

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that participate in the spatiotemporal regulation of messenger RNA and protein synthesis. Aberrant miRNA expression leads to developmental abnormalities and diseases, such as cardiovascular disorders and cancer; however, the stimuli and processes regulating miRNA biogenesis are largely unknown. The transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP) family of growth factors orchestrates fundamental biological processes in development and in the homeostasis of adult tissues, including the vasculature. Here we show that induction of a contractile phenotype in human vascular smooth muscle cells by TGF-β and BMPs is mediated by miR-21. miR-21 downregulates PDCD4 (programmed cell death 4), which in turn acts as a negative regulator of smooth muscle contractile genes. Surprisingly, TGF-β and BMP signalling promotes a rapid increase in expression of mature miR-21 through a post-transcriptional step, promoting the processing of primary transcripts of miR-21 (pri-miR-21) into precursor miR-21 (pre-miR-21) by the DROSHA (also known as RNASEN) complex. TGF-β- and BMP-specific SMAD signal transducers are recruited to pri-miR-21 in a complex with the RNA helicase p68 (also known as DDX5), a component of the DROSHA microprocessor complex. The shared cofactor SMAD4 is not required for this process. Thus, regulation of miRNA biogenesis by ligand-specific SMAD proteins is critical for control of the vascular smooth muscle cell phenotype and potentially for SMAD4-independent responses mediated by the TGF-β and BMP signalling pathways.
Mutations in molecules of the TGF-β or BMP signalling pathways are found among patients with vascular disorders, indicating the essential role of TGF-β or BMP pathways in vascular homeostasis1,2. Both TGF-βs and BMPs are known to be critical modulators of the vascular smooth muscle cell (VSMC) phenotype3-5. Inhibition of TGF-β or BMP signalling in VSMCs decreases the expression of VSMC-specific genes and transforms VSMCs from a fully differentiated or ‘contractile’ phenotype to a dedifferentiated or ‘synthetic’ state4-6.
miR-21 modulates smooth muscle phenotype
We investigated the involvement of miRNAs in the TGF-β-family-mediated modulation of the VSMC phenotype by cloning and comparing the relative abundance of miRNAs expressed in vehicle- and BMP4-treated human primary pulmonary artery smooth muscle cells (PASMCs; Supplementary Fig. 1). The expression level of a selected group of miRNAs was then directly measured by quantitative polymerase chain reaction with reverse transcription (qRT–PCR) after 24 h of BMP4 stimulation (Fig. 1a): mature miR-21 and miR-199a showed a significant increase in expression (5.7-fold and 2.1-fold, respectively) in the presence of BMP4 (P < 0.05). miR-21 was comparably induced by three BMP ligands that stimulate VSMC differentiation (BMP2, BMP4 and BMP7)5 (Supplementary Fig. 2). Thus, a subset of miRNAs is induced by BMP signalling in VSMCs. High expression of miR-21 has also been observed in the vascular wall of balloon-injured rat carotid arteries—an in vivo model recapitulating smooth muscle phenotype switch7.
Figure 1. miR-21 is critical for the modulation of the VSMC phenotype by BMP.
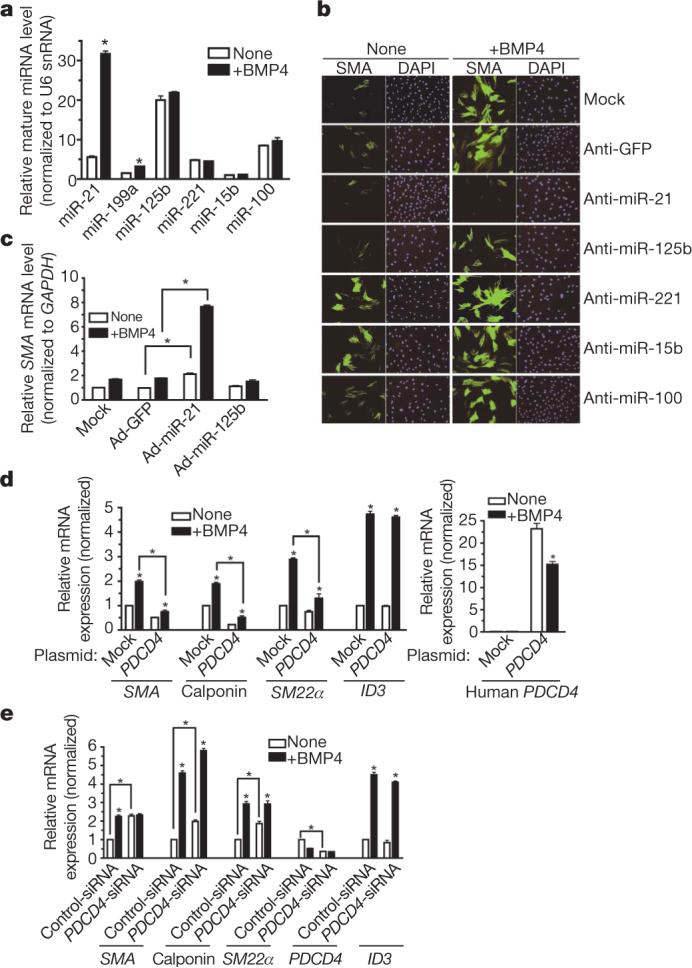
