

Abstract
A mechanism for proton pumping by the B-type cytochrome c oxidases is presented in which one proton is pumped in conjunction with the weakly-exergonic, two-electron reduction of Fe-bound O2 to the Fe-Cu bridging peroxodianion, and three protons are pumped in conjunction with the highly-exergonic, two-electron reduction of Fe(III)-−O-O−-Cu(II) to form water and the active oxidized enzyme, Fe(III)-−OH, Cu(II). The scheme is based on the active site structure of cytochrome ba3 from Thermus thermophilus, which is considered to be both necessary and sufficient for coupled O2 reduction and proton pumping when appropriate gates are in place (not included in the model). Fourteen detailed structures obtained from DFT geometry optimization are presented that are reasonably thought to occur during the four-electron reduction of O2. Each proton pumping step takes place when a proton resides on the imidazole ring of I-His376 and the large active site cluster has a net charge of +1 due to an uncompensated, positive charge formally associated with CuB. Density functional theory (DFT) of four types was applied to determine the energy of each intermediate, and standard thermochemical approaches were used to obtain the reaction free energies for each step in the catalytic cycle. This application of DFT generally conforms with previously suggested criteria for a valid model [P. E. M. Siegbahn & M. A. R. Blomberg (2000) 100 421 - 437] and, shows how the chemistry of O2-reduction in the heme a3-CuB dinuclear center can be harnessed to generate an electrochemical proton gradient across the lipid bilayer.
Introduction
Mitochondrial and bacterial respiration accounts for ∼60 % of global O2 consumption1, of which the activity of cytochrome c oxidases dominates. Enzymes of this type are located in the inner membrane of mitochondria and prokaryotic cells, where they catalyze the reduction of O2 to H2O by the one-electron transfer agent, cytochrome c, and concomitantly translocate protons across the membrane. The result is an electrochemical gradient determined by a higher H+-concentration and/or positive charge density on the OUTside of the membrane and lower H+-concentration and/or positive charge density on the INside of the membrane [See2-9 for review]. Free energy thus captured is subsequently used to drive a variety of cellular processes, most prominently the synthesis of cellular ATP10; defects in these enzymes have also been implicated in human metabolic disorders11.
Types of heme-copper oxidases
Amino acid sequence analyses of the heme-copper oxidase super-family12 reveal the existence of three major- and several sub-classes of cytochrome c oxidases: The A1-type includes the extensively studied enzyme from bovine heart mitochondria and from closely related prokaryotes Paracoccus denitrificans13 and Rhodobacter sphaeroides14. Unique to enzymes in this sub-group is the presence of a glutamate residue at respective sequence numbers 242, 278, and 286, that is part of a conserved -GHPEVY-sequence cluster occupying the same 3-dimensional space in all three enzymes [see14 for a high-resolution structure of this region]. Numerous studies have indicated the importance of this glutamate residue in A1-type oxidase function.
The B-type enzymes are found both in eubacteria and archaea of which the founding member is the cytochrome ba3 from T. thermophilus15-18. These enzymes contain the -SHPIVY- cluster wherein the isoleucine side chain is isopositional with the glutamate residue chacteristic of the A1-type enzymes; in Tt cytochrome ba3, I-Ile235 contributes to the inner surface of the O2-channel19.
Structural considerations
Results from X-ray crystallographic studies indicate that while the A1- and B-type oxidases are highly divergent in their amino acid sequences they are highly convergent in their 3-dimensional structures13, 14, 17, 18, 20, 21. Fig. 1 shows a side-on schematic of the overall structure of Thermus cytochrome ba3, PDB code 1XME18and PDB code 1EHk17.
Fig. 1.
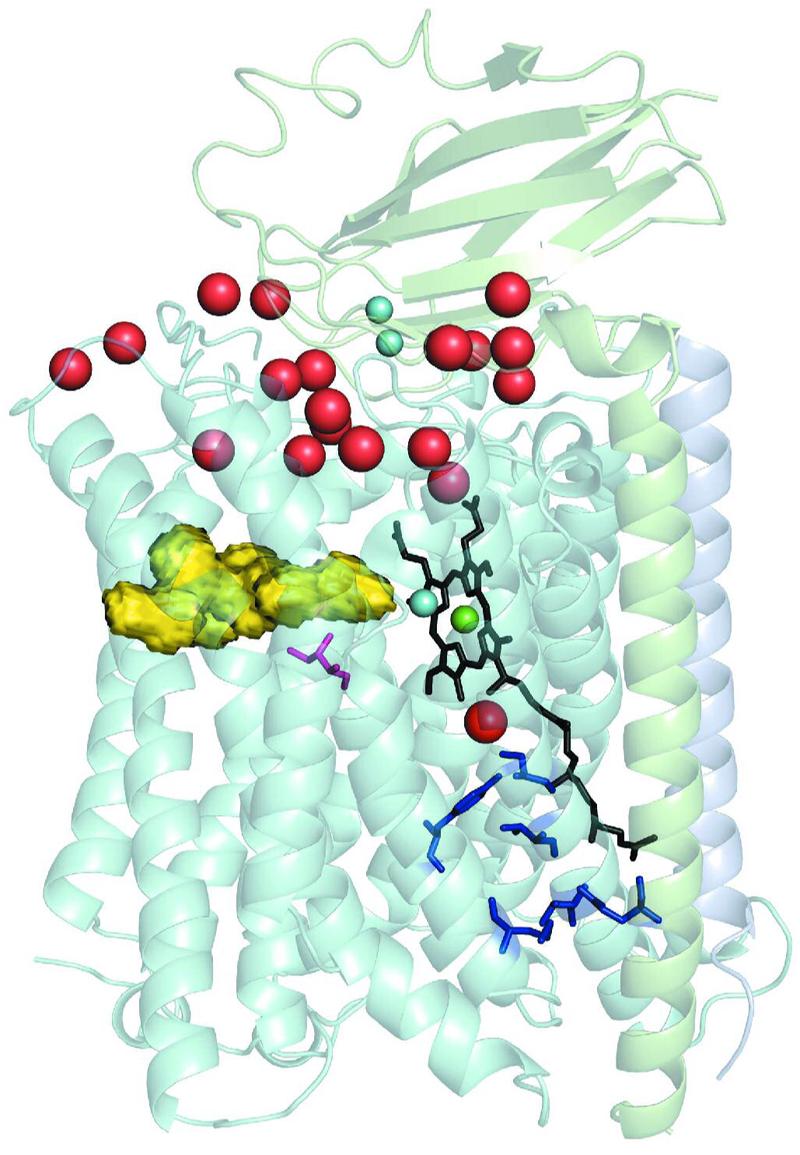
Representation of the Thermus cytochrome ba3 structure using translucent ribbons. The molecule is embedded in a phospholipid bilayer whose OUTside is roughly parallel to a plane through the lowest water molecules (red spheres) and whose INside lies just below the blue spheres near the bottom of the molecule. Subunit II, shown in light gray, is at the top with its single transmembrane element running down the right side of the structure. Subunit IIa, being a very short transmembrane segment running from N-(top) to its C-terminal (bottom), is shown in light mauve. Subunit I, shown in light blue, is composed of thirteen transmembrane segments. Within the helices of subunit I are two heme moeities: The a3-heme is shown in black with its long, hydrophobic, geranylgeranyl tail running from near the center of the molecule downward, the Fea3 at the center of the a3-heme is shown in green, and just to the left is the cyan CuB. To reduce clutter in the picture the b-heme, that lies behind the a3-heme is not shown. The dinuclear CuA center is shown as a pair of small cyan spheres near top and center. The larger red spheres at the top of the figure represent some of the water molecules residing near or in the space between the globular domain of subunit II and the concavity of subunit I into which the globular domain of subunit II intrudes. The yellow object to the left just above center represents the inner surface of the oxygen uptake channel that leads from near the center of the membrane bilayer to the dnc19. The pink colored residue center left and below the oxygen channel represents residue I-Ile235, which is replaced by a glutamate residue in A1-type oxidases. The blue structures near the bottom right of the structure represent the currently identified elements of the K-pathway for proton uptake with the lowest representing II-Glu15 whose Cα atom is situated near the N-terminus of subunit II; other residues shown are listed in the text. The red ball just below the green Fea3-atom corresponds to HOH O32 and might be thought of as the exit terminus of the K-path.
It is important to visualize the functioning of the system in light of the structure. Briefly, four redox centers are present in the enzyme: (1) CuA in subunit II is a homo-dinuclear Cu-dimer that serves as the primary site of electron entry to cytochrome c oxidase [see22 and references therein]; (2) a six-coordinate (6c) low-spin heme (not shown here), which, in the case of cytochromes aa3 is an A-heme and in the case of cytochromes ba3 is a B-heme; (3) a variably coordinated A-type heme (5- to 6c) in both classes of enzymes; (4) and a single Cu ion (CuB) that is ∼4.5 to 5.5 Å distant from Fea3. This Fea3 - CuB complex [(3) and (4)] is here referred to as the dinuclear center [dnc, see23for abbreviations used].
Oxygen molecules in the lipid bilayer enter the hydrophobic channel that leads directly to the reaction chamber19. As electrons move onto O-atoms of O2, protons arrive from the proton K-path, that originates at or near II-Glu15 some 14 Å from the secondary alcohol of heme-a3 (Fig. 1). As water molecules are released from the metal ions, they are repelled by the hydrophobicity of the O2-channel and attracted by the hydrophilicity of the watery region between subunits I and II. Finally, the pumped protons are released above the two propionate side chains of the a3-heme into the water cluster19, 24.
Structural differences between the A1- and B-type enzymes that influence enzyme mechanism studies are the presence of the isoleucine side chain that is isopositional with the central glutamate residue of A1-type enzymes, and the seeming absence of either D- or Q-proton transfer pathways in the B-type enzymes25. Clearly, the central glutamate residue plays an integral role in both electron transfer events and proton translocation by A1-type enzymes [see26-31and references therein], but this cannot be true of the B-type enzymes, which also differ from A1-type enzymes in several chemical behaviors32-34.
Mechanisms of dioxygen reduction
Given their structural and functional similarities, A- and B-type enzymes are likely, at least, to have a common mechanism of dioxygen reduction, and a summary of this process is roughly shown in Fig. 2, where we use the reaction times of Siletsky et al.35 for Thermus cytochrome ba3; four intermediates were reported. Compounds A, P (with P likely composed of both PM and PR), and O were identified as similar to their analogs in A1-type enzymes. However, unlike the A1-type enzymes, the spectral features of F were found to be highly similar to those of P. Unfortunately, optical absorption spectra of heme-a3 do not clearly report on the chemical status of either CuB or Y′OH.
Fig. 2.

General schematic of the catalytic cycle of cytochrome c oxidase, with the time constants, where known, taken from the recent work of Siletsky et al.35. We presume that P in their scheme consists of both PM and PR.
Even for the extensively studied A1-type enzymes, there is great variation in the content of this “wheel” in the oxidase literature9, 31, 36-38. Generally, the reaction of reduced enzyme, R, is initiated by photolysis of the a3-Fe-CO adduct of the enzyme in the presence of dioxygen39, and changes in a wide array of properties are recorded from the nanosecond to the second time frame [see40 and references therein]. The well established chemical steps are R to A, in which the 2-electron reduced dnc binds O2 at Fe(II)a3 with the appearance of optical absorption properties indicative of a heme-a3-Fe-O2 complex A [see40 and references therein]. At 25 C, this reaction occurs in a few μs41. Step A to PM occurs in a few tens of μs, most obviously upon photolysis of the mixed-valent form consisting of the oxidized, low-spin heme-A and the oxidized CuA center while the reduced dnc is occupied with CO that is photolyzed to start the reacton; this experiment is yet to be carried out with ba3. Spectroscopy and 18,18O2 exchange kinetics into bulk water [see42 for details] show that PM is not a peroxide containing compound, as implied in earlier notation, but one in which the O-O bond has already been broken. Moreover, the O-atoms are reduced to the level of two O(-II)-atoms, but neither is protonated to H2O43-46.
As shown in Fig. 2, already in PM, four electrons have been transferred from the two-electron reduced a3-CuB bimetallic site to O2, with newly formed “holes” on Fe(IV), Cu(II), and the His-Tyr conjugate as Y′O●, while two electrons remain associated with the CuA center and with the 6c heme. Next, PM to PR occurs when an electron arrives at the dnc, apparently to reduce Y′O●. PR to F is yet a slower reaction and F still contains the Fea3IV=O structure47. Among A1-type enzymes, a compound F-like substance is formed by exogenous H2O248, and in the case of the A2-enzyme, cytochrome caa3, shows Mössbauer properties characteristic of an intermediate-spin, S = 1, Fe(IV) electronic structure49. The conversion of F to O occurs on a millisecond time scale as the final two electrons, one on the 6c, low-spin heme and one on CuA, reduce Fe(IV) to Fe(III) and restore Y′O● to Y′O− (or Y′OH) thereby yielding the O form of the enzyme. During catalysis, in order to regenerate R, the enzyme must accept additional electrons from cytochrome c.
Approach and rationale
We propose a detailed chemical scheme whereby a B-type oxidase carries out dioxygen reduction and H+-translocation (HT) without the complicating factors of A1-type enzymes. Density Functional Theory (DFT) calculations with four types of theory are presented that nicely replicate the energetics of the overall, catalyzed reaction and provide strong support for the proposed mechanism.
The computation uses an active-site model that is assumed to be uniquely responsible for overall enzyme function. We then carry out a step-by-step reduction of O2 by this entity and compute minimized energies, E0K, of all intermediates proposed in the catalysis. The 298 K free energies of e− (standard hydrogen electrode), H+(aq), O2(aq.) and H2O(liq) were also computed. Thereby we obtain the information needed to calculate theoretical reaction free energies for the each step of the catalytic process [see50, 51 and Appendix B]. The object of the computational experiment is to determine if a chemical path can be articulated for coupled O2-reduction/H+- translocation that does not present with insurmountable energy barriers.
We generally follow the path of O2-reduction shown in Fig. 2 and incorporate others' ideas about the mechanism for breaking the O-O bond52. The vectorial nature, i.e., moving chemical entities from one side of the membrane to the other53, of the proposed chemistry is founded on the presence of reasonably placed INput- and OUTput gates that are not, however, included in the quantum calculations. Sites of proton uptake and release on the active site model are intuitively assigned, and proton “pumping” is incorporated through the device of binding all protons to the active-site structure from a “pH 7 bath” and removing “pumped” protons from a fixed site on the dnc model into a “pH 3 bath”.
Since the 1977 discovery by Wikström2 of proton pumping by the oxidase, there have been numerous ideas published that seek to explain this phenomenon [see2, 9, 31, 36, 37, 54-65 and references therein]; all suffer from the age-old problem that the spectral features of the heme moeities, in general, do not reveal what is going on at CuB. Therefore, what is proposed to be occuring at CuB in any mechanism is necessarily spectulative.
The power of DFT to explore the fundamental properties of increasingly large molecular assemblages, including those containing transition metals66, is now being used to explore and refine the mechanisms of metalloenzymes67-72. Indeed, there are several reports of theoretical studies of H+-translocation by A1-type cytochrome c oxidases [see73,74,75,52, 76-79,80-85,86, 87,88, 89,90,91, 92and references therein]. Several of these works explicitly include the internalized glutamate residue of the A1-type enzymes73, 84, 93. Because experimental methods currently fail to parse the chemical grammar of CuB and to monitor the presence of Y′O●, theoretical approaches provide and will continue to provede useful tools to examine the roles of Fea3, CuB, and Y′O● in cytochrome oxidase mechanism. These pioneering efforts contribute to an evolution of ideas that move us toward the goal of a consensual mechanism for proton pumping, and it is in that context that our work rejoins the recent comment from Kim, Wikström, and Hummer that “Even though many models have been proposed to explain [- - -] proton pumping, the central question remains unanswered: How can redox chemistry be harnessed to move protons against both chemical and potential gradients?94” Here we provide such chemistry.
Defining the problem in chemical terms
Creation and properties of the quantum model
Keeping track of structures and changes
Fig. 3 shows two views of the active-site model. Fig. 4 shows a two-dimensional representation of structure 1 that emphasizes valences and ligation and protonic states. This figure also shows the additional components that form parts of the INput and OUTput gates. A further contraction of 3-D information is shown below in Fig. 5. The shorthand notations used to depict metal valences and ligation states and protonation states are presented in detail in the Supporting Information. These provide the information needed to follow the structural changes that occur between the individual steps of the proposed mechanism.
Fig. 3.
