene product. We are currently exploring the reactivity of these types of intermediates with other electrophiles for the potential development of an asymmetric allene synthesis method.
Experimental Section
General procedure for synthesis of conjugate alkynes 2
To a round bottom flask (50 mL) under argon, dry THF (7 mL) and LiHMDS (4 mL, 1.0 M in THF) were added. The solution was then cooled to −78 °C (acetone-dry ice bath) followed by the addition of β-keto ester (2 mmol). After stirring for 45 min, triflic anhydride (2 mmol) was added slowly over 15 min. The reaction was then stirred overnight slowly warming to room temperature and quenched with saturated NH4Cl solution. The organic layer was separated, the aqueous fraction was extracted with ether, and the organic layers were combined and dried with anhydrous Na2SO4. The product was purified by silica gel chromatography in the usual manner using 1–2% EtOAc in hexanes.
Ethyl hex-2-ynoate (2d)
1H NMR (400 MHz, CDCl3) δ 4.22 (q, J = 7.14 Hz, 2H), 2.31 (t, J = 7.1 Hz, 2H), 1.66-1.57 (m, 2H), 1.31 (t, J = 7.14 Hz, 3H), 1.02 (t, J = 7.38 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 153.8, 89.2, 73.2, 61.7, 21.0, 20.5, 14.0, 13.4; HRMS calc. for C8H12O2 [M+H]+: 141.0910. Found: 141.0899.
Procedure for synthesis of alkynyl ester 8
To a round bottom flask (50 mL) under argon, dry THF (7 mL) and LiHMDS (4 mL, 1.0 M in THF) were added. The solution was then cooled to −78 °C (acetone-dry ice bath) followed by the addition of β-keto ester (2 mmol). After stirring for 45 min, BnBr (2 mmol) was added dropwise followed by warming slowly to room temperature over the course of 6 h. The reaction mixture was then cooled to −78 °C and charged with LiHMDS (2 mL, 1.0 M in THF). After for 45 min, Tf2O (2 mmol) was added slowly over 15 min and the reaction was stirred overnight slowly warming to room temperature and quenched with saturated NH4Cl solution. The organic layer was separated, the aqueous fraction was extracted with ether, and the organic layers were combined and dried with anhydrous Na2SO4. The product was purified by silica gel chromatography in the usual manner using 1–2% EtOAc in hexanes.
Ethyl 5-phenyl-pent-2-ynoate (8)
1H NMR (400 MHz, CDCl3) δ 7.40-7.22 (m, 5H), 4.21 (q, J = 7.2 Hz, 2H), 2.89 (t, J = 7.6 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 154.0, 139.9, 128.8, 128.6, 126.9, 88.5, 73.9, 62.1, 34.1, 21.1, 14.3; HRMS calc. for C13H14O2 [M+H]+: 203.1067. Found: 203.1059.
General procedure for deconjugated alkyne/conjugated allene isomers via monoanionic enyne enolate with ZnCl2 additive
To a round bottom flask (50 mL) under argon, dry THF (7 mL) and LiHMDS (4 mL, 1.0 M in THF) were added. The solution was then cooled to −78 °C (acetone-dry ice bath) followed by the addition of β-keto ester (2 mmol). After stirring for 45 min, triflic anhydride (2 mmol) was added slowly over 15 min. The reaction was maintained at −78 °C with stirring for an additional 1 h followed by the addition of LiHMDS (2 mmol). The mixture was stirred at −78 °C for an additional 30 min followed by the addition of HMPA (7 mmol). After stirring for stirring at −78 °C for an additional 30 min, ZnCl2 (2.4 mmol) was added as an ether solution. The reaction was quenched after an additional 30 min by pouring into an ice-cold stirring biphasic mixture of ether and saturated aqueous NH4Cl. NMR spectra were recorded after workup for each reaction confirming the formation of only conjugated allene which were compared with literature references (except for 10h).
tert-Butyl-2-n-butylallenylester (10h)
1H NMR (400 MHz, CDCl3) δ 5.05 (t, J = 3.03 Hz, 2H), 2.18 (m, 2H), 1.47 (s, 9H), 1.45-1.30 (m, 4H), 0.91 (t, J = 7.19 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 213.6, 166.6, 101.7, 80.7, 30.2, 28.0, 27.7, 22.3, 13.9; HRMS calc. for C12H20O2 [M+H]+: 197.1542. Found: 197.1543.
General procedures for silylated deconjugated alkyne/conjugated allene isomers via dianionic enolate
To a round bottom flask (50 mL) under argon, dry THF (7 mL) and LiHMDS (4 mL, 1.0 M in THF) were added. The solution was then cooled to −95 °C (methanol-liquid N2 bath) followed by addition of β-keto ester (2 mmol). After stirring for 1 h, triflic anhydride (2 mmol) was added slowly over 15 min. Since Tf2O is a solid at this reaction temperature, the slow addition should be accompanied by vigorous stirring. Alternatively, Tf2O can be added as ether solution (not in THF since this solvent reacts to form polymer at room temperature). The reaction mixture was stirred for an additional 1 h at −95 °C followed by the addition of LiHMDS (4 mL, 1.0 M in THF). After an additional 1 h at −95 °C, R3SiCl (6 mmol) was added dropwise followed by warming to room temperature over 1 hour and quenching with saturated NH4Cl solution. The organic layer was separated, the aqueous fraction was extracted with ether, and the organic layers were combined and dried with anhydrous Na2SO4. The product was purified by silica gel chromatography in the usual manner using 1–2% EtOAc in hexanes.