a, The level of expression of miRNAs normalized to U6 small nuclear RNA (snRNA) in PASMCs treated with BMP4 for 24 h (*P < 0.05, n = 4). b, PASMCs transfected with antisense RNA oligonucleotides against different miRNAs or GFP (control). After BMP4 treatment (48 h), cells were stained with anti-SMA antibody (green) and 4,6-diamidino-2-phenylindole (DAPI; blue). c, PASMCs were infected with an adenovirus carrying CMV-driven GFP (control; Ad-GFP), miR-21 (Ad-miR-21) or miR-125b (Ad-miR-125b). The SMA mRNA level was measured after BMP4 treatment (48 h) (*P < 0.05, n = 4). d, 10T1/2 cells were transfected with vector (mock) or a human PDCD4 cDNA construct, followed by BMP4 treatment (24 h). Expression of Sma, calponin, Sm22α, Id3 or human PDCD4 relative to GAPDH mRNA is shown (*P < 0.001, n = 3). e, PASMCs transfected with control siRNA (Control-siRNA) or siRNA for PDCD4 (PDCD4-siRNA). Relative mRNA expression is shown as in d. Error bars represent s.e.m.
The function of miRNAs was tested by transfecting PASMCs with ‘anti-miRs’: 2′-O-methyl-modified RNA oligonucleotides complementary to individual miRNA sequences8. Anti-miR-21 specifically decreased mature miR-21 expression (Supplementary Fig. 3) and effectively reduced both basal and BMP4-induced expression of the smooth muscle cell (SMC) markers smooth muscle α-actin (SMA, also known as ACTA2) and calponin (also known as CNN1) (Fig. 1b and Supplementary Fig. 4a, b), suggesting that miR-21 is necessary for SMC-specific gene expression. Downregulation of different miRNAs showed specific effects: targeting miR-125a and miR-125b inhibited SMC markers (Fig. 1b and Supplementary Fig. 4a, b), whereas depletion of miR-221 and miR-15b stimulated basal SMA expression (Fig. 1b and Supplementary Fig. 4b). Anti-miR-21 also decreased SMA in pluripotent mouse C3H10T1/2 (10T1/2) cells treated with BMP4 (Supplementary Fig. 4c). In gain-of-function experiments, forced expression of miR-21 by infection with an adenoviral miR-21 construct (Ad-miR-21)9 increased SMA protein and mRNA levels in PASMCs (Fig. 1c and Supplementary Fig. 5). Thus, miR-21 is a critical mediator of SMC differentiation by BMP signalling.
PDCD4 is a critical target of miR-21 in vascular smooth muscle
Because miR-21 has been shown to target the tumour suppressor gene PDCD4 and to downregulate its expression in cancer cells10-12, we asked whether PDCD4 mediates the effect of miR-21 in SMCs. Forced expression of miR-21 and reduction of miR-21 by anti-miR-21 in PASMCs decreased and increased PDCD4 mRNA expression, respectively (Supplementary Fig. 6a, b), confirming that PDCD4 is a miR-21 target. BMP4 treatment reduced PDCD4 (∼30%; Supplementary Fig. 6a, b) and anti-miR-21 abolished this effect (Supplementary Fig. 6b), suggesting that PDCD4 is negatively regulated by BMP4 as a result of miR-21 induction. We next examined whether modulation of PDCD4 expression in PASMCs affects SMC marker expression. Transfection of a human PDCD4 expression construct, which includes a miR-21 target sequence in its 3′ untranslated region (UTR)11 (Supplementary Fig. 6c), increased the expression of human PDCD4 in 10T1/2 cells (Fig. 1d, right panel) and inhibited basal and BMP4-induced expression of the SMC markers Sma, calponin and Sm22α (also known as Tagln), but not of Id3, a gene directly regulated by BMP4 (ref. 13), indicating that PDCD4 represses specifically SMC genes, and has no effect on BMP signalling in general (Fig. 1d, left panel). BMP4 treatment still significantly augmented SMC gene expression and decreased ectopic PDCD4 mRNA (P < 0.001), presumably through the 3′ UTR miR-21 target site (Fig. 1d). Conversely, PDCD4 knockdown (∼60%) by siRNA (PDCD4-siRNA) in PASMCs increased the basal expression of SMA, calponin and SM22α approximately twofold (Fig. 1e). BMP4 failed to induce SMA above the basal level when PDCD4 was depleted in the cell (Fig. 1e), whereas the levels of calponin and SM22α were still induced by BMP4 treatment, suggesting that BMP4 induces calponin and SM22α in part through a PDCD4-independent mechanism5,14 (Fig. 1e). In conclusion, PDCD4 is a functional target of miR-21 involved in the BMP-mediated induction of SMC markers in VSMCs.