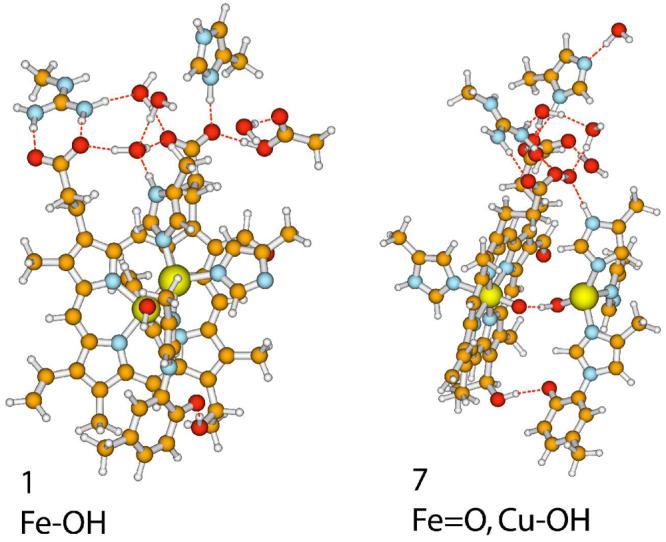
Two views of the quantum model of the ba3-active site. Left. Structure 1,in which Fe(III) coordinates an OH− and Cu(II) binds only the three histidine imidazole rings of the protein. The view is directly down the Fe-O bond with the H of OH pointing toward the bottom. The macrocycle is nominally in the plane of the paper. The Cu-atom appears to the right and above the Fe, and the NE-atom of I-His283, directly above the Cu, is shown coordinated to the Cu while its ND-atom hydrogen bonds with the water network top center. The ND-atom of I-His282 center right is deprotonated in this structure as is the phenol O-atom of I-Tyr237 lower center. The imidazolium cation of I-His376 is shown at the top of the structrure. Right. Structure 7, in which the a3-heme contains the Fe(IV)=O “ferryl” specie and Cu(II) coordinates an HO− can be seen in the center of the active-site model. The proximal ligand to the Fea3 is the imidazole group of I-His384 and is shown center far left. The covalent linkage between I-Tyr237 and I-His233 is shown on the lower right along with the coordination of the I-His233 imidazole ring to CuB. The close interaction of the phenol O-atom with the secondary alcohol of the truncated geranylgeranyl side chain of the a3-heme is shown center bottom. In this structure, a pendant water, upper right, donates an H-bond to the ND-atom of I-His376 (see Fig. 5). Structural files of all intermediates are available in the Supporting Information, which can be downloaded and opened in a viewing program, for example, Molden or Maestro. The structures are best analyzed in conjunction with 2-D plans of Fig.5 also available in Supporting Information.
Fig. 4.
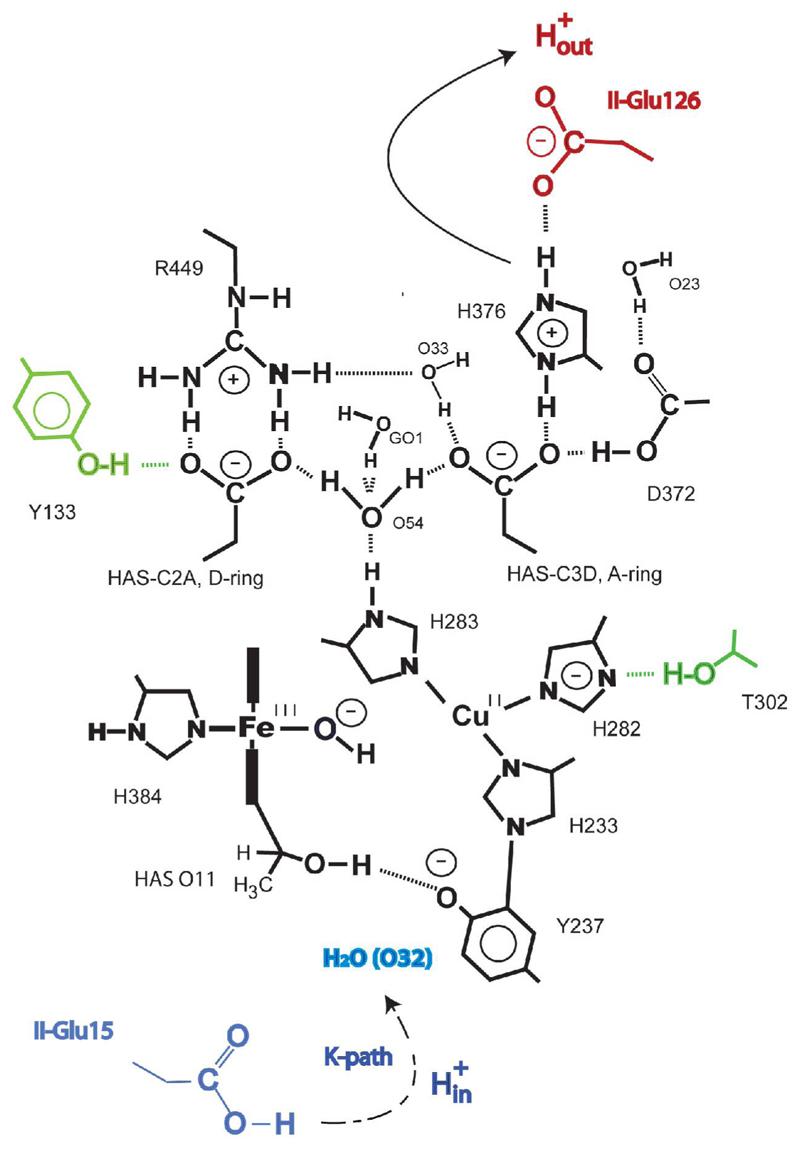
A two-dimensional representation of the active-oxidized (1) form of the catalyst that is useful in keeping track of metal valences, metal ligation, and the various protonation / deprotonation steps that change during the catalytic process. The plan is best understood by placing it side-by-side with Fig. 3. In the lower-left quadrant is an edge-on view of the a3-heme with the Fe(III)-−OH moeity in the middle (see structure 7 of Fig. 3) and the secondary alcohol with its connection to I-Tyr237 at bottom. To the left of the heme plane lies the proximal imidazole ring of I-His384. To the right of the heme Fe-atom lies the Cu, which is coordinated to its three imidazole rings labeled according to their amino acid designation. I-His233 extends downward and makes the covalent link to I-Tyr237. I-His282 extends to the right, and its potential interaction with the OG-atom of I-Thr302 is shown. In the upper left quadrant lies the D-ring propionate and its contact with the guanidinium group of I-Arg449. In the upper right quadrant lies the propionate of the A-ring and its interactions with the imidazolium cation of I-His376. The latter lies directly below II-Glu126 thought to make an additional salt-bridge, H-bonding arrangement with the neutral I-Asp372 carboxyl group. The water cluster lies in the upper half of the plan (see structure 1 of Fig. 3) and shows the connection between it and the “top” histdine of Stuchebrukhov and colleagues83. Five colored moeities shown in this depiction are not included in the quantum model starting clockwise from II-Glu15, I-Thr302, HOH O32, II-Glu15, and I-Tyr133. Similar plans for each intermediate along with their relationship to the two additional plans used in expressing the state of the catalyst in shorthand notation (see Fig. 5).
Fig. 5.
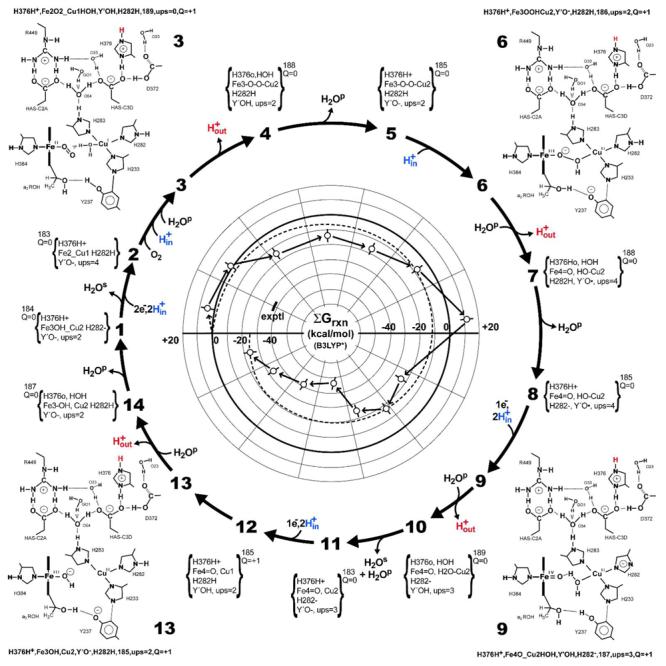
Polar representation of the 14-step catalytic mechanism, showing the stoichiometric reactions, including proton translocation. In each corner, starting in the upper left quadrant, are 2-D plans of structures 3, 6, 9,and 13 that represent the state of the system prior to removal of a proton, H+out, from I-His376. To each of these is attached one-line codes that summarize the protonation status of the four proton binding residues in the dnc; metal valence, ligation and the states of ligand protonation, number of atoms, number of unpaire delectron spins (ups), and overall charge of each intermediate, Q. The small 2-D representations of intermediates 7 and 8 that connect 6 to 9, structures 10, 11,and 12 that connect 9 to 13, and structures 14, 1,and 2 that connect structure 13 to structure 3 are contractions of the one-line codes. The three codes for each structure are provided in the Supporting Information. The central polar plot of the running sum, Σ(ΔGrxn), shows the energetic course of catalysis as computed at the level of B3LYP* (see Fig. 6). A straight line (dashed) is plotted between zero at structure 1, and −38.7 kcal/mol (short line) at the end of the reaction between 14 and the return to 1 corresponds to the experimental reaction free energy for the overall reaction.
Construction details
The initial positions of the side chains potentially involved in catalysis were taken from the 2.3 Å resolution, X-ray structure of as-isolated, recombinant cytochrome ba3 [PDB 1xme18]. The quantum model was extracted from the protein by breaking the CA – CB bonds of the histidine, aspartate, and tyrosine residues and the CD – CG of arginine listed in Table 1. A linking H-atom was added to fill the open valence of the terminal carbon atom, and this C-H distance was initially set to 0.95 Å. The xyz-coordinates of these H-atoms were fixed in subsequent geometry calculations; the C-H bond length was not fixed. The active site complex consists of CuB; I-His233, I-His282, I-His283, I-His376; heme a3 (HAS 801), I-His384; I-Tyr 237, I-Asp372, I-Arg449; the Fea3 and CuB are the only metals involved.
Table 1.
Components and interactions within the quantum model and with the surrounding protein structure.
| His233: ND1 → CuB and NE2 covalently linked to CE2 of I-Y237 |
Tyr237:
|
His282: NE2 → CuB
|
His283: NE2 → CuB
|
Asp372:
|
His376:
|
His384: NE2 → Fea3 of heme As
|
Arg449:
|
HAS801:
|
Four water molecules
|
All structural data taken from PDB entry 1xme.
The xyz positions of the metal ions were generally fixed, however, two sets of coordinates were used. For structures 1, and 11 through 14, those from 1xme were used. Here the Fe – Cu distance is ∼4.4 Å. For structures 2 through 10, coordinates of the chemically reduced, carbonylated structure were used. Here the Fe to Cu distance is 5.2 Å95. Heme a3 was “extracted” by cleaving the bond between HAS C12 and C13 and attaching an H-atom to C12 as described above for the CA atoms. The proximal histidine ligand to Fea3, I-His384NE, was moved ∼0.4 Å toward the Fe, so as to form an initial bond of ∼2.0 Å96. This allowed the Fe-N bond to move within a reasonable range during geometry optimization. Four waters of crystallization: HOH O54, HOH O23, HOH O33, and an HOH added at the position of O1 of the bound glycerol were also included18. Table 1 also summarizes the components of the model, their internal interactions, and their interactions with atoms in the cavity from which the active-site model was “extracted.”
Formal charge considerations
Rich and colleagues97, 98 have argued the importance of maintaining a net zero electric charge on the dnc during O2-reduction. Our model generally follows this principle. We include the cationic guanidinium group of I-Arg449 to compensate the ring D, C2A propionate in a doubly H-bonded arrangement, and the cationic imidazolium group of I-His376 to compensate the C3D propionate of ring A; the latter interaction changes during the computational experiment. I-Asp372 is included as a neutral carboxylic acid, donating an H-bond to the ring A, C3D propionate and accepting an H-bond from HOH O23; this arrangement does not change during the computational experiment. Structure 1, Fig. 4, has a net zero charge: The formal 3+ on Fe(III) are compensated by 2- from the deprotonated pyrrole rings of the porphyrin while the remaining 1+ charge on Fe is compensated by the HO− ligand. The formal 2+ of Cu(II) is compensated in structure 1 by loss of a proton from ND1 of I-His282 to form an imidazolate and from Y′OH to form a phenolate. These seem to be reasonable choices because in each case the deprotonated atom, ND1 or OH, is uniquely positioned to accept a H-bond from a weak Bronsted acid: In the case of Y′OH, the alcohol group on C11 of HAS is only 2.67 Å (heavy atom distance) from the phenol O-atom of I-Tyr237. This structure is used as the active, oxidized form of the quantum model99.
Overall geometry
The isolated model of the dnc can be viewed as consisting of four domains that interact with each other in chemically interesting ways: The Fe domain, the water domain, the copper domain, and the phenol/phenolate/K-path domain (see Figs. 3 and 4).
The Fe-domain is comprised of the heme a3 with the NE atom of I-His384 as the proximal ligand, and its distal pocket for exogenous ligands is analogous to the atomic arrangement observed in dioxygen transporting heme proteins.
The water domain is comprised of a cluster of water and polar amino acid residues some of which form a polar layer marked by the positions of the two propionate groups. Some of these are included in the quantum model. This cluster is neutral and presumed to contain neither H3O+ nor OH− but it is highly polar. At the boundary between Fe- and the water cluster is water O54, which bridges the two propionates by H-bonding to an O-atom on each carboxylate. This is clearly visible in Fig. 3. Water O54 coordinates to the glycerol O1 “water mimic” in the Hunsicker-Wang structure18 and forms an H-bond with protonated ND of the NE-CuB-coordinated I-His283. In this arrangement, water O54 is surrounded by a shell of four H-bonds in a roughly tetrahedral arrangement, and the ring of I-His283 serves as a bridge from CuB to the water cluster. We suspect, as have others74, 83, that water O54 plays a key role in moving protons into the space between subunits I and II; although in our model it is never protonated to H3O+. Water O33 bridges from the NH of I-Arg449 to the carboxyl (C=O) O2D atom of HAS-C3D propionate, while the other O-atom of this carboxylate accepts an H-bond from the free acid on OD1 of I-Asp372. The carboxyl group of I-Asp372 accepts an H-bond from water O23. ND of His376 is also in H-bonding distance of the propionate atom HAS-C3D/O1D.
The Cu-domain includes two histidine imidazole side chains (I-His282 and I-His283) that are NE ligated to CuB. A third ligating imidazole ring, ND of I-His233, is additionally NE-alkylated at CE2 of I-Tyr237. The three N-atoms coordinated to CuB form a plane above the position of the Cu that is slightly canted toward rings A and D of the a3-macrocycle100 and the distance between the Cu and the Fe varies from ∼4.5 to 5.5 Å depending on valence and ligation states, as derived from available X-ray structures. This arrangement leaves a coordination site on CuB roughly facing a coordination site on Fea3, both of which are open to binding small exogenous ligands. The side chain of I-Val236 contacts the CB atom of I-Tyr237 and ring C of HAS, thereby providing additional steric blocking. Trans to the open coordination position on the CuB, however, are hydrophobic residues that prevent solute access to the “back side” of CuB. For example, the indole ring of I-Trp229 is stacked on the ring of I-His283. Both I-W229 and I-Val236 play a role in the functioning of the O2-channel where they appear to “guide” the incoming O2 molecule into the reaction chamber19.
The phenol/phenolate/K-path domain completes the ring of domains and contains the covalent link between NE of I-His233 and C2 of I-Tyr233. The geometry of this region places the phenol O-atom of I-Tyr233 in H-bonding distance to the secondary OH group (HAS O11) of the hydroxyethyl-geranylgeranyl side chain of heme As.
Continuing away from the dnc, HAS O11 is in H-bonding distance to HOH 32, which although not included in the model, is 2.69 Å from the OG atom of I-Ser309 continuing “down” to II-Glu15, which apparently serves as the entrance to the K-pathway (Fig. 4). In our scheme, protons from the pH 7 proton bath are added to Y′O− at will.
Other interactions of the active-site model with the protein
Examination of the of the hydrophobic cavity obtained after “extraction” of the quantum model reveals a few additional interactions that may be important in enzyme function (see Fig. 4): Protons are likely to enter from the K-pathway. The distance from the secondary alcohol (HAS O11-atom) to HOH O32 is 2.67 Å and the distance from HAS O11-atom to the phenol OH of Tyr237 is 2.69 Å. Water O32 might justifiably have been included in the quantum model [see Kaukonen74], even though little is known about how protons actually move into the reaction chamber.
Atom OG of I-Thr302 and the backbone carbonyl O-atom of I-Val279 are both within H-bonding distance of the ND1 atom of I-His282 (see Table 1), similarly the OG atom of I-Thr302 is within H-bonding distance to the carbonyl O-atom of I-His298 resulting in a complicated network of H-bonds that may or may not support the chosen protonation states. While our energetic data support the idea that I-His282 can be deprotonated, the involvement of these residues in regulating the protonation state of I-His282 deserves further consideration. The carboxylate group of the HAS O1A-atom (propionate of ring D) is in H-bonding distance of the phenol O-atom of I-Tyr133, neatly mirroring the interaction of the OD2 atom of the ring A HAS propionyl carboxylate interaction with the protonated OD1-atom of I-Asp372. The interactions with I-Thr302 and I-Tyr233 were not included in the model.
From its bottom (Fig3left and Fig. 4), up to a rough plane formed by the two propionates and I-Asp372, this cavity is quite hydrophobic (except for HOH O32 and OG of I-Thr302), suggesting a relatively low dielectric environment. Above this plane, moving toward the interface between subunits I and II, the dielectric environment may be closer to that of water. We estimate the size of the water cluster at ∼8 – 12 molecules, which could vary during catalysis. One side of the water cluster interacts with the propionate assemblage while the other side interacts with hydrophilic residues of subunit II. Ion exchange reactions, ingress of small molecule inhibitors (including those as large as glycerol18), and egress of protons and water molecules most likely occur along paths in the crevice between subunits I and II19.