Ethyl-2-triisopropylsilylhexa-2,3-dienoate (18b)
1H NMR (400 MHz, CDCl3) δ 5.24 (t, J = 6.61 Hz, 1H), 4.16 (q, J = 7.05 Hz, 3H), 2.20-1.05 (m, 2H), 1.34-1.18 (m, 9H), 1.07 (d, J = 7.40 Hz, 18H); 13C NMR (100 MHz, CDCl3) δ 216.2, 168.6, 91.5, 89.6, 60.7, 20.9, 18.6, 17.7, 14.2, 12.2; HRMS calc. for C17H32O2Si [M+H]+: 297.2244. Found: 297.2236.
Supplementary Material
Spectroscopic data for all new compounds, detailed experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank the National Institutes of Mental Health (66963-01) and General Medical Science (073621-01), and NSF (0311369) for financial support. We also thank Dr. Manishabrata Bhowmick for helpful discussions.
References
- 1.For recent examples see: Shi M, Tang X-Y, Yang Y-H. J. Org. Chem. 2008;73:5311. doi: 10.1021/jo800608h. Huang X, Shen R, Zhang T. J. Org. Chem. 2007;72:1534. doi: 10.1021/jo062376m. Hopkins CD, Guan L, Malinakova HC. J. Org. Chem. 2005;70:6848. doi: 10.1021/jo050886v. Ma S. Chem. Rev. 2005;105:2829. doi: 10.1021/cr020024j.
- 2.For recent examples see: Creech GS, Kwon O. Org Lett. 2008;10:429. doi: 10.1021/ol702462w. Brawn RA, Panek JS. Org. Lett. 2007;9:2689. doi: 10.1021/ol070936d.
- 3.Xu B, Hammond GB. Angew. Chem. Int. Ed. 2008;47:689. doi: 10.1002/anie.200704221. [DOI] [PubMed] [Google Scholar]
- 4.Lepore SD, He YJ, Damisse P. J. Org. Chem. 2004;69:9171. doi: 10.1021/jo0487754. [DOI] [PubMed] [Google Scholar]
- 5.Lepore SD, He YJ. J. Org. Chem. 2005;70:4546. doi: 10.1021/jo050218+. [DOI] [PubMed] [Google Scholar]
- 6.Lepore SD, Khoram A, Bromfield DC, Cohn P, Jairaj V, Silvestri MA. J. Org. Chem. 2005;70:7443. doi: 10.1021/jo051040u. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hargrove RJ, Stang PJ. J. Org. Chem. 1974;39:581. [Google Scholar]; (b) Brummond KM, Gesenberg KD, Kent JL, Kerekes AD. Tetrahedron Lett. 1998;39:8613. [Google Scholar]
- 8.Brummond KM, Dingess EA, Kent JL. J. Org. Chem. 1996;61:6096. doi: 10.1021/jo9610265. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fleming I, Ramarao C. Org. Biomol. Chem. 2004;2:1504. doi: 10.1039/b401435a. [DOI] [PubMed] [Google Scholar]; (b) Fleming I, Harley-Mason J. J. Chem. Soc. 1963:4771. [Google Scholar]
- 10.Although not appropriate for the methodology in the present work, the diastereoselective preparation of enol triflates from acetoacetate derivatives using aqueous bases has been recently described: Babinski D, Soltani O, Frantz DE. Org. Lett. 2008;10:2901. doi: 10.1021/ol8010002.
- 11.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769. for a recent example see: Voituriez A, Ferreira F, Perez-Luna A, Chemla F. Org. Lett. 2007;9:4705. doi: 10.1021/ol701952y.
- 12.(a) Bieber LW, da Silva MF. Tetrahedron Lett. 2007;48:7088. [Google Scholar]; (b) Spinella A, Caruso T, Coluccini C. Tetrahedron Lett. 2002;43:1681. [Google Scholar]
- 13.Vinyl triflate 3a, 4, and 5 were surprisingly stable to isolation using silica gel chromatography.
- 14.Eaton PE, Stubbs CE. J. Am. Chem. Soc. 1967;89:5722. [Google Scholar]
- 15.Chamorro E, Toro-Labbe A, Fuentealba P. J. Phys. Chem. A. 2002;106:3891. [Google Scholar]
- 16.As opposed to arylethyl halides which would be required for additions involving weak propiolate nucleophiles to form compounds such as 8.
- 17.Although an aldol reaction of methyl 2-butynoate using aluminum tris(tripenylphenoxide) has resulted in γ-substitution: Saito S, Shiozawa M, Yamamoto H. Angew. Chem. Int. Ed. 1999;38:1769. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1769::AID-ANIE1769>3.0.CO;2-0.
- 18.Seebach D. Angew. Chem. Int. Ed. 1988;27:1624. and references cited therein. [Google Scholar]
- 19.see: Rosi M, Sgamellotti A, Floriani C. Theochem. 1998;431:33. and references cited therein.
- 20.Petasis NA, Teets KA. J. Am. Chem. Soc. 1992;114:10328. [Google Scholar]
- 21.Wilcox CS, Babston RE. J. Org. Chem. 1984;49:1451. [Google Scholar]
- 22.see: van Maanen HL, Kleijn H, Jastrzebski JTBH, Lakin MT, Spek AL, van Koten G. J. Org. Chem. 1994;59:7839. and references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic data for all new compounds, detailed experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.