TGF-β and BMP signalling promote miR-21 processing
TGF-β, another inducer of the contractile phenotype3-5, stimulated the expression of both miR-21 and miR-199a to a level comparable to BMP4 (Fig. 2a) with similarly fast kinetics (2 h; Supplementary Fig. 7), indicating that TGF-β and BMPs both support a contractile phenotype by means of an increase of miR-21.
Figure 2. Post-transcriptional regulation of miR-21 biosynthesis by TGF-β.
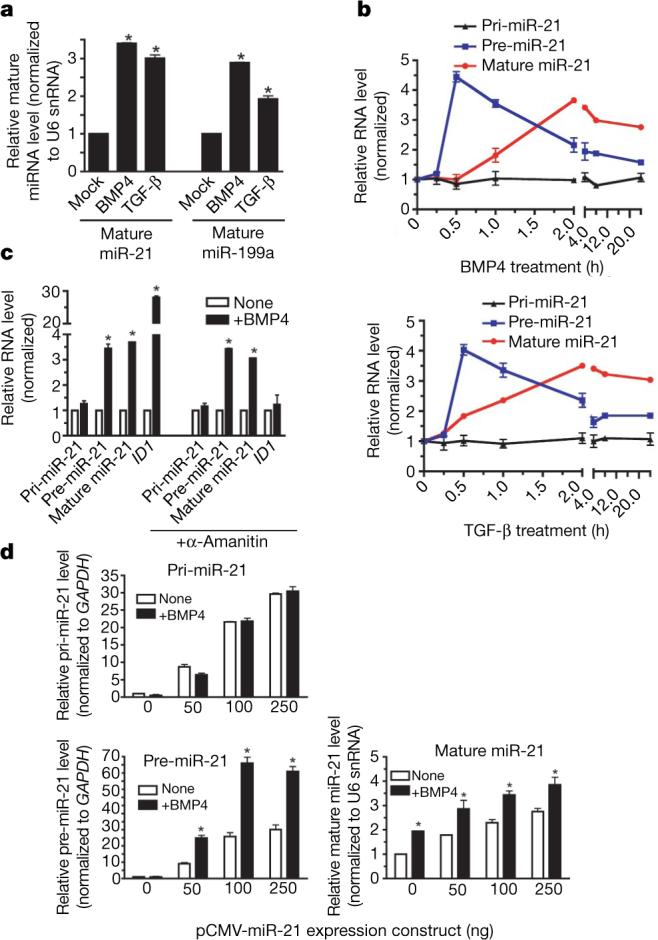
a, Expression of mature miR-21 and miR-199a normalized to U6 snRNA in PASMCs stimulated with BMP4 or TGF-β (24 h; *P < 0.05; n = 3). b, Time course of pri-miR-21, pre-miR-21 or mature miR-21 expression in PASMCs on stimulation with BMP4 (upper panel) or TGF-β (lower panel). c, PASMCs pretreated with α-amanitin were stimulated with BMP4 (5 h). Expression of pri-miR-21, pre-miR-21 and mature miR-21 or ID1 is shown (*P < 0.05; n = 3). d, Relative expression of pri-miR-21, pre-miR-21 and mature miR-21 derived from increasing amounts of human miR-21 expression construct (pCMV-miR-21) transfected into 10T1/2 cells (*P < 0.05; n = 3). Error bars represent s.e.m.