Proton gating
The scheme proposed here for coupled dioxygen reduction / proton translocation is driven by coupling non-vectorial chemistry in the dnc to protons being taken up by Y′O− at pH 7 and removed from H376H+ at pH 3. The validity of this approach follows from the fundamental nature of the transmembrane proton gradient, namely, that ΔμH+ is the sum of the free energy due to the ion gradient and the concentration gradient, i.e., ΔμH+ = Δψ − RT(ΔpH)/ℑ, where ℑ is the Faraday constant and Δψ is the charge gradient10, 53. The enzyme embedded in a closed membrane vesicle “vectorializes” this chemistry by using input and output gates that limit back-flow of protons from the OUTside to the INside of the membrane. While the functioning of these gates is poorly understood, the combination of 3-D structural information, site-directed mutations, and comparisons to bacteriorhodopsin allow useful hypotheses. Because the overall reaction can be at least partially reversed in closed vesicles, the gates must be microscopically reversible101.
The input gate
The X-ray structure reveals a possible proton pathway [as first noted by Soulimane et al.17] from II-Glu15 to the dnc, and mutational studies25 suggest that the K-pathway consists nominally of I-S309, I-T312, I-T315, I-248Y and I-244Y and is the only route for protons from the INside to enter the reaction chamber; the distance between the heme alcohol HAS O11 and II-Glu15 OE1, which is ∼14 Å, gives one a sense of the linear distance the protons must move to enter the dnc. Experimentally, mutation of II-Glu15 to either Gln or Ala results in an inactive enzyme with a full complement of redox co-factors (Y. Chen and J. A. Fee, unpublished).
The carboxyl group of II-Glu15 resides in a relatively hydrophobic pocket in which the OE1 atom of II-Glu15 is 3.51 Å from the SD atom of Met264. It is reasoned that the free acid of II-Glu15 resides in this pocket ready to donate its proton to a more basic atom “upstream,” possibly via a Grotthuss mechanism [see102-104 and references therein] along a “local area network of hydrogen bonded water molecules and amino acid side chains105.” The X-ray structure suggests that upon giving up the proton, its side-chain may rotate about the CB - CG and CG - CD bonds to swing out of its hydrophobic pocket into the aqueous INside, where it can bind a proton before returning to the pocket. Like Asp96 of bacteriorhodopsin106 II-Glu15 is considered to be relatively basic and therefore almost always protonated107.
The exit gate
The X-ray structure shows strong H-bonding between the NE2 atom of I-His376 imidazole ring and the OE2 atom of the II-Glu126 carboxylate (2.79 Å). I-His376 is H-bonded to the O1D atom of propionate (HAS-C3D), and being further constrained by the side chains of residues I-Asn377 and I-Ala286, this side-chain has little space in which to move. By contrast, the carboxylate of II-Glu126 is able to rotate about its CB - CG and CG - CD bonds thereby being unconstrained to swing into the OUTside of the molecule where there are numerous water molecules (glycerol binds here) and charged side-chains, including I-Arg225. It is reasoned that the pKa of II-Glu126, when pointing OUT, is low, i.e., it is a relatively strong acid. Only when the H+ on I-His376H+ has a lower affinity for its H+ will it be passed to II-Glu126, which is reasoned to rotate out, lose the proton, and rotate back to form a salt-bridge with a newly protonated His376H+. In this fashion, I-His376 and II-Glu126 may serve to gate protons OUT of the molecule while preventing back-flow of protons from the OUTside into the reaction chamber. This is highly similar to the situation with Asp85 of bacteriorhodopsin108, however, see109. The extended features of the INput and OUTput gates, though important to understanding the model, are not included in the quantum calculations.
Methods
Computational details
Gas phase electronic energies of small molecules
Dioxygen, dihydrogen, H2O, and H2O2 structures were optimized in the gas phase at the levels of PW91 and OPBE using ADF2006.01 as described below66, 110, 111. The sub-key FREQ was used at optimal geometry to obtain thermochemical information at 298 kelvin (see below). Because ADF programs do not currently support geometry optimization at the hybrid levels of B3LYP and B3LYP*, geometry optimization was carried out using the Jaguar program112, which also provides 298 kelvin thermochemical data. Subsequently, single point calculations at the levels of B3LYP and B3LYP* were done in ADF2007.01 to obtain zero kelvin energies with respect to ADF reference fragments. The standard ADF reference fragments are spin restricted atoms66, 110, 111. For our purposes, the important point is that for balanced chemical equations these reference fragment energies drop out when chemical energy differences ΔE and ΔG are calculated. The data are provided in the Supporting Information.
Free energies of small molecules
Free energies at 298 kelvin in aqueous solution of H2,O2,H2O, and H2O2 were obtained as follows (see also Appendices A and B). For liquid water, the absolute free energy of water in water at 298 kelvin as computed in PW91 is Gxc(H2O)liq298(eV) = Exc(H2O)0Kgas + ZPVE + U298 − T•S plus the solvation free energy of H2O by water (−0.089 eV). Here Exc(H2O)0Kgas is the zero kelvin energy of gaseous H2O computed in PW91, ZPVE is the zero point vibrational energy of gaseous water [see113], U298 is the internal energy of the water molecule at 298 kelvin, and T•S is 298 K times the molar entropy of gaseous H2O. Term by term, for the case of PW91, Gpw91(H2O)liq298(eV) = −14.1394 + 0.5582 + 0.0766 − (298.15 × 45.17e-3 / 23.06) − 0.089 = −14.1549 eV with respect to the ADF fragments (see below). Similarly, Gxc(O2)aq298(eV) = Exc (O2)0Kgas + ZPVE + U298 − T•S plus a term to change the historical reference state of O2 from one atmosphere of pure gas to the more biochemically relevant 1 M O2 in water [ΔG = −RTln (1.25e-3 M /1 M)) = + 0.172 eV, see114]. Term by term, in the case of PW91, Gpw91(O2)aq298(eV) = −9.6712 + 0.094 + 0.0664 − (298.15 × 46.89e-3 / 23.06) + 0.172 = −9.9450 eV with to respect to the ADF fragments. More detail can be found in Appendix B, and the resulting data are presented in the Results section.
Free energies of the ions e− and H+
Our computations include half reactions of the type, e− + H+ + I → I-H, where I is an intermediate of catalysis. Here a combination of theory and experiment is used to obtain appropriate 298 kelvin free energies for e− and H+ that relate to the ADF reference fragments. Typically, we have used the combined calculational/experimental value of the standard (or normal) hydrogen electrode (SHE)50, i. e., G(SHE) = −4.36 eV and an expression for Gxcwater(H+) = Exc0K(H+)gas + ΔGsolv(H+) − TΔSgas(H+) + 3/2 RT + pV, where, for example, in the case of PW91: Epw910K(H+)gas = +12.5150 eV; ΔGsolv(H+)= −11.37 eV [−262.1 Kcal/mol, which is the best DFT determined, free-energy of solvation of the proton in water115]; −TΔSgas(H+) = −0.34 eV is the entropy of 1 mole of gaseous H+ at 298 kelvin; 3/2 RT is the translational energy of a mole of protons in the gas phase and pV (=RT) is the work term to account for the one-to-two volume change in the gas phase reaction (BH → B− + H+ where B− is any Bronsted base); and to determine Gxcwater(H+)at any pH, −0.059 pH must be added to the above expression. As will be considered in the Results section, more satisfactory outcomes were obtained for reaction energies of half reactions when Gxcwater(H+), calculated in the above manner, was referenced to the normal hydrogen electrode: 2 G(SHE)expt + 2 Gxcwater(H+) =G(H2)298gas (see Appendix A).
Free energy of proton pumping
The value of ΔGo for proton pumping at 298 kelvin does not depend on input from theoretical calculations and is given by ΔGo = −0.059 × (ΔpH = − 4) corresponding to + 0.236 eV per proton “pumped” from a medium of pH 7 to one of pH 3 (an uphill step of 5.54 kcal/mol-proton).
Geometry optimization
Density functional calculations were performed using the Amsterdam Density Functional packages (ADF-2005.01, ADF-2006.01, and ADF2007.01) 66, 110, 111. Initially, the parametrization of Vosko, Wilk, and Nusair (VWN) for the local density approximation (LDA) term116, and the corrections of Perdew and Wang (PW91)117 were used for the nonlocal exchange and correlation terms. The geometries of all intermediates were optimized in the COSMO (conductor-like screening model) solvation model in ADF [see118, 119 and references therein], somewhat arbitrarily using a dielectric constant of a simple ketone (18.5). (It was found to be important to use the sub-key, SCF var ALL, when implementing the SOLVATION key in ADF.) In this type of calculation, the substance of interest is embedded in a molecular-shaped cavity surrounded with the chosen dielectric. The radius of the probe that defines the dimensions of the cavity was in all cases 1.4 Å. The energy of the interaction between the solvent and the solvated molecule is thus calculated, and the zero kelvin energy obtained is that of a molecule in a solvent of the given dielectric constant. During geometry optimization triple-zeta plus polarization (TZP) basis set was applied to the Fe and Cu atoms and the double-zeta plus polarization (DZP) basis set for other atoms. The inner cores of C(1s), N(1s), O(1s), Fe(1s,2s,2p), and Cu(1s,2s,2p) were treated by the frozen core approximation.
The total number of unpaired spins (ups) was assumed to be the maximum available to the system, and ferromagnetic coupling was assumed for simplicity. Aside from states 4, 5, and 6 where the Fe(III)-O22−-Cu(II) and Fe(III)-−O2H-Cu(II) bridges are strong and may be antiferromagnetic, other states are likely to exhibit only weak interaction between the Fe and the Cu. In addition, states 7 and 8 may involve antiferromagnetic linkage of Cu(II) and Y′O●. For the active oxidized intermediate, 1,in which CuB is in the Cu(II) state, both high-spin and corresponding broken-spin states of the dimetallic complex were considered for both high- and low-spin Fe(III) [see99 and120 for details of spin-flipping]. Future studies should consider alternative spin-states.
Because of the large size of the intermediates (183 to 189 atoms), modest relaxation of electronic and geometric structure optimization criteria were permitted, including a decrease in the sub-key, lshift 0.3 err=2e-6, rather than the default 1e-6 and CONVERGE 5e-4 rather than the default 1e-4 hartrees. The accuracy parameter for the numerical integration grid was 4.0. These criteria were maintained for all geometry optimized structures. All raw data are presented in the Supporting Information in Tables 1-SI, 2_SI, and 3_SI.
When computing optimum geometries in PW91 of structures from which the proton was removed from H376H+, it was apparent that there was significant resistance to convergence. Fortunately, adding a pendant HOH as an H-bond donor to NE of neutral I-His376 in these four structures greatly decreased the number of iterations, both SCF and GO, required to achieve optimum geometry. In this fashion, optimized structures 4, 7, 10, and 14, each having this pendant HOH, were obtained. All structures can be found in the Supplementary Material as .xyz files. This paper deals with the fourteen structures obtained in PW91 and shown schematically in Fig. 5. These structures were used in subsequent, single-point SCF calculations at three different levels of theory.
Calculation of reaction free energies
The reaction free energy of each transformation was calculated from ΔG = Σ G(products) − Σ G(reactants), in which, for example, the conversion of 1 to 2 in Fig. 5, 2 e− + 2 H+(pH7)ZPVE + 1 ® 2 + H2Os is calculated{G(2) + G(H2Os) − [G(1) + 2 G(SHE) + 2 G(H+)(pH7)}. Taking numerical values for each G from Table 2 (see Results) and Table 2_SI, that were computed in PW91, we obtain {+6.5138 + (−14.7361) − [0.0000 + (−4.56 × 2) + 0.2200 × 2} = 0.4577 eV = +10.55 kcal/mol. See also Table 3_SI and Appendix B of Supporting Information. Data are presented in the Results section.
Table 2.
Free energies at 298 kelvin in eV obtained at four levels of theory and referenced to the ADF fragment. Data that was used in the calculation of reaction free energies.
| Exchange correlation potential | ||||||||
|---|---|---|---|---|---|---|---|---|
| Species | PW91 | OPBE | B3LYP | B3LYP* | ||||
| G298 in eV referenced to ADF fragments | ||||||||
| e− (pH 7)a | −4.36 | −4.36 | −4.36 | −4.36 | ||||
|
e− (pH 7, cyt. c2+)a |
−4.56 | −4.56 | −4.56 | −4.56 | ||||
| H+gas(corr)b | 12.5672 | 12.5664 | 12.1597 | 12.2908 | ||||
| H+aq (pH 7) | 0.5080 | 0.5074 | 0.1007 | 0.2318 | ||||
| H+aq (pH 3) | 0.7440 | 0.7434 | 0.3367 | 0.4678 | ||||
| AZPVEb | 0.288 | 0.284 | 0.288 | 0.285 | ||||
|
H+aq (pH 7) −Δ ZPVE |
0.2200 | 0.2234 | −0.1873 | −0.0532 | ||||
|
H+aq (pH 3) −Δ ZPVE |
0.456 | 0.4594 | 0.0487 | 0.1828 | ||||
|
H2 at 1 atm gas |
−6.8758 | −6.8791 | −7.6926 | −7.4304 | ||||
|
O2 at 1 atm gas |
−10.1190 | −10.4793 | −11.8255 | −10.9137 | ||||
|
1 M O2 in water |
−9.9450 | −10.3065 | −11.6535 | −10.7417 | ||||
|
H2O at 1 atm gas |
−14.0886 | −14.1346 | −15.6997 | −14.9567 | ||||
|
Liquid H2Op |
−14.7361 | −14.7943 | −16.3640 | −15.6212 | ||||
|
Liquid H2Os |
−14.7361 | −14.7943 | −16.3640 | −15.6212 | ||||
|
H2O2 at 1 atm gas |
−17.9326 | −17.8934 | −20.5087 | −19.3356 | ||||
Search for the “best” exchange correlation potential
The relatively large size of the structures subjected to quantum calculations precluded an extensive search among available functionals for those that might yield the most coherent and chemically reasonable output. We used the PW91 geometry optimized structures mentioned above to obtain single-point, zero-kelvin energies at different levels of theory. Based on the suggestions of other researchers, we tested OPBE121-124, a non-hybrid functional often used as an alternative to PW91117, and two hybrid functionals, B3LYP [see125 and references therein], and B3LYP*126 that has already demonstrated favorable response to systems having multiple spins residing in orbitals of similar energies [see127 and references therein]. In OPBE, as for PW91, the VWN parameterization for the LDA were used for the non-local exchange and correlation terms. Spin unrestricted, single point SCF calculations were carried out at the levels of B3LYP and B3LYP* were carried out using ADF2007.01. In ADF, these calculations do not allow frozen cores and use different basis sets from those used by the non-hydrid exchange correlation potentials. From these calculations the cosmo (dielectric constant = 18.5) zero kelvin energies of all intermediates were obtained for each intermediate with each functional and made relative to state 1. Data are presented in the Supporting Information.
Results
Primary electronic energies
Small molecules and structures of the quantum model
The computed, 298 kelvin gas phase free energies of H2, O2, H2O, and H2O2 are presented in Table 1_SI of the Supporting Information. The computed, zero kelvin gas phase energies of the fourteen structures of the proposed catalytic cycle are presented in Table 2_SI of the Supporting Information. These data are referenced to the ADF fragments are used to compute the various reaction free energies described below. Values of Gxcwater(H+) obtained at the four levels of theory are also referenced to the ADF fragments. As noted in Methods, Gxcwater(H+) was also referenced to the normal hydrogen electrode: 2 G(SHE)expt + 2 Gxcwater(H+) = G(H2)298gas. All experimentally relevant, absolute free energies at 298 kelvin used in the subsequent calculations are listed in Table 2.
Mulliken charge and spin populations
Additional insight into correspondence between results from the different levels of theory are the Mulliken charge and spin populations listed in the output files of ADF calculations. The charge and spin populations are consistent with the parameters provided with the ADF input file: number of atoms, total charge and sign, and number of unpaired spins. These data are presented in Tables 4_SI to 9_SI.
Thermochemical results
Overall reaction catalyzed by cytochrome oxidases
The half reactions of concern are [1 to 4] along with the relevant free energy changes obtained from experiment and referenced to the standard hydrogen electrode at 1 M H+ and 1 M O2 [see10, 128 for experimental data concerning dioxygen potentials. The mid-point potential of Thermus cytochrome c552 is 0.2 V with respect to the standard hydrogen electrode129. See also114.
| [1] |
| [2] |
| [3] |
the sum of which is the reaction catalyzed by the cytochrome c oxidases
| [4] |
or using the Nernst equation, ΔGo′ = −nℑΔEo′ = ∼ −39.2 Kcal/mol at 298 K, which is close to the value of −38.7 kcal/mol obtained from thermochemical calculations and presented in Table 3. (Note that the change in standard state causes reaction [2] to appear more favorable when compared to the standard state for pure O2 being 1 atm gas.) We are now able to consider the catalytic process and make direct comparisons to the energetics of the overall reaction.
Table 3.
Reaction free energies of small molecules in kcal/mol at four levels of theory with comparison to experiment
| Reaction | PW91 | OPBE | B3LYP | B3LYP* | Exp'tl | ||
|---|---|---|---|---|---|---|---|
| [5] O2)gas + H2)gas --> H2O2)gas | |||||||
| −21.67 | −12.3 | −22.84 | −22.86 | −25.25 | |||
| [6] H2O2)gas + H2)gas --> 2 H2O)gas | |||||||
| −76.63 | −80.64 | −73.75 | −72.58 | −84.01 | |||
| [7] O2)gas + 2 H2)gas --> 2 H2O)gas | |||||||
| −98.3 | −93.01 | −96.59 | −95.45 | −109.3 | |||
| [8] H2O2)gas + 2 e−(SHE) + 2 H+)aq --> 2 H2O)liq | |||||||
| −80.69 | −84.84 | −77.87 | −74.04 | −88.11 | |||
| [9] O2)aq + 4 e-(SHE) + 4 H+)aq --> 2 H2O)liq | |||||||
| −106.28 | −101.16 | −104.68 | −103.54 | −117.4 | |||
| [4] O2 )aq + 4 e- (from cyt c2+) + 8 H+)aq(pH7) --> 2 H2O)liq + 4 H+)aq(pH3) | |||||||
| −27.97 | −22.85 | −26.37 | −25.23 | −38.7 | |||
In Eq. 8 and 9, pH = 0 and [O2]aq = 1 M. The free energy difference between Eq. 6 and Eq. 8 is due solely to the different standard states of H2O)gas (1 atm) vs. H2O)liq (55 M). Similarly, the free energy difference between Eq. 7 and Eq. 9 is due to the different standard states of O2)gas (1 atm) vs. O2)aq (1 M) and H2O)gas vs. H2O)liq.
Reation free energies
Small molecules
Table 3 compares the reaction free energies of gas phase reactions in which O2 and H2 combine to form H2O or H2O2 at 298 kelvin. For reaction [5] of Table 3, the formation free energy of gaseous H2O2 in PW91, B3LYP, and B3LYP* give values close to ∼ −22 kcal/mol, somewhat below the experimental value of −25.25 kcal/mol, while OPBE gives the anomalous ΔG of −12.3 kcal/mol. In reaction [6], in which H2 combines with H2O2 to yield H2O, all four functionals fall in the range of −72.6 to −80.6 kcal/mol, while their deviation from the experimental value of −84.0 kcal/mol is +4 to +8 kcal/mol. Similarly, in reaction [7] of Table 3, the four functionals fall in the range −93.0 to −98.3 kcal/mol but are up to ∼16 kcal/mol more positive than the experimental value of −109.3 kcal/mol.
For the half-reactions [8], [9], and [4], it was important to reference G(H+)aq to the standard hydrogen electrode condition that 2 G(SHE) + 2 G(H+)aq → G(H2)gas = zero (see Appendix A). In so doing, the reaction free energies for the four functionals fall in the same ranges observed in the above isodesmic reactions and are similarly more positive than experimental values (Table 3). In the case of the reduction and protonation of aqueous O2 by e− and H+ (Reaction 9), the four functionals give reaction free energies in the range −101.16 to −106.28 kcal/mol and are satisfactorily close to the experimental value of −117.4 kcal/mol. For the overall reaction (Reaction [4]) catalyzed by our model cytochrome c oxidase, the range of reaction free energies is −22.9 to −28.0 kcal/mol, sufficiently close, for our purposes, to the experimental value of −38.7 kcal/mol.
We accept these deviations from experiment as due largely to shortcomings at the density functional level to accurately compute the energies of H2, O2, H2O, and H2O2, and proceed to carry out calculations in which e−, H+, O2, and H2O interact with the larger structure proposed as the core of the B-type cytochrome c oxidases.
Individual steps of catalysis
The reaction free energy for each conversion was calculated as described above and given in Table 4 for each level of theory and plotted in Fig. 6A. These are the central results of the computational “experiment.” Note that the sum of the individual reaction free energies corresponds to the computed reaction free energy of the overall reaction, as shown in the bottom rows of Table 4 and in Table 3. Note also how the running sum of the reaction free energies relate to the average energy change line in Fig. 6B.
Table 4.
Reaction free energies in kcal/mol of the individual steps in the proposed mechanism.a
| PW91 | OPBE | B3LYP | B3LYP* | |||
|---|---|---|---|---|---|---|
| 2 e− (0.2 eV)b + 2 H+in (pH7)c + | 1 → 2 | + H2Os | +10.55 | −14.39 | +0.54 | +4.79 |
| O2d + H2Op + 1 H+in(pH7) | 2 → 3 | −28.61 | +15.78 | +7.64 | −3.82 | |
| 3 → 4 | + H+out (pH3)e | − 6.21 | −11.56 | −27.70 | −15.45 | |
| 4 → 5 | + H2Op | + 2.24 | +0.05 | +10.64 | +2.14 | |
| H+in (pH7) + | 5 → 6 | −2.74 | −0.05 | −7.14 | −4.22 | |
| H2Op + | 6 → 7 | + H+out pH3) | + 14.88 | +14.50 | +12.15 | +8.83 |
| 7 → 8 | + H2Op | + 9.96 | +8.11 | +20.67 | +19.55 | |
| 1 e− (0.2 eV) + 2 H+in (pH7) + | 8 → 9 | − 31.54 | −30.58 | −36.86 | −35.30 | |
| H2Op + | 9 → 10 | + H+out (pH3) | +10.47 | +12.60 | +7.62 | +7.86 |
| 10 → 11 | + H2Op + H2Os | −8.37 | −18.99 | −9.00 | −7.20 | |
| 1 e− (0.2 eV) + 2 H+in (pH7) + | 11 → 12 | −19.35 | −20.19 | −20.96 | −23.16 | |
| 12 → 13 | −0.62 | +1.74 | +0.26 | +5.77 | ||
| H2Op + | 13 → 14 | + H+out (pH3) | +9.17 | +7.86 | +11.01 | +7.97 |
| 14 → 1 | + H2Op | +11.96 | +12.27 | +4.80 | +6.81 | |
| Σ 4 e− (0.2 eV) + O2 + 8 H+in (pH7) | 1 → 1 | 4 H+out (pH3) + 2 H2Os | −28.21 | −22.87 | −26.32 | − 25.46 |
| Overall rxn: 4 e− (0.2 eV) + O2 + 8 H+in (pH7) → 4 H+out(pH3) + 2 H2Os | −27.97 | −22.85 | −26.37 | −25.23 | ||
See text for explanation of energetics.
Relative to the standard hydrogen electrode.
Representing the proton taken up on the “inside” of the ba3 molecule, carries its ZPVE.
Referenced to 1 M O2 in water rather than to 1 atm pure gas.
Representing the proton “pumped” to the “outside” of the ba3 molecule, carries its ZPVE.
Indicating a passive water carrying with it a full ZPVE.
indicating a “scalar” water also carrying its full ZPVE. See Table 3.
Fig. 6.
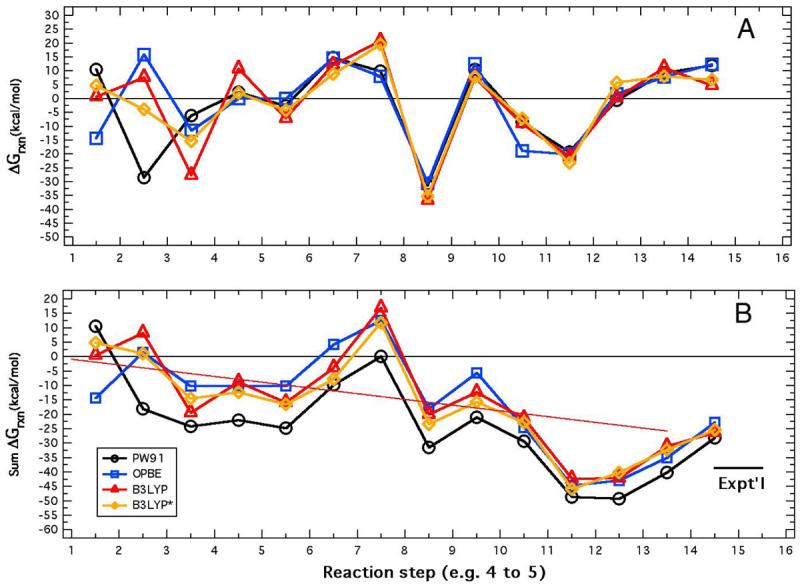
Visual comparison of individual ΔGrxn (A)and ΣΔGrxn (B) values obtained using the four functionals: PW91, OPBE, B3LYP, and B3LYP* taken from Table 4 along a trajectory from 1 to 14 to 1 ending at zero in plot A and ending in the average overall reaction free energy, −26.1 kcal/mol, and comparison with the experimental value of −38.7 kcal/mol (short horizontal line). The sloping line in B is made between 0.00 kcal/mol at state 1 and −26.1 kcal/mol at state 14.
There is generally excellent correspondence of all functionals from step 3 to 4 onward and rather poor correspondence between the non-hybrid functionals in steps 1 to 2 and 2 to 3. The hybrid potentials, B3LYP and B3LYP*, show the best correspondence among the 14 steps of the cycle. Indeed, the small variation obtained from computations on structures of the size and complexity as those considered here justify the inference that the theory chosen is adequate for the purpose. Overall, we suggest that the B3LYP* functional126, 130 shows the smoothest reaction free energy profile (see Fig. 6B), and the remainder of the paper refers primarily to results obtained using this functional.
Discussion
Description and rationalization of the model
General features
The motivating ideas are that: (1) Uptake of protons is coupled to the reduction of the metal sites. Protons enter from a bath pre-set at pH 7 via the K-path, but only when Y′O− is sufficiently basic to “call” for a proton. The stoichiometry of the overall reaction requires that, on average, each electron taken into the reaction chamber is accompanied by two protons. At two points in the cycle, the system is primed for proton pumping by reactions having a 1e− to 2 H+ uptake stochiometry, which increases the overall charge on the system from Q = 0 to Q = +1. Crucial to this stoichiometyric requirement is the ability of I-His282 to become deprotonated in structures 1 and 8 through 11. Whereas steps 1 to 2, 2 to 3, and 5 to 6 exhibit 2 e− + 2H+in uptake or simple proton uptake. In this sense, proton pumping is coupled to electron transfer31. The protons to be pumped out are moved from the imidazolium group of I-His376 (for a typical unaltered imidazolium the pKa is ∼7) into a bath pre-set at pH 3. This ΔpH of four units corresponds to an endergonic barrier of ∼5.4 kcal/mol (∼230 mV) and is in line with experimental estimates of the proton gradient94.
(2) Electrons enter CuA from cytochrome c552 one at a time and are then transferred to heme b one at a time, with further similar transfer from heme b to heme a3 [see22, 91and references therein]. Dioxygen reduction can occur in 2-electron steps. Formally, the reactions: O2 + 2e− + 2H+ → H2O2 and H2O2 + 2e− + 2H+ → 2H2O, occuring at pH 7 in water, are accompanied, respectively, by moderate (−10 kcal/mol O2 at 1 M) and by extreme (−69 kcal/mol H2O2) free energy changes, respectively. In the scheme presented in Fig. 5, pumping of the first proton is accomplished by harnessing the free energy change after the first 2-electron reduction and O2 binding in (step 3 → 4), whereas the second, third, and fourth proton pumping steps harness the free energy release associated with the second 2-electron reduction step. In this process, the oxidizing equivalents of the two O(-I)-atoms of the bridging peroxodianion are transformed into two new oxidizing centers on the protein: formallhy Fe(IV) and Y′O●. This completes the 4-electron reduction of the two O(0)-atoms of dioxygen to two O(-II) atoms.
(3) The free energy of O2-reduction is not fully dissipated until the scalar O(-II)-atoms are protonated to the level of water (H2Os). Of the four scalar protonation steps, one is avoided by formation of Cu(II)-−OH, which is protonated to water later in the scheme, and two protonation steps are avoided by the fact that Fe(IV)=O is a weak base131as long as the proximal histidine ring (I-His384) remains neutral. Therefore, not only does Fe(IV)=O represent a potentially strong oxidant, it also carries with it the potential for exergonic protonation of O(-II) that will later manifest itself. These concepts are in line with the comments of Siegbahn and colleagues67 that during the catalytic reaction, the enzyme should minimize proton affinity while maintaining electron affinity.
(4) The negative free energy change subsequently released by reducing and protonating Y′O● and Fe(IV)=O, as the catalyst returns to its initial state, is captured twice (steps 6 to 7 and 9 to 10) in the creation of formal, uncompensated, positive charges at CuB (Fig. 5). Uncompensated charge in the low dielectric cavity can be expected to cause a general “acidification” of the system, which in each instance results in the loss of a proton into the pH 3 proton bath. The Fe(IV)=O moeity itself may also have a direct role in proton pumping since the formally charged unit [Fe=O]2+ attracts electron density from the heme edges, particularly from the two propionate side chains therefore augmenting the positive potential of the I-Arg449 monocation on the dissociation of H376H+. In the current form of the model, all steps in which H376H+ donates its proton to the pH 3 bath are either moderately exergonic or moderately endergonic (see Table 4). The process of removing a proton from an imidzolium cation bears some resemblance to the 1994 idea of Wikström et al.57.
(5) Only five atoms change their valence and/or protonation state during the proposed cycle. (a) The Fe-atom is limited to high-spin Fe(II), low-spin Fe(III)-−OH (or peroxodianion), and intermediate-spin Fe(IV)=O. (b) The Cu-atom is limited to Cu(I), Cu(II), Cu(II)-OH− (or peroxodianion) and Cu(II)-OH2. (c) Three residues change protonation or redox states during the catalytic reaction. These are the phenol ring of I-Tyr237, indicated as Y′OH, Y′O−, or Y′●, the imidazole ring of I-His282, indicated as H282− or H282H, and I-His376, indicated at H376° or H376H+. I-His283 [the “top” histidine of Stuchebrukhov and colleagues84, 85] does not change its protonation state.
(6) Because proton movements are likely to be rapid, although they occur by ill-defined mechanisms, any of the selected tautomeric forms of the cluster is likely to reflect just one component of an equilibrium distribution. Previous theoretical studies have suggested that water molecules have access to the reaction chamber, where they are important mediators of proton-coupled electron transfers78, 132. However, as noted above, specific hydrophobic sidechains are packed around the active site, making it difficult for water from either side of the membrane to penetrate into the dnc.
Individual steps
The model is discussed in terms of what are deemed: reasonable chemistry, as obtained from decades of global discussion of oxygen reduction mechanisms [see52, 128, 133; the magnitude and interpretation of individual reaction free energies; and possible relationships to experimental information. Each sub-section is headed by the reaction itself, in bold, and the reaction free energy taken from the B3LYP* column of Table 4 and rounded to the first digit, in parentheses. Maps of individual 2-D plans and the two other shorthand notations used to keep track of the redox and protonation status as well as a downloadable electronic .xyz file for each structure are provided in the Supplementary Material.
1 + 2 e− + 2 H+(pH7) →2 (ΔGrxn = ∼ +5 kcal/mol)
Structure 1 (H376H+,Fe3OH_Cu2, Y′O−,H282−,184,ups=2,Q=0) where ups is the number of unpaired spins and Q is the overall charge on the complex, see Fig. 4) corresponds to the oxidized form (O in Fig. 2) of the enzyme as it is thought to exist during catalysis99explains the rationale for choosing this as the active, oxidized form of the enzyme. It is comprised of a low-spin ferric heme to which a hydroxyl anion is coordinated in the 6th coordination site, while the cupric CuB is coordinated only to N-atoms of I-His233, I-His282 (in its imidazolate form), and I-His283 coordinated as the neutral imidazole form, and the tyrosine phenol, covalently linked to I-His233, is deprotonated.
Conversion of 1 to structure 2 (H376H+,Fe2_Cu1,Y′O−,H282H,183,ups=4,Q=0) involves reducing Fea3 from low-spin Fe(III) to high-spin Fe(II) and CuB from Cu(II) to Cu(I) coupled to uptake of two protons at pH 7 (2 H+ / 2 e−). One of these protons is bound to the Fe-bound HO− group and is released as the first scalar water, i.e., one derived from the O2 molecule, while the second proton goes to form H282H°, and the tyrosine residue remains deprotonated (Y′O−) in order to retain the overall charge of zero. The Fe-atom is high-spin ferrous (ups=4) and, as expected, it has been pushed slightly out of the porphyrin plane. The oxidized forms of Type-A1 cytochrome c oxidases are known from experiment to take up more than one proton upon reduction98, 134. Note that in Fig. 2, two electrons in the O to R transition reduce the other metals centers: oxidized-CuA to reduced-CuA and the low-spin heme, a3+ to a2+ for cytochromes aa3 and b3+ to b2+ for cytochrome ba3.
Using the B3LYP* level of theory, the overall free energy change of this step is moderately endergonic, being respectably close to the recent paper of Sousa et al.135 on the redox potentials (vs. SHE) of Thermus ba3, which reports that the mid-point potential of heme b at pH 7 is ∼0.21 V, being similar to that of Thermus cytochrome c552 (∼0.2 V), while the mid-point potential of heme a3 is ∼0.29 V at pH 7. Their data would predict ΔGrxn ∼ −2 kcal/mol for step 1 to 2. States 1 and 2 are experimentally accessible and remain the subject of continuing spectroscopic and chemical investigations.
2 + O2 + H2O + H+ in(pH7) →3 (ΔGrxn = ∼ −4 kcal/mol)
In step 2 → 3 (H376H+,Fe2O2_Cu1HOH,Y′OH,H282H,189,ups=0,Q=+1). Structure 2 takes up O2, one water molecule, and an additional proton from the pH 7 bath to form structure 3; see 2-D plan in Fig. 5. In the geometry optimized structure, one proton of the Cu(I) bound water molecule is 2.27 Å from the Fe-bound O-atom and 2.12 Å from the non-metal bound O-atom; its presence apparently stabilizes the Fe-O2 complex. This H-bonding to the O2 molecule prevents “rotation” of the non-bonding O-atom of O2 toward the Cu and subsequent “in silico formation” of a metal bridging O-O species during geometry optimization.
Structure 3 corresponds most closely to Compound A of Fig. 2, and it recently has been detected in ba3 by Siletsky et al.35 and in unpublished work from the Einarsdóttir and Fee laboratories. In the oxygenated form of the Fea3-CuB dnc (see Fig. 5), the oxy-complex has an overall charge, Q = +1, due to the added proton on Y′OH. This charge is formally associated with the Cu(I) complex while the Fe(II)-O2 heme moeity is formally neutral, and in accordance with its similarity to that in oxy-hemoglobin136, 137, the oxy complex has no unpaired spins. The B3LYP* reaction free energy of this step (Table 4) might be compared to an experimental measurement of isolated O2 affinity for the a3-site in A1-type oxidases, i.e., not involving other interactions, for example with CuB [see Riitsma et al.45, 138 ∼ −5 kcal/mol].