The biogenesis of miRNAs initiates with the transcription of the miRNA gene and proceeds with the cropping of the primary transcript (pri-miRNA) into a hairpin intermediate (pre-miRNA) by the nuclear ∼650 kilodalton microprocessor complex, comprised in humans of the RNase III DROSHA15, the DiGeorge syndrome critical region gene 8 (DGCR8)16,17, and the DEAD box RNA helicases p68 and p72 (also known as DDX17)18. The pre-miRNA is then exported from the nucleus and processed into a ∼22-nucleotide miRNA duplex by the cytoplasmic RNase III DICER19-21. Regulation of miRNA expression has been documented at the transcriptional level, but little is known about the stimuli and molecules regulating post-transcriptional processing19,21-26. BMPs and TGF-βs control gene expression through the SMAD proteins, which embody the qualities of both signal transducers and transcriptional modulators27,28, but are not known to affect RNA processing. Therefore, we examined the accumulation of primary miR-21 gene transcripts (pri-miR-21), pre-miR-21 and mature miR-21 on BMP or TGF-β treatment in an expression time course (Fig. 2b), expecting to find a transcriptional induction of pri-miR-21 transcripts in response to factor stimulation29. However, although we observed induction of mature miR-21 and pre-miR-21 2 h after BMP4, BMP2 or TGF-β treatment, we detected no significant change in the expression of pri-miR-21 (P < 0.05) after factor addition (Fig. 2b and Supplementary Fig. 8a), suggesting that induction of miR-21 by BMP4, BMP2 or TGF-β occurs at a post-transcriptional step. Likewise, BMP4-mediated induction of both pre-miR-21 and mature miR-21 was resistant to inhibition of RNA polymerase II by α-amanitin, whereas induction of the BMP4 transcriptional target gene ID1 (ref. 30) was abolished (Fig. 2c). Furthermore, a luciferase reporter construct containing the miR-21 gene promoter was not activated by BMP4 or TGF-β treatment, whereas it was induced by its known regulator STAT3 (Supplementary Fig. 9)31.
A dose-dependent increase of all three forms of miR-21 was observed on transfection in mouse 10T1/2 cells of pCMV-miR-21, a plasmid in which human pri-miR-21 is transcribed from the cytomegalovirus (CMV) promoter32 (Fig. 2d), indicating an expression level proportional to the episomal DNA copies. However, BMP4 could further induce pre-miR-21 and mature miR-21, but not pri-miR-21 (Fig. 2d), indicating that the miR-21 promoter or genomic locus is not required for post-transcriptional induction of miR-21 by BMP4. The plasmid-derived miR-21 induced by BMP4 was functional, because it repressed a miR-21 sensor construct containing complementary binding sites for the miR-21 sequence at the 3′ UTR of a luciferase reporter gene (Supplementary Fig. 8b). Furthermore, expression of CMV-transcribed miR-21 induced SMA mRNA and protein in 10T1/2 cells in a dose-dependent manner, and was further increased by BMP4 stimulation (Supplementary Fig. 10a, b). Thus, the BMP4 pathway promotes the expression of precursor and functional mature miR-21 through a post-transcriptional, genome-independent mechanism.
SMADs interact with the RNA helicase p68
We investigated the molecular pathway leading to miR-21 induction by RNA interference (RNAi) knockdown (∼80%, SMAD-siRNA) of the BMP-specific receptor-specific SMAD proteins (R-SMADs) expressed in PASMCs (SMAD1 and SMAD5; Fig. 3a, bottom panel, and Supplementary Fig. 11). SMAD-siRNA abolished BMP4 induction of both pre-miR-21 and mature miR-21, whereas the level of expression of pri-miR-21 was not affected (Fig. 3a, top panel). Induction of SMA and of the BMP transcriptional target ID3 was also inhibited by SMAD1 and SMAD5 depletion, as expected (Fig. 3a, bottom panel). Therefore, R-SMADs are required for pre-miR-21 stimulation by BMP4.
Figure 3. Interaction of SMADs with p68, a component of the DROSHA complex.
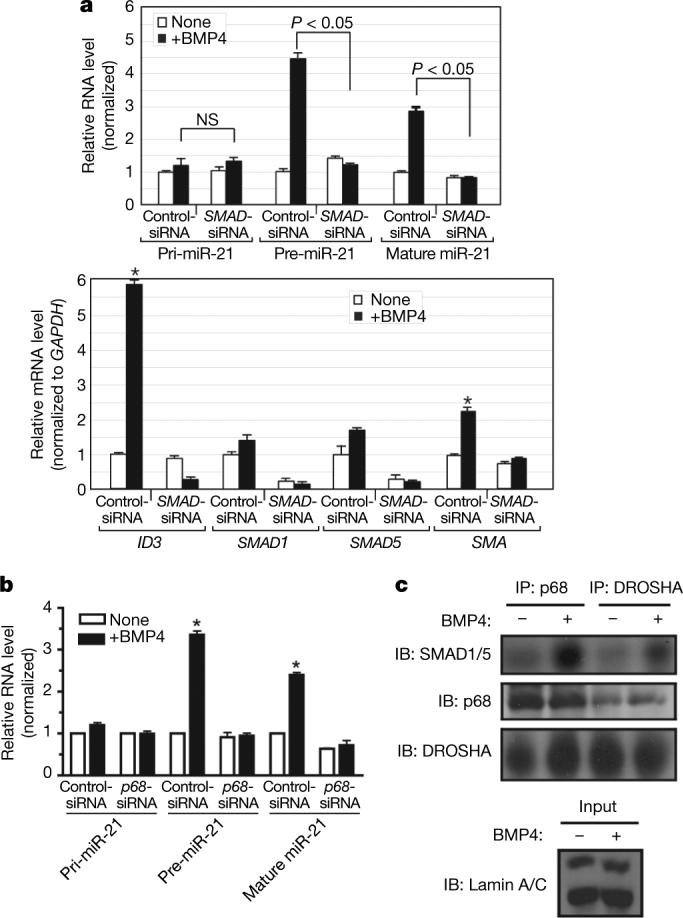