The generally negative free energy of O2 binding suggests strong affinity of the active site structure for O2, and we capitalized on this to add a third proton, Y′O− → Y′OH, that increases the overall charge from 0 to +1. This catalytic intermediate is now activated for proton pumping having two available electrons and three available protons. Assuming, for this step, that O2 binding is rate limiting, specifically that the H+ and H2O bind after O2, then step 2 → 3 would be independent of pH as found experimentally for the bovine oxidase41. Formal reduction of O2 begins in the next step and continues through step 6 → 7 whereupon both the O-atoms of O2 become reduced to the (−II) valence state.
3 →4 + H+out (ΔGrxn = ∼ −15 kcal/mol)
Step 3 to 4 (H376°,HOH,Fe3OOCu2,Y′OH,H282H,185,ups=2,Q=0) is the first proton pumping step. As noted above, it is coupled to the internal 2-electron transfer within Fe(II)-O2 and Cu(I) to form Fe(III)-−O-O−-Cu(II) with loss of the proton on H376H+ into the pH 3 proton bath. This DFT energy supports the idea that the proton on H376H+ can, during certain transformations of the cluster, become strongly acid, relative to the pH 3 proton bath. As will be evident when considering the next step, binding and internal transfer of the passive water molecule, H2Op, contributes to the favorable energetics of proton pumping in this step.
A bridging peroxide intermediate, corresponding to states 4, 5, and 6 in our proposed catalytic cycle, is very likely to form in all oxidases. However, transient, isotope-edited resonance Raman studies of A1-type enzymes have failed to identify signals from any intermediate down stream of Compound A that are attributable to O-O stretching43, 139, 140; it is thus likely to have a very short life-time (< ∼1 μs) and is not expected to accumulate to any degree during catalysis. Our results for the energetics of steps 3 to 4, 4 to 5, 5 to 6, and 6 to 7 will give a short-lived Fe(III)-−O-O−-Cu(II) consistent with experiment, at least for the free energy differences between these intermediates. Evaluation of transition states may, of course, produce additional barriers, a subject for further investigations.
4 →5 + H2O (ΔGrxn = ∼ +2 kcal/mol)
In step 4 → 5 (H376H+,Fe3OOCu2,Y′O−,H282H,185,ups=2,Q=0) the proton on Y′OH is moved to NE2 of I-His376 thereby restoring H376H+, displacing the pendant HOH, and creating a basic Y′O− near the exit of the K-path. This is a weakly unfavorable reaction with small positive ΔGrxn = ∼ +2 kcal/mol. However, if an H-bond between the carboxylate of II-Glu126 and the imidazolium ring of His376+ were included (see Fig. 4), it is likely that this step would become more favorable. This clearly requires further study.
5 + H+in (pH7) →6 (ΔGrxn = ∼ −4 kcal/mol)
Step 5 → 6 (H376H+,Fe3OOHCu2,Y′O−,H282H,186,ups=2,Q=+1) involves at least two steps whereby a proton is brought into the reaction chamber from the K-path via Y′OH to form the Fe(III) to Cu(II) bridging hydroperoxide anion (HOO−). Blomberg and Siegbahn and co-workers79 in their DFT studies of O-O bond breaking discussed this step in great detail, and concluded that protonation of one of the O-atoms is prerequisite for breaking the O-O bond of the peroxide dianion and that one or more H2O may also be required to transfer the proton from Y′OH to the metal bridging peroxide. Examination of the structure of ba3 (PDB Code 1XME) shows that CG1 and CG2 of the I-Val236 side-chain make contact with rings B and C of the a3-heme, CG2 contacts the ring of I-Tyr237, and CG1 points toward CuB. The position of this side chain provides an extremely hydrophobic environment and leaves little space for water in this region. Thus, the role of water in protonation of the bridging peroxide needs further examination. While, in the next step there are proton coupled electron transfers141, 142, here we simply protonate the bridging peroxide from the pH 7 bath (see structure 6 in Fig. 5), and proceed with a geometry optimization calculation. During this calculation there is no tendency for the O-O bond of the HOO− anion to break.
The computed reaction free energy for this step is moderately exergonic and only weakly dependent on the exchange correlation potential (see Table 4), compared to other steps. These results suggest that barriers to the movement of protons among the different sites in the quantum model may be quite small. In addition to protonation of the bridging peroxide, the overall charge on the model increases from Q = 0 to +1, thereby priming the system for the second proton pumping step. Again, because of the experimental absence of intermediates containing an O-O bond, this intermediate is not likely to accumulate to any extent during re-oxidation.
6 + H2O→ 7 + H+out (ΔGrxn = ∼ +9 kcal/mol)
Step 6 → 7 (H376°HOH,Fe4O,Cu2OH,Y′O●,H282H,188,ups=4,Q=0) represents the second-internal, two-electron transfer step, analogous to the two-electron reduction of H2O2 → 2 H2O for the uncatalyzed reaction, with the potential loss of a great amount of energy as heat. However, the enzyme captures this potential free energy first by releasing a proton into the pH 3 bath, which accounts for the second pumped proton, and then by creating two new oxidizing equivalents: one as Fe(III) → Fe(IV) and one from Y′O−, which provides the second electron needed to break the O-O bond of the bridging hydroperoxide anion to form Y′O● and Cu(II)-−OH. There is experimental support for the formation of Y′O● in the bovine enzyme [see143 and references therein], and our theoretical results provide additional evidence for its occurence in the oxidase mechanism.
Both the non-hybrid functionals, PW91 and OPBE, and the hybrid functionals, B3LYP and B3LYP*, predict a moderately positive ΔGrxn for this step (see Table 4). As in the first proton pumping step, a water molecule is allowed to donate an H-bond to NE2 of I-His376° after deprotonation to account for the second proton pumping step. Because Y′O● must be a very weak base, it will block proton uptake from the K-path. The calculations show that the spin of the formal Y′O● is largely distributed over the atoms of I-Y237 and I-His233, with only ∼ 20 % being located on the phenolic O-atom (see Supporting Information). Intermediate 7 may accumulate to some extent, but it is likely to be EPR silent because the intermediate-spin Fe(IV)=O lacks a signal, and the tyrosine free radical and the Cu(II) are likely to be spin-coupled.
Both atoms of the original O2 molecule have now been reduced to the (−II) valence. One has been protonated to the level of (Cu(II)-−OH) and is thereby partially exhausted, whereas the O(−II)-atom bound to Fe(IV) remains to be protonated in subsequent steps. Similarly, much of the oxidative potential of the original O2 molecule remains in the form of Fe(IV)=O and Cu(II)/Y′O●. The following steps show how this free energy may be captured.
7 →8 + H2O (ΔGrxn = ∼ +20 kcal/mol)
Step 7 → 8 (H376H+,Fe4O_Cu2,Y′O●,H282−,185,ups=4,Q=0) involves the transfer of a proton from NE2 of H282H to form H376H+ with release of the pendant water, occurring with an average reaction free energy of ∼ +15 kcal/mol (over all DFT functionals) while proton uptake via the K-path is effectively blocked by Y′O●. Using unperturbed pKa values for H+ + ImH → ImH2+ on I-H376 (pKa ∼7) and Im− + H+ → ImH on I-H282 (pKa ∼12), this step would be endergonic by only 5 × 1.37 = ∼6.9 kcal/mol. The source of the endergonicity is thus not clear, although the missing I-Thr302 will stabilize the imidazolate form of I-His282. More effectively, coupling steps 7 → 8 and 8 → 9 would give an overall exergonic process by coupling proton transfer to reduction of the latent oxidizing equivalents, Y′O●.
8 + e− + 2 H+(pH7) →9 (ΔGrxn = ∼ −35 kcal/mol)
Step 8 → 9 (H376H+,Fe4O_Cu2HOH,Y′OH,H282−,187,ups=3,Q=+1) involves one-electron reduction of Y′O● and uptake of two protons. One of the protons goes to the HO− ligand of Cu(II) to form a Cu-bound H2O molecule, which donates an H-bond to Fe(IV)=O, while the second proton goes to form Y′OH. Y′O● is clearly the strongest oxidant in the reaction chamber at this stage, so it will receive the electron in conjunction with the uptake of a proton via the K-path. The presence of Y′O● blocks proton influx from the K-path thereby precluding electron/proton flow to the Fe(IV)=O moeity (see below). This is a very exergonic step, attesting to the instability of the Y′O● radical, and it leaves an uncompensated charge at CuB thereby priming the active site to pump the third proton. Thus, with the uptake of the third electron, the potential free energy in one of the two transient oxidizing species has been captured.
Intermediates 9, 10, and 11 represent the three-electron state, and correspond most closely to PR of Fig. 2. Experimentally, if any one these accumulates to any degree, it may be detected because each is potentially EPR active at Cu(II). Any one fits the rationale for the detection of a paramagnetic/oxygen EPR signal found during reoxidation of the bovine enzyme by Hansson et al.144 in the early 1980's, and by Morgan et al.38. A search for such an intermediate in ba3 is therefore recommended in studies of the type carried out by de Vries, Ludwig and co-workers145.
9 + H2O→ 10 + H+(pH3) (ΔGrxn = ∼ +8 kcal/mol)
The third proton is pumped in Step 9 → 10 (H376°HOH,Fe4O_Cu2HOH,Y′OH,H282−,189,ups=3,Q=0) in which a proton is removed from H376H+ at pH 3 and is replaced by a water molecule that donates an H-bond in its place. The moderate endergonicity of this step once again indicates that uncompensated charge on CuB drives acidification and it is this acidification that makes possible energetically favorable loss of the proton on H376H+. One transient oxidizing equivalent remains to drive the last proton pumping event.
10 →11 + 2 H2O (ΔGrxn = ∼ −7 kcal/mol)
Step 10 → 11 (H376H+,Fe4O_Cu2,Y′O−,H282−,183,ups=3,Q=0) is similar to step 4 → 5 and involves internal proton transfer from Y′OH to H376° to form H376H+ with loss of the pendant H2O, plus the loss of the scalar water associated with the Cu(II); all charges are compensated, Q = 0, and all functionals are moderately to strongly exergonic. However, this step should be assessed with some caution as there is no compelling reason to remove the scalar water from CuB at this stage (see below).
11 + e− + 2 H+(pH7) →12 (ΔGrxn = ∼ −23 kcal/mol)
Step 11 → 12 (H376H+,Fe4O,Cu1,Y′OH,H282H,185,ups=2,Q=+1) involves reduction of Cu(II) by the fourth electron and the uptake of two protons (2 H+ / e−). This represents yet another proton coupled electron transfer step where the first proton arrives via the K-path to convert Y′O− to Y′OH then moves to H282− to form the neutral imidazole ligand, H282H, while the second proton also arrives from the K-path to reform Y′OH. Q is increased from zero to +1, thereby priming the system for the fourth proton pumping step. The un-compensated charge is formally localized on Cu(I) because both I-His282H and Y′OH are neutral. This step is strongly exergonic because of Cu(II) to Cu(I) redox chemistry plus protonation of the H282− and Y′O− anions. Experimental evidence for the formation of a state in which the iron exists as Fe(IV)=O and the CuB as Cu(I) has been reported by Zazlavsky et al.47. Were it to accumulate, intermediate 12 would be EPR silent in ordinary perpendicular mode.
12 →13 (ΔGrxn = ∼ +6 kcal/mol)
In Step 12 → 13 (H376H+,Fe3OH,Cu2,Y′O−,H282H,185,ups=2,Q=+1) both internal electron and proton transfer occur. In steps 10 to 13, the system has received all the electrons needed to reduce O2 to the level of H2O, and the presence of Fe(IV)=O represents the last of the transient oxidants formed in step 6 to 7. Although not considered in detail in this work, the reduction of Fe(IV)=O by Cu(I) probably requires that a water molecule is coordinated to Cu(I) for the purpose of donating an H-bond to the oxoanion on the iron atom. In yet another proton-coupled, electron-transfer process, Fe(IV)=O2−-H2O-Cu(I) ⇔ Fe(III)-−OH -- HO−-Cu(II) occurs. Following this proton transfer from Y′OH to HO--Cu(II) to yield Y′O- and H2O-Cu(II) the second scalar water can probably be eliminated. In this fashion, the last transient oxidizing equivalent is reduced. Although this process is weakly endergonic, it leaves the system primed for the fourth proton pumping step, which occurs in the next step. Intermediate 13 is likely to be EPR silent, because it should have an electronic structure similar to the oxidized enzyme.
13 + H2O →14 + H+(pH3) (ΔGrxn = ∼ +8 kcal/mol)
Step 13 → 14 (H376°HOH,Fe3OH_Cu2,Y′O−,H282H,187,ups=2,Q=0) involves removal of the proton on H376H+ into the pH 3 bath, thus representing the fourth proton pumping step, and binding a pendant H2O. All functionals agree that this step is weakly endergonic.
14 →1 + H2O (ΔGrxn = ∼ +7 kcal/mol)
Step 14 → 1 involves reprotonation of I-His376NE2 by loss of the proton from Cu(II) bound H282H in a endergonic process that restores the catalyst. Events in this step are comparable to those in step 7 → 8.
General comments
Examination of the reaction pathway energetics reveals novel concepts that merit further comment.
Coupled uptake of electrons and protons
Uncontrolled availability of electrons to the reaction chamber could uncouple dioxygen reduction from proton pumping. This is apparently not the case for step 1 to 2 in which two protons accompany two electrons, and there is no choice about where these particles will bind. However, in step 8 to 9, the incoming electron has a choice of binding to either Y′O• or Fe(IV)=O. While it is true that Fe(IV)=O is a strong oxidant when a proton is available to form the Fe(III)-bound hydroxide, its one-electron reduction to Fe(III)-bound oxodianion is likely to be energetically unreasonable. Therefore, in the absence of a proton, Fe(IV)=O is probably quite stable. In state 8, Y′O• also has low proton affinity, and its presence serves to block proton uptake from the K-path. However, one-electron conversion of Y′O• to Y′O− seems reasonable. Depending on the overall basicity of the active-site molecule, Y′O− may then be protonated to Y′OH. Accordingly, the most reasonable target for the incoming electron will be Y′O•.
A similar scenario appears to hold for step 11 to 12 where the incoming electron can bind to either Fe(IV)=O or Cu(II)-(H282−). The computational results indicate that this step is quite exergonic, such that steps that involve formation of H282H° and Y′OH are energetically feasible. There seem to be no distinguishing features that would cause the electron to bind to CuB rather than Fe(IV)=O, and we have not tested all alternatives involving Fe(IV) reduction to Fe(III). However, step 11 to 12 is strongly exergonic and 12 to 13 is weakly endergonic, which argues in favor of the sequence we propose.
Proton pumping
All proton pumping steps are either mildly exergonic or mildly endergonic. One may infer from these data that H376H+ has become acidic with respect to the pH 3 proton bath into which the proton is “pumped.” In each such step, the active-site molecule has a single positive charge, Q = +1, that is formally localized on CuB. Each step of proton pumping is preceded by a step in which one of O2, bridged peroxide, or the transients Y′O● or Fe(IV)=O is reduced and protonated. These steps are indeed, the fundamental steps in energy transduction, i.e., the conversion of electron reduction potential into protonation potential.
Endergonic steps
Concomitant with loss of the proton from H376H+, passive water molecule is brought into the system that binds to H376° donating an H-bond to the non-bonded electron pair of H376°. Steps subsequent to proton pumping are sometimes endergonic, which likely arises from breaking the interaction between the NE-atom of I-His376 and the pendant HOH associated with the “post pump” structures. While future refinements of the model must address this situation, it is possible for the system to circumvent potentially kinetic barriers by combining with a subsequent, highly exergonic step. An example is the endergonicity of step 7 to 8 that can be circumvented going directly to 9.
7 + 1 e− + 2 H+(pH7) → 9 + H2Op (ΔGrxn = ∼ −2 kcal/mol)
This combines the unfavorable transfer of a proton from the neutral H282H in conjunction with unfavorable loss of the pendant HOH, with the highly favorable uptake of the third electron which reduces Y′O● to Y′O−. The latter is accompanied by the uptake of two H+ at pH 7 (2 H+ / e−) leading to formation of Y′OH and Cu(II)-OH2 and creating an un-compensated charge on Cu(II) that raises Q from 0 to +1. So primed, the system can carry out the third proton pumping event. This approach to getting around endergonic barriers has been discussed in terms of constructing life processes that are physiologically functional146.
For the cytochrome ba3, there are at least two ways in which kinetic barriers of the magnitude presented here might be circumvented. First, the lower bounds pose only a small problem, assuming a maximum contribution of ∼+20 kcal/mol (see B3LYP* column of Table 4) to small activation energies. Second, the protein may provide additional interactions with the quantum model that lower the kinetic barriers of these steps. For example, the H-bond network involving OG of I-Thr282, ND1 of I-His282, and CO of I-His298 may stabilize the Cu(II) bound imidazolate form of I-His282. In a similar vein, an H-bond between the carboxylate from II-126Glu and the H376H+ may facilitate this proton transfer (see Table 1 and Fig. 4).