a, PASMCs were transfected with control siRNA (Control-siRNA) or a mixture of siRNAs for SMAD1 and SMAD5 (SMAD-siRNA). After BMP4 treatment (2 h), the expression of pri-miR-21, pre-miR-21 and mature miR-21 was compared (top panel). As controls, expression of ID3, SMAD1, SMAD5 and SMA is shown (bottom panel). NS, not significant (P > 0.05). b, PASMCs were transfected with control siRNA (Control-siRNA) or siRNAs for p68 (p68-siRNA). Expression of pri-miR-21, pre-miR-21 and mature miR-21 was examined after BMP4 treatment (2 h) (*P < 0.05; n = 3). c, Nuclear extracts prepared from PASMCs treated with BMP4 (2 h) and subjected to immunoprecipitation with anti-p68, anti-DROSHA antibody, or non-specific IgG (control), followed by immunostaining with anti-SMAD1/5, anti-p68 or anti-DROSHA antibody. Nuclear extracts were immunostained with anti-lamin A/C antibody (control). IB, immunoblot; error bars represent s.e.m.
We postulated that the requirement of SMADs for pre-miR-21 induction might entail a direct involvement of SMADs in the DROSHA microprocessor complex on the basis of a previous report of a constitutive interaction between the carboxy-terminal MH2 domain of SMAD1 and the RNA helicase p68 (ref. 33), a critical subunit of the DROSHA microprocessor complex18. To examine whether p68 is involved in the regulation of miR-21 expression by BMP4, p68 was downregulated in PASMCs by siRNA (∼70%, Supplementary Fig. 12). Expression of pri-miR-21 and the BMP4 target gene ID3 (ref. 13) did not change significantly (Supplementary Fig. 12b), but induction of pre-miR-21 and mature miR-21 by BMP4 was completely abolished (Fig. 3b), indicating an essential role of p68 in the TGF-β and BMP-regulated synthesis of pre-miR-21.
We found that the interaction between exogenous SMAD1 and p68 is BMP4-inducible in Cos7 cells (Supplementary Fig. 13a). In in vitro glutathione S-transferase (GST) pull-down, p68 interacts both with BMP-specific SMAD1 or SMAD5 and with TGF-β-specific SMAD3, suggesting that induction of pre-miR-21 by TGF-β may also involve an R-SMAD–p68 complex (Supplementary Fig. 14a). No interaction was observed between p68 and the cofactor SMAD4 (Supplementary Fig. 14a) or the inhibitor SMAD6 (data not shown). The interaction between R-SMADs and p68 was resistant to RNase A treatment, suggesting that R-SMADs and p68 interact in the absence of pri-miRNAs (Supplementary Fig. 15). We also confirmed that the carboxyl-terminal MH2 domain of SMAD1 is sufficient to pull down p68 (ref. 33), whereas the amino-terminal MH1 domain does not bind p68 (Supplementary Fig. 14b). Thus, by binding p68, SMAD1 may be recruited to the DROSHA microprocessor complex. Indeed, on BMP4 stimulation, endogenous SMAD1 or SMAD5 could be coimmunoprecipitated with DROSHA from PASMCs (Fig. 3c) or Cos7 extracts expressing tagged DROSHA and SMAD1 (Supplementary Fig. 13b). The interaction of R-SMADs with DROSHA was markedly reduced by RNase A treatment (Supplementary Fig. 15), suggesting that the association of R-SMADs with DROSHA, unlike the R-SMADs–p68 complex, may be facilitated by miRNA transcripts. Therefore, after ligand stimulation, SMADs associate with the DROSHA microprocessor complex by means of interaction with p68, ultimately promoting accumulation of specific pre-miRNAs.
Ligand-induced association of R-SMADs with pri-miRNAs
To test whether the R-SMAD–p68–DROSHA complex assembles specifically on pri-miR-21, we performed an RNA-chromatin immunoprecipitation (ChIP) analysis on Cos7 cells co-transfected with pCMV-miR-21 and Flag-tagged SMAD1, SMAD3 or SMAD2. The association of SMAD1 (but not SMAD2 or SMAD3) with pri-miR-21 was induced threefold on BMP4 stimulation for 2 h (Fig. 4a and Supplementary Fig. 16a), whereas TGF-β increased binding to pri-miR-21 by SMAD2 and SMAD3, but not by SMAD1, indicating that the association between R-SMADs and pri-miR-21 is specifically regulated by ligand stimulation (Fig. 4a).