Previous models
Over the decades there have been many hypotheses regarding the proton pumping mechanism of A1-type cytochrome c oxidases [see2, 9, 31, 36, 37, 54-64 and references therein). These studies and numerous others are the foundation of current research on cytochrome c oxidases. In the realm of theoretical studies, Siegbahn and Blomberg tackled the problem of how the O-O bond is broken in the bridging peroxodianion complex. This work, done in collaboration with the late Jerry Babcock, demonstrated the need for a proton to break the O-O bond of the peroxide52, 78; we have included this in our mechanism. The work of Yoshioka and colleagues introduced the concept that water may have access to the dnc75, 132, although close examination of the I-Val236 side chain in the structure of ba3 reveals very little space for water molecules beween the Fe, Cu, and the phenol group of I-Tyr237. The recent work of Kaukonen74 supports the idea that I-Tyr237 (ba3 numbering) can participate in both proton transfer as well as electron transfer. The work of Stuchebrukhov and colleagues83, 84 provided evidence for oxidative acidification of the Cu-imidazole rings, although Pommes and co-workers dispute this idea86, 87. Together, the theoretical studies (see Introduction) extend and clarify models derived from experimental studies. The present work builds on these previous works and extends them by suggesting (a) that the dnc of cytochrome c oxidase is constrained to carry out 2-electron reductions of O2, (b) introducing new chemistry that accounts for the 1 e− / 2 H+ ratio inherent in the overall stoichiomentry of the oxidase catalyzed reaction, and (c) suggesting an experimentally testable, chemical mechanism for proton pumping.
How might the model be falsified?
Indirect coupling
Our most general assumption is that the chemistry of O2 reduction and proton pumping is carried out entirely by the active-site model chosen for study. If, for example, proton pumping were to be coupled indirectly to O2 reduction via conformation changes in the protein, then the model cannot be correct. Although such mechanisms have been discussed64, 147, there is no evidence that they actually occur. Yoshikawa and co-workers148 have pointed to small changes in structure that occur upon reduction of the bovine enzyme and interpreted these to be important for proton uptake. However, these changes appear to be unique to bovine aa3 as they occur in a part of the structure where struture-based, amino-acid sequence is not conserved (e.g., ba3 lacks I-His503 and I-Asp91 aligns with a deletion in the ba3 structure). In addition, similar structural changes have not been observed in the Pd enzyme149 or the cytochrome ba395. Moreover, because the thermodynamic results themselves indicate that the active-site molecule, as utilized here, is able to carry out the known chemistry of ba3 (and possibly the A1-type oxidases) without resort to indirect coupling, the assumption of direct coupling would appear reasonable. However, small conformational changes of the type noted by Muramoto et al.64 may be important for opening and closing the input and output gates.
Mutational studies
Mutation of any amino acid residue of the active-site structure that creates a proton pumping enzyme that in addition has a “complete and perfect” 3-dimensional structure, would also tend to falsify the model. This, already excludes residues serving as ligands to the two metals, mutations of which are known to alter the structure in the immediate vicinity of the active site. Mutations at I-Arg449 & 450 reveal a mixed picture, with the general conclusion that I-Arg449 can be replaced with lysine in the Rs enzyme whereas I-Arg450 can be replaced with a variety of residues with causing significant loss of both ET and HT activity; work with E. coli bo3 also reveals mixed results150, 151. In our model, I-Arg449 serves to neutralize the presumed negative charge on the D-ring propionate and plays no direct role in either electron transfer or proton pumping activity. Results from ongoing mutational studies of ba3 in the Fee and Gennis laboratories reveal a complicated picture but do not falsify the model25.
Choices of e− / H+ distributions
A further assumption of the model is that the electron/proton distributions, chosen on the basis of intuition, are, at each step, the most thermodynamically stable arrangement. To the extent that this is not the case, the model is vulnerable to unidentified thermodynamic traps that would break the catalytic cycle and cause the accumulation of the catalyst in a single form. Remarkably, only one step in the scheme of Fig. 5, 7 → 8 (ΔGrxn = ∼+20 kcal/mol, using B3LYP*) falls in this category. This step involves moving a proton from either Y′OH or H282H to reprotonate H376° HOH and move the HOH molecule back into water. To first order, part of this endergonicity may be associated with the inherently different pKa values of imidazole and phenol. Roughly, taking the pKa of the former equal to 7 and that of the latter equal to 12, corresponds to an energonic contribution of only ∼7 kcal/mol. Because the pKa of Y′OH is lower than the isolated tyrosine molecule152, the ΔG of this internal proton transfer may actually be more favorable than indicated by this approximation; clearly other factors need to be considered. These barriers may also be eliminated by choosing a different proton distribution. For example, the proton on H282H in 4 could be moved to H376° in 5 leaving Y′OH in its protonated state. Similarly, in step 7 → 8, the proton on H282H could be moved to Cu(II)-−OH to form Cu(II)-OH2 rather than to H376° to form H376H+.
Additional interactions with the protein
A legitimate shortcoming of the model is the failure to include additional protein components that could affect the “smoothness” of the thermodynamic trace shown in Fig.6 (shown also in the center of Fig. 5). These include: (a) water O32 that lies only 2.89 Å from the secondary alcohol (HAS O11) of the geranylgeranyl side chain of heme a3 and might be expected to affect the proton binding properties of the I-237Tyr phenol moeity; (b) the NE atom of I-His282 lies near enough (2.81 Å) to the OG O-atom of I-Thr302 and to other residues in the neighborhood to affect its proton binding properties; (c) the phenol hydroxyl moeity of I-Tyr133 lies only 2.55 Å from the O1A atom of the ring D propionate and mirrors I-Asp372, which donates a H-bond to the O1D atom of the ring A propionate; and (d) incorporating an interaction between the OE2 atom of deprotonated II-Glu126 and the NE2 atom of protonated I-His376 (H376H+)may lower the above mentioned barrier in step 7 → 8. Including these groups in the active-site molecule would, however, greatly increase the computational costs and further increase the risk of encountering false minima during geometry optimization. Nevertheless, their presence will be important in the future refinement of the model.
Novel features of the model
The model incorporates several interesting features that, when considered together, have not been fully considered in either experimentally derived or computationally supported mechanisms. These are:
Oxygen reduction
It has been known for some time that, depending on conditions, there are numerous paths along which O2 can be reduced to 2 H2O including both radical and non-radical routes128. It is now generally agreed that heme-copper oxidases avoid the release of free radicals, the weakly reducing superoxide and the highly oxidizing hydroxyl, probably because they are not on the O2-reduction path dictated by the enzyme. Moreover, a radical path tends to focus nearly all of the oxidative potential of O2 to water in the HO• to water step while allowing the O2 to HO• conversion to be roughly isoenergetic. Our observations indicate that the active-site prefers carry out two-electron redox chemistry while capturing the inherent oxidative potential of O2 to 2 H2O in four unique proton-coupled, electron-transfer steps, all the time avoiding free radical release.
Currently, there is no spectral evidence for a bridging peroxide structure, but such an intermediate is likely to be spectrally similar to the oxidized form of the enzyme. Thus, were it to form and decay in the time frame of A to P in Fig. 2, while not changing its “color,” it would not be detected by optical absorbance changes in heme-a3. However, an O-O stretch band might be observed using transient resonance Raman techniques that depend on excitation into the oxidized heme absorption bands, but this approach has not been successful153. We reason that a metal-bridging peroxodianion is an essential intermediate in reoxidation and agree with Blomberg and co-workers52 that, in order to actually cleave the O-O bond, one of the O-atoms must be protonated. This increases the positive charge on the active-site and causes the loss of a proton from H376H+ thereby capturing the free energy of the first two-electron reduction of O2 (Fig. 5).
There is strong experimental evidence for the the second 2-electron reduction step, and the thermochemical results reported here indicate that the structure of the dnc has evolved to carry out this type of chemistry while optimizing the capture of the peroxide to water oxidative potential. We questioned why Cu(II) does not itself promote O-O bond cleavage to form and found that Cu(III)=O containing analogs of our model are extremely unstable (J. A. Fee, unpublished observations, see also52 and references therein). The covalently linked I-His233 - I-Tyr237 modification provides a means to form a strong oxidant other than Cu(III)=O. The extended bonding from Fea3:(HO2−):CuB to the phenol O-atom of tyrosine to the secondary alcohol moeity on C-11 of heme-a3 allows the second 2-electron reduction of O22− to simultaneously form ferryl “Fe(O)2−” and a stabilized Y′O•. The thermochemical results reported here suggest that this rearrangement of atoms and electrons occurs isoenergetically (see steps 5 to 6 and 6 to 7 in Table 4). The practical meaning of this result is that essentially all the oxidative potential in the O2 to H2O2 and H2O2 to 2 H2O transformations is captured: partly as a proton gradient and partly as two new oxidizing agents in the form of Fe(IV)=O and Y′O•. The experimental evidence lends general support to this chemistry, and the theoretical calculations support and extend this major feature of the oxidase mechanism. The overall efficiency of proton pumping under the conditions described here is remarkably high154.
Proton/electron uptake ratio
The stoichiometry of the enzyme in its natural habitat requires, on average,
where two protons are taken up on the INside and one finds its way to the scalar water while the other is released to the OUTside. It has been recognized for some time56 that this must be true in mitochondria. And, any viable mechanism for the enzyme must account for the fact that, when the enzyme is extracted from the membrane with detergents, its inherent polarity is retained. Under this condition, proton pumping chemistry supports a futile cycle during which a proton taken up by the K-path on the INside of the soluble enzyme is replaced by diffusion of a proton from the OUTside, around the protein molecule, through the homogeneous bath that connects the IN- and OUTsides. The chemistry is therefore identical in both membrane bound and detergent solubilized forms of the oxidase. From the point-of-view of physical chemical experiments with the detergent solubilized enzyme, however, the pH dependence of the above half-reaction is changed to
which is generally supported by both enzyme activity and pH dependent redox potentials41, 135.
The model suggests reasonable paths for 1 e− to 2 H+ reactions. Beginning with the active-oxidized state (1), several steps are involved in sequestering the two electrons and the four protons needed to carry out the second 2-electron reduction of oxygen to Fe(IV)=O, Y′O●, and Cu(II)-−OH. In step 8 to 9, reduction of Y′O● is coupled to formation of Y′OH while the second proton binds to Cu(II)-−OH to form the second scalar water (see Fig. 5). Similar movements of protons occur in the 1 e− to 2 H+ process of step 11 to 12. In each case, I-His282, I-His376, and I-Tyr237, along with the metals and the metal-ligated atoms of less than fully protonated water molecules, mediate the characteristic Nernstian behavior of a 1 e− / 2 H+ process (for example, the behavior of the Rieske protein [see50, 155and references therein].
Control of electron transfer into the dnc
There are two steps along the path shown in Fig. 5 where an electron entering the dnc could uncouple oxygen reduction from proton pumping. Electron transfer among cytochrome c,the CuA center, and the low-spin heme is rapid and nominally independent of pH, and an electron residing on either of these is available to the dnc via a conserved electron transfer path directly connecting the 6c heme to the dnc91, 156. There are distinct experimental conditions under which the enzyme may behave differently. The flow-flash experiment, in which the enzyme contains four electrons and the necessary protons (via proton wires) to reduce O2 to water, represents the electron-rich condition and is the condition under which almost all rapid kinetic studies of cytochrome c oxidase mechanism have been carried out. It is also the condition most conducive of spurious electron transfer that could uncouple proton pumping. For the most part, this does not occur, and there is overwhelming evidence that the enzyme pumps protons under these conditions9, 31, 36. The mechanism presented here suggests that events of electron transfer from the 6c heme may actually be predetermined by the nature of the intermediates themselves. For example, in step 5 to 6, a third electron could reduce the bridging hydroperoxide anion to form Fe(I)=O and Cu(II)-−OH thereby shunting the system from state 6 to state 9. If this were to occur, Y′O● would not be formed and intermediate 9 would most likely receive the fourth electron into the Fe(IV)=O moeity. This would completely uncouple proton pumping from the normal O2-reduction cycle. Similarly in step 8 to 9, of the three reducible centers Y′O● seems the most favorably disposed to accept an electron simply because it can be reduced directly to Y′O− without requiring a proton. Again, in step 11 to 12, three-coordinate Cu(II) may be reduced to Cu(I) without the need for a proton, thereby making it the most favorably disposed of the two remaining oxidizing centers for reduction. This leaves intermediate F of Fig. 2, in accordance with numerous experimental data, as the final intermediate requiring a proton to recycle the catalyst. These ideas can be straight forwardly pursued by theoretical examination, and possibly by experiment.
The second condition under which oxidase activity has been studied is that of paucity of electrons and excess O2. Gupte and Hackenbrock157, 158 showed that maximum oxygen respiration by mitochondria and mitochondrial inner membranes, under normal aeration, depend on the diffusion characteristics of the cytochrome c that ferries electrons from Complex III to cytochrome c oxidase. The rate limiting step in O2 reduction, under this condition, is that at which reduced cytochrome c diffuses to and transfers its electron to CuA. Certainly, one way to minimize spurious electron flow into the dnc is to control electron flow into the cytochrome c. Chance159 reviewed experiments to measure rates of tisue respiration encompassing available O2 concentrations. The enzyme is zero-order in O2 from air saturation down to oxygen concentrations on the order of a few nanomolar. At these lower concentrations of O2, tissues accumulate reduced cytochrome c oxidase indicating a surfeit of reducing equivalents. It seems highly unlikely that the enzyme actually “leaks” at these very low [O2], as discussed above, although this is not known. The model presented here assumes that Fe(IV)=O and Y′O● are formed prior to uptake of the 3rd and 4th electrons. How might this be regulated?
In order to retain proton pumping under the condition of surfeit electrons, it is necessary that the reducing electron on the 6c heme “perceive” the redox potential of a unique site within the dnc that is prepared to receive it. This process will be controlled by the laws that regulate electron transfer [see160, 161as well as proton coupled electron transfer142 and references therein]. The validity of the model is thus predicated on the idea that YO● has the highest “reduction probablility” in structure 8, therefore it receives an electron instead of Fe(IV)=O or Cu(II)-−OH. Similarly, in structure 11, the tris-coordinated Cu(II) is likely to have a higher reduction probablility than Fe(IV)=O. A recent report from Collman and colleagues162 describes the observation of Fe(IV)=O, phenol-O●, Cu(II) in a small-molecule cytochrome c oxidase model under conditions of limited electron availability. This and similar systems may open the door to experimental tests of these ideas.
The chemical act of proton pumping
One of the vexing questions about proton pumping by the oxidases has been how to couple proton pumping steps to the chemical, “free energy releasing” steps of O2 reduction while achieving a stoichiometry of H+out/e−in = 1. The model provides an answer to that question by demonstrating that an uncompensated positive charge (formally on CuB) in the proposed chemistry can drive the release of a proton into the pH 3 bath from four structurally different intermediates of the cycle. The underlying force of this common step is “oxidative acidification,” which results in peripheral groups, such as I-His376, becoming subject to deprotonation. While the proton might be “taken” from any of the proton-active groups, the structural reasons for selecting H376H+ are appealing. Nevertheless, there are potentially numerous subtleties about how the uncompensated positive charge on the cluster is actually coupled to proton translocation. One consideration must be the bipolar nature of the cavity, which should promote movement of the excess, Q = +1, charge from the less polar region toward the more polar boundary region described by I-Arg449, I-His376, and the two propionates. This asymmetry of the environment suggests that future attention be given to the electrostatic properties of the cluster and the functioning of the input and output gates.
Supplementary Material
Acknowledgements
We dedicate this communication to the memory of Jerry Babcock. JAF thanks Dr. Wen-Ge Han for her help with all aspects of the day-to-day running of programs, constructing and editing files and general assistance with computer technologies. Dr. William S. Young deserves great credit for maintaining and assisting interactions with the super computer facilities at Scripps. Dr. Vladimir Pelmentchekov wrote and generously shared a script to generate ADF.in files containing any number of fixed atoms. Mr. Mitch Luna prepared Fig. 1 using PYMOL.
Supported by USPHS grants GM35342 (JAF), GM39914 (DAC), and GM43278 (LN).
Footnotes
Supporting Information Statement
Appendices describing how to reference G(H+)aq to the standard hydrogen electrode and how to calculate reaction free energies, fourteen figures showing two-dimensional maps indicating atoms, charges, and protonation status of each intermediate in the catalytic cycle; two tables showing primary results of theoretical calculations for small and large molecules; an Excel file showing the numerical calculation of catalytic reaction free energies at four levels of theory; six tables showing Mulliken charge and spin populations on selected atoms of the catalytic intermediates; two figures showing the charge and spin population changes on Fe, Cu, and Y′O in each intermediate. Lastly, the geometry of each catalytic intermediate is provided in a compressed tar file.