Figure 4. Association of SMADs with pri-miRNA promotes processing by DROSHA.
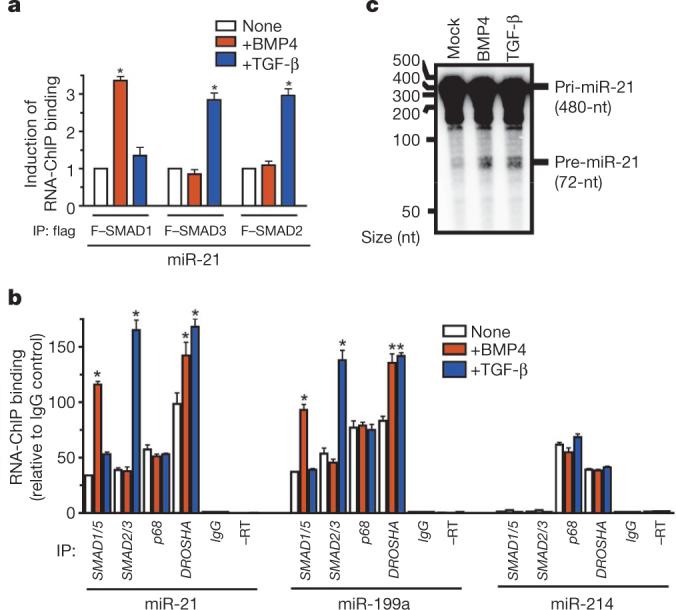
a, Cos7 cells transfected with pCMV-miR-21 and Flag–SMAD1, Flag–SMAD3 or Flag–SMAD2, followed by BMP4 or TGF-β treatment (2 h). RNA-ChIP performed with anti-Flag antibody or non-specific IgG (control), followed by PCR amplification with miR-21 primers (*P < 0.05, compared to no treatment; n = 4). IP, immunoprecipitation. b, After treatment of PASMCs with BMP4 or TGF-β (1 h), endogenous SMAD1/SMAD5, SMAD2/ SMAD3, p68 or DROSHA were immunoprecipitated and subjected to PCR analysis with miR-21, miR-199a or miR-214 primers. As controls, RNA samples untreated with reverse transcriptase (–RT) or immunoprecipitated with non-specific IgG (IgG) were subjected to PCR (*P < 0.05 compared to none; n = 4). c, In vitro pri-miRNA processing assay performed by incubating pri-miR-21 substrate with the nuclear extracts prepared from Cos7 cells treated with vehicle, BMP4 or TGF-β (2 h). nt, nucleotide; error bars represent s.e.m.
R-SMADs also interacted in a ligand-specific manner with pri-miR-21 in PASMCs (Fig. 4b), whereas p68 constitutively associated with pri-miR-21 and the recruitment of DROSHA was moderately enhanced by either TGF-β or BMP4 (Fig. 4b). Similar results were obtained for miR-199a (Fig. 4b). The significant increase (P < 0.05) we observed in the association of DROSHA with pri-miR-21 and pri-miR-199a (Fig. 4b) suggests that binding of SMADs to the pri-miRNA might stabilize the association between DROSHA and the pri-miRNA. We detected a constitutive association of pri-miR-214 with p68 and DROSHA, but no interaction with SMADs (Fig. 4b), confirming that pre-miR-214 is not regulated by BMP or TGF-β signals (Supplementary Fig. 17). Thus, recruitment of SMADs to the p68–DROSHA complex is pri-miRNA-specific.
A SMAD1 mutant that was non-phosphorylatable on BMP stimulation (SMAD1(3SA)) retained the ability to interact with pri-miR-21 (Supplementary Fig. 16a). Furthermore, bacterially expressed unphosphorylated GST–SMAD fusion proteins are able to interact with p68 (Supplementary Figs 14 and 15), indicating that receptor-mediated phosphorylation of R-SMADs is not essential for the association with pri-miRNA and suggesting that BMPs may affect the association between SMAD1 and pri-miRNAs primarily by controlling SMAD nuclear localization.
Pull-down experiments using partially purified GST–SMAD fusion proteins as bait confirmed that SMAD1, SMAD3 and SMAD5 can interact with pri-miR-21. Interestingly, both the MH1 and the MH2 domains of SMAD1 bound to pri-miR-21 (Supplementary Fig. 18). Because MH1 does not interact with p68 (Supplementary Fig. 14b), it is possible that MH1 interacts either with pri-miR-21 directly or with other miR-21-binding proteins.