References
- 1.Guy RD, Fogel ML, Berry JA. Photosynthetic fractionation of the stable isotopes of oxygen and carbon. Plant Physiol. 1993;101:37–47. doi: 10.1104/pp.101.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wikström MKF. Proton pump coupled to cytochrome c oxidase in mitochrondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 3.Babcock GT, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–308. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 4.Rich PR. Towards an understanding of the chemistry of oxygen reduction and proton translocation in the iron-copper respiratory oxidases. Aust. J. Physiol. 1995;22:479–486. [Google Scholar]
- 5.Ferguson-Miller S, Babcock GT. Heme/Copper terminal oxidases. Chem. Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 6.Michel H, Behr J, Harrenga A, Kannt A. Cytochrome c oxidase: Structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 1998;27:329–356. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]
- 7.Richter O-MH, Ludwig B. Cytochrome c oxidase - structure, function, and physiology of a redox driven molecular machine. Rev. Physiol. Biochem. Pharmacol. 2003;147:47–74. doi: 10.1007/s10254-003-0006-0. [DOI] [PubMed] [Google Scholar]
- 8.Brunori M, Giuffre A, Sarti P. Cytochrome c oxidase, ligands and electrons. J. Bioinorg. Chem. 2005;99:324–326. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Bränden G, Gennis RB, Brzezinski P. Transmembrane proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 2006;1757:1052–1063. doi: 10.1016/j.bbabio.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 10.Nicholls DG, Ferguson SJ. Bioenergetics3. Academic Press Ltd.; London: 2002. [Google Scholar]
- 11.Zee JM, Gerum DM. Defects in cytochrome oxidase assembly in humans: lessons from yeast. Biochem. Cell Biology. 2006;84:859–869. doi: 10.1139/o06-201. [DOI] [PubMed] [Google Scholar]
- 12.Pereira MM, Santana M, Teixeira M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta. 2001;1505:185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 13.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376(6542):660–9. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 14.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Nat. Acad. Sci. U.S.A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmermann BH, Nitsche CI, Fee JA, Rusnak F, Munck E. Properties of a copper-containing cytochrome ba3: a second terminal oxidase from the extreme thermophile Thermus thermophilus. Proc. Nat'l. Acad. Sci. U. S. A. 1988;85(16):5779–83. doi: 10.1073/pnas.85.16.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keightley JA, Zimmermann BH, Mather MW, Springer P, Pastuszyn A, Lawrence DM, Fee JA. Molecular genetic and protein chemical charactertization of the cytochrome ba3 from Thermus thermophilus HB8. J. Biol. Chem. 1995;270:20345–20358. doi: 10.1074/jbc.270.35.20345. [DOI] [PubMed] [Google Scholar]
- 17.Soulimane T, Buse G, Bourenkov GB, Bartunik HD, Huber R, Than ME. Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunsicker-Wang LM, Pacoma RL, Chen Y, Fee JA, Stout CD. A novel cryoprotection scheme for enhancing the diffration of crystals of the recombinant, integral-membrane cytochrome ba3 from Thermus thermophilus. Acta Cryst. 2005;D61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 19.Luna VMM, Chen Y, Fee JA, Stout CD. Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: Structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry. 2008;47:4657–4665. doi: 10.1021/bi800045y. [DOI] [PubMed] [Google Scholar]
- 20.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272(5265):1136–44. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 21.Svensson-Ek M, Abramson J, Larsson G, Törnath S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 22.Farver O, Chen Y, Fee JA, Pecht I. Electron transfer among the CuA-, heme b- and heme a3-centers of Thermus thermophilus cytochrome ba3. FEBS Lett. 2006;580:3417–3421. doi: 10.1016/j.febslet.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Abbreviations used: ADF, Amsterdam Density Functional, a computer program; B3LYP and B3LYP*, hybrid exchange correlation potentials; DFT, density functional theory; ET, electron transfer; HT, proton transfer; GO, geometry optimization; PW91 and OPBE, non-hybrid exchange correlation potentials; LS, low-spin; SCF, self consistent field; SHE, standard hydrogen electrode; ups, unpaired spins; UT, internal energy of a molecule at temperature T; ZPVE, zero point vibrational energy.
- 24.Schmidt B, McCracken J, Ferguson-Miller S. A discrete water exit pathway in the membrane protein cytochrome c oxidase. Proc. Nat'l. Acad. Sci. U. S. A. 2003;100:15539–15542. doi: 10.1073/pnas.2633243100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang H, Chen Y, Fee JA, Gennis RB. Evidence for a single proton uptake pathway in the cytochrome ba3 of Thermus thermophilus. 2008 (In preparation) [Google Scholar]
- 26.Verkhovskaya MI, Garcia-Horsman A, Puustinen A, Rigaud J-L, Morgan JE, Verkhovsky MI, Wikström M. Glutamic acid 286 in subunit I of cytochrome bo3 in involved in proton translocation. Proc. Nat'l. Acad. Sci. U. S. A. 1997;94:10128–10131. doi: 10.1073/pnas.94.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsubaki M, Hori H, Mogi T. Glutamate-286 mutants of cytochrome bo-type ubiquinol oxidase from Escherichia coli: influence of mutations on the binuclear center structure revealed by FT-IR and EPR spectroscopies. FEBS Lett. 1997;416:247–250. doi: 10.1016/s0014-5793(97)01218-0. [DOI] [PubMed] [Google Scholar]
- 28.Ädelroth P, Karpefors M, Gilderson G, Tomson FL, Gennis RB, Brzezinski P. Proton transfer from glutamate 286 determines the transition rates between oxygen intermediates in cytochrome c oxidase. Biochim. Biophys. Acta. 2000;1459:533–539. doi: 10.1016/s0005-2728(00)00194-8. [DOI] [PubMed] [Google Scholar]
- 29.Nyquist RM, Heitbrink D, Bolwien C, Gennis RB, Heberle J. Direct observation of protonation reactions during the catalytic cycle of cytochrome c oxidase. Proc. Nat'l. Acad. Sci. U. S. A. 2003;100:8715–8720. doi: 10.1073/pnas.1530408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Namslauer A, Pawate AS, Gennis RB, Brzezinski P. Redox-coupled proton translocation in biological systems: Proton shuttling in cytochrome c oxidase. Proc. Nat'l. Acad. Sci. U. S. A. 2003;100:15543–15547. doi: 10.1073/pnas.2432106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belevich I, Verkhovsky MI, Wikström M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 32.Surerus KK, Oertling WA, Fan C, Gurbiel RJ, Einarsdottir O, Antholine WE, Dyer RB, Hoffman BM, Woodruff WH, Fee JA. Reaction of cyanide with cytochrome ba3 from Thermus thermophilus: spectroscopic characterization of the Fe(II)a3-CN.Cu(II)B-CN complex suggests four 14N atoms are coordinated to CuB. Proc. of the Nat'l. Acad. Sci. U. S. A. 1992;89(8):3195–9. doi: 10.1073/pnas.89.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kannt A, Soulimane T, Buse G, Becker A, Bamberg E, Michel H. Electrical current generation and proton pumping catalyzed by the ba3-type cytochrome c oxidase from Thermus thermophilus. FEBS Lett. 1998;434:17–22. doi: 10.1016/s0014-5793(98)00942-9. [DOI] [PubMed] [Google Scholar]
- 34.Einarsdóttir Ò, Killough PM, Fee JA, Woodruff WH. An infrared study of the binding and photodissociation of carbon monoxide in cytochrome ba3 from Thermus thermophilus. J. Biol. Chem. 1989;264:2405–2408. [PubMed] [Google Scholar]
- 35.Siletsky SA, Belevich I, Jasaitis A, Konstantinov AA, Wikström M, Soulimane T, Verkhovsky MI. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta. 2007 doi: 10.1016/j.bbabio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Faxen K, Gilderson G, Adelroth P, Brzezinski P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature. 2005;437:286–289. doi: 10.1038/nature03921. [DOI] [PubMed] [Google Scholar]
- 37.Konstantinov AA. Cytochrome c oxidase as a proton-pumping peroxidase: Reaction cycle and electrogenic mechanism. J. Bioenerg. Biomemb. 1998;30:121–129. doi: 10.1023/a:1020571930850. [DOI] [PubMed] [Google Scholar]
- 38.Morgan JE, Verkhovsky MI, Palmer G, Wikström M. Role of the Pr intermediate in the reaction of cytochrome c oxidase with O2. Biochemistry. 2001;40:6882–6892. doi: 10.1021/bi010246w. [DOI] [PubMed] [Google Scholar]
- 39.Blackmore RS, Greenwood C, Gibson QH. Studies of the primary oxygen intermediates in the reaction of fully reduced cytochrome oxidase. J. Biol. Chem. 1991;266:19245–19249. [PubMed] [Google Scholar]
- 40.Einarsdottir O. Fast reactions of cytochrome oxidase. Biochim. Biophys. Acta. 1995;1229:129–147. doi: 10.1016/0005-2728(94)00196-c. [DOI] [PubMed] [Google Scholar]
- 41.Hallén S, Nilsson T. Proton transfer during the reaction between fully reduced cytochrome c oxidase and dioxygen: pH and deuterium isotope effects. Biochemistry. 1992;31:11853–11859. doi: 10.1021/bi00162a025. [DOI] [PubMed] [Google Scholar]
- 42.Fabian M, Wong WW, Gennis RB, Palmer G. Mass spectrometric determination of diolygen bond splitting in the “peroxy” intermediate of cytochrome c oxidase. Proc. Nat'l. Acad. Sci. U. S. A. 1999;96:13114–13117. doi: 10.1073/pnas.96.23.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proshlyakov DA, Pressler MA, Babcock GT. Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc. Nat'l. Acad. Sci. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitagawa T, Ogura T. Time-resolved resonance Raman investigation of oxygen reduction mechanism of bovine cytochrome c oxidase. J. Bioenerg. Biomemb. 1998;30:71–79. doi: 10.1023/a:1020511612194. [DOI] [PubMed] [Google Scholar]
- 45.Han S, Ching Y-C, Rousseau DL. Cytochrome c oxidase: Decay of the primary oxygen intermediate involves direct electron transfer from cytochrome a. Proc. Nat'l. Acad. Sci. 1990;87:8408–8412. doi: 10.1073/pnas.87.21.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han S, Takahashi S, Rousseau DL. Time dependence of the catalytic intermediates in cytochrome c oxidase. J. Biol. Chem. 2000;275:1910–1919. doi: 10.1074/jbc.275.3.1910. [DOI] [PubMed] [Google Scholar]
- 47.Zaslavsky D, Smirnova IA, Adelroth P, Brzezinski P, Gennis RB. Observation of a novel transient ferryl complex with reduced CuB in cytochrome c oxidse. Biochemistry. 1999;38:2307–2311. doi: 10.1021/bi9822832. [DOI] [PubMed] [Google Scholar]
- 48.Fabian M, Palmer GP. Redox state of peroxy and ferryl intermediates in cytochrome c oxidase catalysis. Biochemistry. 1999;38:6270–6275. doi: 10.1021/bi982541v. [DOI] [PubMed] [Google Scholar]
- 49.Fee JA, Zimmermann BH, Nitsche CI, Rusnak F, Münck E. The Cytochrome caa3 from Thermus thermophilus. Chem. Scripta. 1988;28A:75–78. [Google Scholar]
- 50.Ullmann GM, Noodleman L, Case DA. Density functional calculation of p K(a) values and redox potentials in the bovine Rieske iron-sulfur protein. J. Biol. Inorg. Chem. 2002;7(6):632–9. doi: 10.1007/s00775-002-0342-6. [DOI] [PubMed] [Google Scholar]
- 51.Irikura KK. Essential Statistical Thermodynamics. In: Irikura KK, Frurip DJ, editors. Computational thermochemistry: Prediction and estimation of molecular thermodynamics. Vol. 677. American Chemical Society; Washington D. C.: 1996. pp. 402–418. [Google Scholar]
- 52.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. Modeling cytochrome oxidase: A quantum chemical study of the O-O bond cleavage mechanism. J. Am. Chem. Soc. 2000;122:12848–12858. [Google Scholar]
- 53.Mitchell P. Chemiosmotic coupling and energy transduction. Glynn Research Ltd.; Bodmin: 1968. p. 111. [Google Scholar]
- 54.Gelles J, Blair DF, Chan SI. The proton-pumping site of cytochrome c oxidase: a model of its structure and mechanism. Biochim. Biophys. Acta. 1986;853:205–236. doi: 10.1016/0304-4173(87)90002-4. [DOI] [PubMed] [Google Scholar]
- 55.Mitchell P. A new redox loop formality involving metal-catalysed hydroxide-ion translocation. A hypothetical Cu loop mechanism for cytochrome oxidase. FEBS Letters. 1987;222(2):235–45. doi: 10.1016/0014-5793(87)80378-2. [DOI] [PubMed] [Google Scholar]
- 56.Wikström MFK. Identification of the electron transfers in cytochrome oxidase that are coupled to proton-pumping. Nature. 1989;338:776–778. doi: 10.1038/338776a0. [DOI] [PubMed] [Google Scholar]
- 57.Wikström M, Bogachev A, Finel M, Morgan JE, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky MI. Mechanism of proton translocation by the respiratory oxidases. The histidine cycle. Biochim. Biophys. Acta. 1994;1187:106–111. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 58.Wikström M. Mechanism of proton translocation by cytochrome c oxidase: a new four-stroke histidine cycle. Biochim. Biophys. Acta. 2000;1458:188–198. doi: 10.1016/s0005-2728(00)00068-2. [DOI] [PubMed] [Google Scholar]
- 59.Wikström M, Verkhovsky MI. Proton translocation by cytochrome c oxidase in different phases of the catalytic cycle. Biochim. Biophys. Acta. 2002;1555:128–132. doi: 10.1016/s0005-2728(02)00267-0. [DOI] [PubMed] [Google Scholar]
- 60.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 61.Michel H. Cytochrome c oxidase: Catalytic cycle and mechanisms of proton pumping, a discussion. Biochemistry. 1999;38:15129–15140. doi: 10.1021/bi9910934. [DOI] [PubMed] [Google Scholar]
- 62.Wikström M. Proton translocation by cytochrome c oxidase: a rejoinder to recent criticism. Biochemistry. 2000;39:3515–3519. doi: 10.1021/bi9925322. [DOI] [PubMed] [Google Scholar]
- 63.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The Low-Spin Heme of Cytochrome C Oxidase as the Driving Element of the Proton-Pumping Process. Proc. Nat. Acad. Sci. U.S.A. 2003;100:15304. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muramoto K, Hirata K, Shinzawa-Itoh K, Yoko-o S, Yamashita Y, Aoyama H, Tsukihara T, Yoshikawa S. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc. Nat. Acad. Sci. U.S.A. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belevich I, Bloch DA, Belevich NP, M. W, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Nat. Acad. Sci. U.S.A. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.te Velde G, Bicklehaupt EM, Baerends EJ, Fonseca-Guerra C, van Gisbergen JA, Snijders JG, Ziegler T. Chemistry with ADF. J. Computational Chem. 2001;22:931–967. [Google Scholar]
- 67.Siegbahn PEM, Blomberg MRA. Transition-metal systems in biochemistry studied by high-accuracy quantum chemical methods. Chem. Rev. 2000;100:421–437. doi: 10.1021/cr980390w. [DOI] [PubMed] [Google Scholar]
- 68.Johansson MP, Kaila VRI, Laakkonen L. Charge parametrization of the metal centers in cytochrome c oxidase. J. Comput. Chem. 2007;29:753–767. doi: 10.1002/jcc.20835. [DOI] [PubMed] [Google Scholar]
- 69.Blomberg MRA, Siegbahn PEM. A quantum chemical approach to the study of reaction mechanisms of redox active metalloenzymes. J. Phys. Chem. B. 2001;105:9375–9386. [Google Scholar]
- 70.Noodleman L, Han W. Structure, redox, pKa, spin. A golden tetrad for understanding metalloenzyme energetics and reaction pathways. J. Biol. Inorg. Chem. 2006;11:674–694. doi: 10.1007/s00775-006-0136-3. [DOI] [PubMed] [Google Scholar]
- 71.Neese F. A critical evaluation of DFT, including time-dependent DFT, applied to bioiinorganic chemistry. J. Biol. Inorg. Chem. 2006;11:702–711. doi: 10.1007/s00775-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 72.Siegbahn PEM. The performance of hybrid DFT for mechanisms involving transition metal complexes in enzymes. J. Biol. Inorg. Chem. 2006;11:695–701. doi: 10.1007/s00775-006-0137-2. [DOI] [PubMed] [Google Scholar]
- 73.Pisliakov AV, Sharma PK, Chu ZT, Haranczyk M, Warshel A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc. Nat. Acad. Sci. U.S.A. 2008;105:7726–7731. doi: 10.1073/pnas.0800580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaukonen M. Calculated reaction cycle of cytochrome c oxidase. J. Phys. Chem. B. 2007;111:12543–12550. doi: 10.1021/jp070578w. [DOI] [PubMed] [Google Scholar]
- 75.Mitani M, Inoue M, Yoshioka Y. A B3LYP study on the mechanism of second H2O formation in a fully reduced cytochrome c oxidase. Chem. Phys. Lett. 2007;440:296–301. [Google Scholar]
- 76.Siegbahn PEM, Blomberg MRA. Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim. Biophys. Acta. 2007;1767:1143–1156. doi: 10.1016/j.bbabio.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 77.Blomberg MRA, Siegbahn PEM. Different types of biological proton transfer reactions studied by quantum chemical methods. Biochim. Biophys. Acta. 2006;1737:969–980. doi: 10.1016/j.bbabio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 78.Blomberg MRA, Siegbahn PEM, Wikström M. Metal-bridging mechanism for O-O bond cleavage in cytochrome c oxidase. Inorg. Chem. 2003;42:5231–5243. doi: 10.1021/ic034060s. [DOI] [PubMed] [Google Scholar]
- 79.Blomberg MRA, Siegbahn PEM, Babcock GT, Wikström M. O-O bond splitting mechanism in cytochrome oxidase. J. Inorg. Biochem. 2000;80:261–269. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 80.Quenneville J, Popovic DM, Stuchebrukhov AA. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim. Biophys. Acta. 2006;1757:1035–1046. doi: 10.1016/j.bbabio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 81.Popovic DM, Stuchebrukhov AA. Proton exit channels in bovine cytochrome c oxidase. J. Phys. Chem. B. 2005;109:1999–2006. doi: 10.1021/jp0464371. [DOI] [PubMed] [Google Scholar]
- 82.Quenneville J, Popovic DM, Stuchebrukhov AA. Redox-dependent pKa of CuB histidine ligand in cytochrome c oxidase. J. Phys. Chem. B. 2004;108:18383–18389. [Google Scholar]
- 83.Popovich DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome c oxidase. J. Am. Chem. Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 84.Popovic DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Lett. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 85.Popovic DM, Quenneville J, Stuchebrukhov AA. DFT/Electrostatic calculations of pKa values in cytochrome c oxidase. J. Phys. Chem. B. 2005;109:3616–3626. doi: 10.1021/jp046535m. [DOI] [PubMed] [Google Scholar]
- 86.Fadda E, Chakrabarti N, Pomes R. Acidity of a Cu-bound histidine in the binuclear center of cytochrome c oxidase. J. Phys. Chem. B. 2005;109:22629–22640. doi: 10.1021/jp052734+. [DOI] [PubMed] [Google Scholar]
- 87.Fadda E, Chakrabarti N, Pomes R. Reply to “Comment on acidity of a Cu-bound histidine in the binuclear center of cytochrome c oxidase. J. Phys. Chem. B. 2006;110:17288–17289. doi: 10.1021/jp052734+. [DOI] [PubMed] [Google Scholar]
- 88.Kamiya K, Boreo M, Tateno M, Shiraishi K, Oshiyama A. Possible mechanism of proton transfer through peptide groups in the H-pathway of the bovine cytochrome c oxidse. J. Am. Chem. Soc. 2007;129:9663–9673. doi: 10.1021/ja070464y. [DOI] [PubMed] [Google Scholar]
- 89.Takano Y, Nakamura H. Quantum mechanical study of the proton transfer via a peptide bond in the novel proton translocation pathway of cytochrome c oxidase. Chem. Phys. Lett. 2006;430:149–155. [Google Scholar]
- 90.Xu J, Sharpe MA, Qin L, Ferguson-Miller S, Voth GA. Storage of an excess proton in the hydrogen-bonded network of the D-pathway of cytochrome c oxidase: Identification of a protonated water cluster. J. Am. Chem. Soc. 2007;129:2910–2913. doi: 10.1021/ja067360s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tan M-L, Balabin I, Onuchic JO. Dynamics of electron transfer pathways in cytochrome c oxidase. Biophys. J. 2004;86:1813–1819. doi: 10.1016/S0006-3495(04)74248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moore DB, Martiniz TJ. Ab initio study of coupled electron transfer/proton transfer in cytochorme c oxidase. J. Phys. Chem. A. 2000;104:2367–2374. [Google Scholar]
- 93.Popovic DM, Stuchebrukhov AA. Two conformational states of Glu242 and pKas in bovine cytochrome c oxidase. Photochem. Photobiol. Sci. 2006;5:611–620. doi: 10.1039/b600096g. [DOI] [PubMed] [Google Scholar]
- 94.Kim YC, Wikström M, Hummer G. Kinetic models of redox-coupled proton pumping. Proc. Nat. Acad. Sci. U.S.A. 2007;104:2169–2174. doi: 10.1073/pnas.0611114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Experimentally, the Fe to Cu distance in ba3-oxidase increases from ∼4.4 Å in the “as isolated” structure to ∼5.2 Å in the chemically reduced, carbonylated form of the enzyme [B. Liu, Y. Chen, J. A. Fee, and C. D. Stout (unpublished observations)]. In the “4.4 Å structures,” the bridging peroxo intermediates (4 and 5) exhibited the unusual behavior, during scf/geometry optimization (see Methods), of producing a very short O-O distance, more typical of dioxygen (∼1.2 Å) than of hydrogen peroxide (∼1.4 Å), whereas in the “5.2 Å structure,” geometry optimized structure 4 shows Fe-O at 1.97, Cu-O at 2.12, and O-O at 1.35 Å. One might expect the metal-metal distance to change somewhat during catalysis and that fixing the Fe-Cu distance might also introduce unaccounted strain. However, there is at present no realistic way to permit such motions in the theoretical model.