In summary, BMPs and TGF-β stimulate the expression of a specific subset of miRNAs by inducing the formation of a complex comprising R-SMAD proteins, pri-miRNAs and subunits of the microprocessor complex such as DROSHA and p68.
Finally, we examined the possibility that ligand treatment may facilitate DROSHA-mediated production of pre-miRNA. In vitro pri-miRNA processing assays were performed by incubating radio-labelled pri-miR-21 substrate (480 nucleotides) with nuclear extracts from Cos7 cells treated with vehicle, BMP4 or TGF-β. Ligand treatment resulted in ∼25% increase (BMP4, 28.5% ± 1.9% (mean ± s.e.m.; TGF-β, 24.2% ± 1.4%; triplicate experiments) in the production of a 72-nucleotide product corresponding to pre-miR-21, compared to incubation with extracts from mock-treated cells (Fig. 4c). This result suggests that ligand-induced association of SMADs with the DROSHA complex increases pri-miR-21 cropping into pre-miRNA.
SMAD4-independent regulation of miRNA processing
Two observations led us to speculate that SMAD4 may be dispensable for the regulation of miR-21 processing: the lack of interaction between p68 and SMAD4 (Supplementary Fig. 14a), the common SMAD required for most transcriptional responses to BMP and TGF-β signalling; and the ability of the SMAD1(3SA) mutant, which does not form a complex with SMAD4 (ref. 34), to associate with pri-miR-21 (Supplementary Fig. 16a). Transfection of an siRNA against SMAD4 (SMAD4-siRNA) in PASMCs markedly reduced SMAD4 protein (∼90%, Supplementary Fig. 18a) and RNA (Fig. 5a), as well as the transcriptional inducibility of the BMP target gene ID3 (ref. 13; from 18-fold to 3-fold), as expected (Supplementary Fig. 18b). However, SMAD4-siRNA did not affect the induction of pre-miR-21 or mature miR-21 by BMP4 (Fig. 5a), in contrast with the result obtained from downregulation of R-SMADs (Fig. 3a). Therefore, SMAD4 is not required for the stimulation of processing of miR-21 by BMP4 in PASMCs.
Figure 5. SMAD4-independent mechanism of maturation of pri-miRNA.

a, Level of expression of pri-miR-21, pre-miR-21 and mature miR-21 or SMAD4 after treatment with BMP4 (2 h) in PASMCs transfected with control siRNA (Control-siRNA) or SMAD4 siRNA (SMAD4-siRNA). b, Level of expression of pri-miR-21, pre-miR-21 and mature miR-21 or PAI-1 in human SMAD4-negative breast carcinoma MDA-MB-468 cells stimulated with TGF-β (0.5 h) (*P < 0.05; n = 3). c, MDA-MB-468 cells were treated with TGF-β (1 h) before RNA-ChIP. Endogenous proteins were precipitated with anti-SMAD1/SMAD5, anti-SMAD2/SMAD3 or anti-DROSHA antibodies, followed by PCR analysis with a miR-21 primer (*P < 0.05, compared to none; n = 3). d, MDA-MB-468 cells were infected with adenovirus carrying dominant-negative type I TGF-β receptor (dnALK5), an inhibitor of TGF-β signalling, before TGF-β treatment (1 h). The amount of pri-miR-21, pre-miR-21 and mature miR-21 was examined (*P < 0.05, compared to none; n = 3). Error bars represent s.e.m.
Cancer cells in which the canonical TGF-β pathway is impaired, such as the SMAD4-negative MDA-MB-468 cells, lack the ability to transcriptionally regulate most TGF-β target genes35,36 but retain some TGF-β responses, such as nuclear translocation of R-SMADs, increased cell migration and epithelial-to-mesenchymal transition36-38. We investigated whether miR-21 stimulation by TGF-β can still occur in MDA-MB-468 cells as it does in PASMCs depleted of SMAD4. A rapid induction of pre-miR-21 and mature miR-21 was observed on TGF-β stimulation in MDA-MB-468 cells, without a change in the levels of pri-miR-21 or of the SMAD4-dependent TGF-β target gene plasminogen activator inhibitor-1 (PAI-1, also known as SERPINE1) mRNA35,36 (Fig. 5b). Similar results were obtained by BMP4 treatment of MDA-MB-468 and SMAD4-expressing breast carcinoma MCF7 cells (Supplementary Fig. 19) or by TGF-β treatment of SMAD4-positive breast carcinoma MDA-MB-231 cells (Supplementary Fig. 20). Stimulation of pri-miRNA processing by TGF-β does not necessarily lead to an increase in mature miRNA: unlike MDA-MB-468 cells (Fig. 5b), MDA-MB-231 cells display little increase of mature miR-21 after TGF-β stimulation despite strong induction of pre-miR-21 (Supplementary Fig. 20), suggesting the existence of another regulatory step of miRNA maturation after pri-miRNA cleavage by the DROSHA microprocessor. An RNA-ChIP analysis confirmed that in MDA-MB-468 cells the association of R-SMADs with the primary transcripts of miR-21 and miR-199a (but not miR-214) is ligand-inducible (Fig. 5c and Supplementary Fig. 21). Therefore, SMAD4 is not necessary for ligand-mediated processing of pri-miRNAs, and some of the SMAD4-independent responses observed in ligand-stimulated cells may be mediated by regulation of miRNA biogenesis by the TGF-β or BMP pathways.