- 96.Dr. B. Liu has re-refined the original data of Dr. L. Hunsicker-Wang finding that the data support an I-His384 NE to Fe distance of 2.2 Å as opposed to the value of 2.4 Å in the original publication. (B. Liu, J. A. Fee, and C. D. Stout, unpublished observations.).
- 97.Mitchell R, Mitchell P, Rich PR. Protonation states of the catalytic intermediates of cytochrome c oxidse. Biochim. Biophys. Acta. 1992;1101:188–191. [PubMed] [Google Scholar]
- 98.Mitchell R, Rich PR. Proton uptake by cytochrome c oxidase on reduction and on ligand binding. Biochim. Biophys. Acta. 1994;1186:19–26. doi: 10.1016/0005-2728(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 99.The initial Mossbauer characterization of ba3-oxidase revealed the presence of low-spin Fe(III)15, and preliminary DFT calculations indicated that Cu(II)-−OH was greatly unstable compared with Fe(III)-−OH (J. A. Fee, unpublished observations). Therefore, we started our study with Fe(III)-−OH, Cu(II) as the probable structure of the active-oxidized form.
- 100.A normal emanating from CuB intersects the approximate plane of Fe(N)4 at ∼ 1.6 Å from Fe. This structural feature may be important in guiding the details of O2-reduction.
- 101.Wikström M. Energy-dependent reversal of the cytochrome oxidase reaction. Proc. Nat. Acad. Sci. U.S.A. 1981;78:4051–4054. doi: 10.1073/pnas.78.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Agmon N. The Grotthuss mechanism. Chem. Phys. Lett. 1995;244:456–462. [Google Scholar]
- 103.Hynes JT. The protean proton in water. Nature. 1999;397:565–566. [Google Scholar]
- 104.Marx D, Tuckerman ME, Hutter J, Parrinello M. The nature of the hydrated excess proton in water. Nature. 1999;397:601–603. [Google Scholar]
- 105.Heberle J. A local area network of protonated water molecule. Biophys. J. 2004;87:2105–2106. doi: 10.1529/biophysj.104.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Szsraz S, Oesterhelt D, Ormos P. pH-induced structural changes in bacteriorhodopsin studied by Fourier transform infrared spectroscopy. Biophys. J. 1994;67:1706–1712. doi: 10.1016/S0006-3495(94)80644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.In the structure of the Rs cytochrome aa3, I-His411 (homologous to I-His376 of ba3) coordinates to a Mg2+-cation which also coordinates II-Glu254 (homologous to II-Glu126 of ba3). It is possible that “oxidative acidification” promotes loss of a proton from this Mg-complex into the water region between the two subunits. This is also the site where water molecules are known to leave the dnc151.
- 108.Balashov SP, Imasheva ES, Govindjee R, Ebrey TG. Titration of aspartate-85 in bacteriorhodopsin: What it says about chromophore isomerization and proton release. Biophys. J. 1996;70:473–481. doi: 10.1016/S0006-3495(96)79591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.The structure of this region in the A1-type Rhodobacter enzyme14 shows that the analogous II-Glu101 is swung out of what is likely its normal position and maintained in this position by coordination to Cd2+, an inhibitor, that was included in the crystallization medium.
- 110.ADF2007.01. SCM . Theoretical Chemistry. Vrije Universiteit, A.; Netherlands, T.: 2006. http://www.scm.com. [Google Scholar]
- 111.Fonseca-Guerra C, Snijders JG, te Velde G, Baerends EJ. Towards an order-N DFT method. Theor. Chem. Acc. 1998;99:391–403. [Google Scholar]
- 112.Jaguar, version 7.0. Schrodinger, LLC; New York, NY: 2007. 2007. [Google Scholar]
- 113.Irikura KK. Experimental vibrational zero-point energies: Diatomic molecules. J. Phys. Chem. Ref. Data. 2007;36:389–397. [Google Scholar]
- 114.A redox potential expressed in volts is not per se a free energy. It can be converted to a free energy in kcal/mol by the Nernst equation: ΔG = − n × (23.060 kcal/mol-volt) × ΔE(volt) where n is the number of electrons involved and a positive value of ΔE referenced to SHE describes a favorable (ΔG < 0) process. It is important to distinguish this from the situation where free energies, ΔG, are already expressed in eV and can be converted to ΔG in kcal/mol by: G(kcal/mol) = G(eV) × 23.060 kcal/mol-eV.
- 115.Zhan C-G, Dixon DA. Absolute hydration free energy of the proton from first-principles electronic structure calculations. J. Phys. Chem. A. 2001;105:11534–11540. [Google Scholar]
- 116.Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980:1200–1211. [Google Scholar]
- 117.Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C. Atoms, molecules, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B. 1992;46:6671–6687. doi: 10.1103/physrevb.46.6671. [DOI] [PubMed] [Google Scholar]
- 118.Klamt A, Jones V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996;105:9972–9981. [Google Scholar]
- 119.Pye CC, Ziegler T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999;101:396–408. [Google Scholar]
- 120.Noodleman L, Baerends EJ. Electronic structurre, magnetic properties, ESR, and optical spectra for 2-Fe ferredoxin models by LCAO-Xalpha valence bond theory. J. Am. Chem. Soc. 1984;106:2316–2327. [Google Scholar]
- 121.Handy NC, Cohen AJ. Left-right correlation energy. Mol. Phys. 2001;99:403–412. [Google Scholar]
- 122.Swart M, Groenhof AR, Ehlers AW, Lammerstma K. Validation of exchange-correlation functionals for spin states of iron complexes. J. Phys. Chem. A. 2004;108A:5479–5483. [Google Scholar]
- 123.Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 124.Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1997;78:1396. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 125.Watson MA, Handy NC, Cohen AJ. B3LYP. J. Chem. Phys. 2003;119:6475–6481. [Google Scholar]
- 126.Reiher M, Salomon O, Hess BA. Reparametrization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Accts. 2001;107:48–55. [Google Scholar]
- 127.Conradie J, Ghosh A. DFT calculation on the spin-crossover complex Fe(salen)(NO): A quest for the best functional. Phys. Chem. Lett. 2007;B111:12621–12624. doi: 10.1021/jp074480t. [DOI] [PubMed] [Google Scholar]
- 128.George P. The fitness of oxygen. In: King TE, Mason HS, Morrison M, editors. Oxidases and Related Systems. Vol. 1. John Wiley & Sons; New York: 1965. pp. 3–33. [Google Scholar]
- 129.Fee JA, Chen Y, Todaro TR, Bren KL, Patel KM, Hill MG, Gomez-Moran E, Loehr TM, Ai J, Thöny-Meyer L, Williams PA, Stura E, V. SS, McRee DE. Integrity of Thermus thermophilus cytochrome c552 synthesized by Escherichia coli cells expressing the host-specific cytochrome c maturation genes, ccmABCDEFGH: Biochemical, spectral and structural characterization of the recombinant protein. Protein Science. 2000;9:2074–2084. doi: 10.1110/ps.9.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Salomon O, Reiher M, Hess BA. Assertion and validation of the performance of the B3LYP(star) functional for the first transition metal row and the G2 test set. J. Chem. Phys. 2002;117:4729–4737. [Google Scholar]
- 131.Behan RK, Green MT. On the status of ferryl protonation. J. Inorg. Biochem. 2006;100:448–459. doi: 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 132.Yoshioka Y, Kawai H, Yamaguchi K. Theoretical study of role of H2O molecule on initial stage of reduction of O2 molecule in active site of cytochrome c oxidase. Chem. Phys. Lett. 2003;374:45–52. [Google Scholar]
- 133.Sawyer DT. Oxygen Chemistry. Oxford University Press; New York: 1991. [Google Scholar]
- 134.Forte E, Scandurra FM, Richter O-MH, D'Itri E, Sarti P, Brunori M, Ludwig B, Giuffre A. Proton uptake upon anaerobic reduction of the Paracoccus denitrificans cytochrome c oxidase: A kinetic investigation of the K354M and D124N mutants. Biochemistry. 2004;43:2957–2963. doi: 10.1021/bi035863u. [DOI] [PubMed] [Google Scholar]
- 135.Sousa FL, Verissimo AF, Baptista AM, Soulimane T, Teixeira M, Pereira MM. Redox properties of Thermus thermophilus ba3: different electron-proton coupling in oxygen reductases? Biophys. J. 2008 doi: 10.1529/biophysj.107.122614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pauling L. Magnetic properties and structure of oxyhemoglobin. Proc. Nat. Acad. Sci. U.S.A. 1977;74:2612–2613. doi: 10.1073/pnas.74.7.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weiss JJ. Nature of the iron-oxygen bond in oxyhaemoglobin. Nature. 1964;202:83–84. doi: 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- 138.Riistama S, Puustinen A, Garcia-Horsman A, Iwata S, Michel H, Wikström H. Channelling of dioxygen into the respiratory enzyme. Biochim. Biophys. Acta. 1996;1275:1–4. doi: 10.1016/0005-2728(96)00040-0. [DOI] [PubMed] [Google Scholar]
- 139.Babcock GT. How oxygen is activated and reduced in respiration. Proc. Nat. Acad. Sci. U.S.A. 1999;96:12971–12973. doi: 10.1073/pnas.96.23.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Proshlyakov DA, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Appelman EH, Kitagawa T. Selective resonance Raman observation of the “607 nm” form generated in the reaction of oxidized cytochrome c oxidase with hydrogen peroxide. J. Biol. Chem. 1994;269:29385–29388. [PubMed] [Google Scholar]
- 141.Chang CJ, Chang MCY, Damrauer NH, Nocera DG. Proton-coupled electron transfer: a unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/breaking reaction of water and oxygen. Biochim. Biophys. Acta. 2004;1655:13–28. doi: 10.1016/j.bbabio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 142.Hatcher E, Soudackov A, Hammes-Schiffer Nonadiabatic proton-coupled electron transfer reactions: Impact of donor-Accpetor vibrations, reorganization energies, and couplings on dynamics and rates. J. Phys. Chem. B. 2005;109:18565–18574. doi: 10.1021/jp052909f. [DOI] [PubMed] [Google Scholar]
- 143.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Oxygen activation and reduction in respiration: Involvement of redox-acive tyrosine-244. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 144.Hansson Ö, Karlsson B, Aasa R, Vänngård T, Malmström BG. The structure of the paramagnetic oxygen intermediate in the cytochrome c oxidase reaction. EMBO J. 1982;1:1295–1297. doi: 10.1002/j.1460-2075.1982.tb01313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wiertz FGM, Richter O-MH, Ludwig B, de Vries S. Kinetic resolution of a tryptophan-radical intermediate in the reaction cycle of Paracoccus denitrificans cytochrome c oxidase. J. Biol. Chem. 2007;282:31580–31591. doi: 10.1074/jbc.M705520200. [DOI] [PubMed] [Google Scholar]
- 146.Moser CC, Page CC, Dutton PL. Darwin at the molecular scale: selection and variance in electron tunnelling proteins including cytochrome c oxidase. Phil. Trans. R. Soc. 2006;361:1295–1305. doi: 10.1098/rstb.2006.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Michel B, Bosshard HR. Oxidation of cytochrome c by cytochrome c oxidase: Spectroscopic binding studies and steady-state kinetics support a conformational transition mechanism. Biochemistry. 1989;28:244–252. doi: 10.1021/bi00427a034. [DOI] [PubMed] [Google Scholar]
- 148.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 149.Harrenga A, Michel H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J. Biol. Chem. 1999;274:33296–33299. doi: 10.1074/jbc.274.47.33296. [DOI] [PubMed] [Google Scholar]
- 150.Puustinen A, Wikström M. Proton exit from the heme-copper oxidase of Escherichia coli. Proc. Nat'l. Acad. Sci. U.S. A. 1999;96:35–37. doi: 10.1073/pnas.96.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qian J, Mills DA, Geren L, Wang K, Hoganson CW, Schmidt B, Hiser C, Babcock GT, Durham B, Millett F, Ferguson-Miller S. Role of the conserved arginine pair in proton and electron transfer in cytochrome c oxidase. Biochemistry. 2004;43:5748–5756. doi: 10.1021/bi036279o. [DOI] [PubMed] [Google Scholar]
- 152.Himo F, Noodleman L, Blomberg MRA, Siegbahn PEM. Relative acidities of ortho-substituted phenols as models for modified tyrosines in proteins. J. Phys. Chem.A. 2002;106A:8757–8761. [Google Scholar]
- 153.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/a-ketoglutarate dioxygenase. J. Am. Chem. Soc. 2003;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 154.The efficiency of the overall process can be expressed as ΔGpump / (ΔGoverall + ΔGpump), which is ∼0.46 for the theoretical process and ∼0.36 if the experimental, overall reaction free energy of −38.7 kcal/mol is considered.
- 155.Zu Y, Fee JA, Hirst J. Complete thermodynamic characterization of reduction and protonation of the bc(1)-type Rieske [2Fe-2S] center of Thermus thermophilus. J Am Chem Soc. 2001;123(40):9906–7. doi: 10.1021/ja016532c. [DOI] [PubMed] [Google Scholar]
- 156.Ramirez BE, Malmstrom BG, Winkler JR, Gray HB. The currents of life: the terminal electron-transfer complex of respiration [comment] Proc. Nat. Acad. Sci. U.S.A. 1995;(26):11949–51. doi: 10.1073/pnas.92.26.11949. coll. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gupte SS, Hackenbrock CR. Multidimensional diffusion modes and collision frequencies of cytochrome c with its redox partners. J. Biol. Chem. 1988;263:5241–5247. [PubMed] [Google Scholar]
- 158.Gupte SS, Hackenbrock CR. The role of cytochrome c diffusion in mitochondrial electron transport. J. Biol. Chem. 1988;263:5248–5253. [PubMed] [Google Scholar]
- 159.Chance B. Reaction of oxygen with the respiratory chain in cells and tissues. J. Gen. Physiol. 1965;49(1P2):163. doi: 10.1085/jgp.49.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gray HB, Malmstrom BG. Long-range electron transfer in multisite metalloproteins. Biochemistry. 1989;28:7499–7505. doi: 10.1021/bi00445a001. [DOI] [PubMed] [Google Scholar]
- 161.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 162.Collman JP, Devaraj NK, Decreau RA, Yang Y, Yan Y-L, Ebina W, Eberspacher TA, Chidsey CED. A cytochrome c oxidase model catalyzes oxygen to water reduction under rate-limiting conditions. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.