TGF-β increases miR-21 expression in breast carcinoma
The expression of mature miR-21 is augmented in different types of tumours and tumour-derived cell lines, including breast carcinoma MCF7, MDA-MB-231 and MDA-MB-468 cells11,39-43. Because TGF-β expression is often increased in cancer cells, where it promotes epithelial-to-mesenchymal transition and metastatic behaviour44-48, we postulated that the increased levels of miR-21 may in part be caused by autocrine TGF-β signalling. A dominant-negative TGF-β type I receptor (ALK5, also known as TGFBR1)49, which harbours a mutation in the kinase domain, was expressed in MDA-MB-468 cells to inhibit TGF-β signalling. Both the basal and the TGF-β-induced expression of pre-miR-21 were greatly reduced, whereas the pri-miR-21 level was unchanged (Fig. 5d). These results indicate that autocrine TGF-β signalling contributes to the high basal expression of miR-21 in cancer cells.
Discussion
This study underscores several unexpected findings. First, the TGF-β superfamily of growth factors triggers VSMC differentiation by increasing the expression of a subset of miRNAs. This induction occurs post-transcriptionally, probably at the level of processing of primary transcripts by the DROSHA microprocessor complex. Ligand-specific SMAD proteins bind to the DROSHA microprocessor subunit p68 to facilitate pre-miRNA accumulation. Finally, we identified a previously unknown mechanism by which the TGF-β pathway may promote the metastatic and invasive potential of cancer cells through modulation of biosynthesis of oncogenic miRNAs such as miR-21, which in turn targets tumour suppressor genes PDCD4 and tensin homologue deleted on chromosome 10 (PTEN)10,11.
Open questions remain regarding, for example, the exact number and identity of pri-miRNAs regulated by SMADs and the determinants of specificity in their selection. The MH1 domain of R-SMADs binds DNA by specifically recognizing a sequence element27,50; we observed that the MH1 domain of SMAD1 associates with pri-miR-21 despite its inability to interact with p68: it is possible to speculate that the SMAD MH1 domain may recognize an RNA sequence or structural element, and thus provide specificity in the selection of BMP and TGF-β target miRNA. The exact role of the SMAD–p68 interaction in the DROSHA microprocessor complex also remains unsolved. Association of SMAD with the DROSHA complex is likely to contribute to various aspects of pri-miRNA processing, such as facilitating the specific recognition and stable binding of DROSHA to pri-miRNAs, increasing the RNase activity of DROSHA, directing the cleavage of pri-miRNAs to a precise sequence, or modulating the stability of pre-miRNA. In summary, our findings open new avenues to the study of TGF-β-family signalling pathways and miRNA biogenesis regulation.
METHODS SUMMARY
Cell culture
Cos7, C3H10T1/2, MDA-MB-468, MDA-MB-231 and MCF7 cells (American Type Culture Collection) were maintained in DMEM supplemented with 10% FBS (Sigma). Human primary PASMCs were purchased from Lonza (CC-2581;http://www.lonzabioscience.com/Lonza_Catnav.oid.734.prodoid. PASMC) and were maintained in Sm-GM2 media (Lonza) containing 5% FBS.
Real-time RT–PCR
Total RNA was extracted by Trizol (Invitrogen) and subjected to reverse transcription using a first-strand cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. The quantitative analysis of the change in expression levels was calculated by real-time PCR machine (iQ5, BioRad)29. For detection of mature miRNAs, the TaqMan MicroRNA assay kit (Applied Biosystems) was used according to the manufacturer's instructions. An average of three experiments each performed in triplicate with standard errors is presented.
RNA-ChIP
RNA-ChIP was performed as described previously18. An average of three experiments each performed in triplicate with standard errors is presented.
In vitro pri-miRNA processing assays
The in vitro pri-miRNA processing assay was performed as described previously26.
Statistical analysis
The results presented are the average of at least three experiments each performed in triplicate with standard errors. Statistical analyses were performed by ANOVA, followed by Tukey's multiple comparison test or by Student's t test as appropriate, using Prism 4 (GraphPAD Software Inc.). P values of <0.05 were considered significant and are indicated with asterisks.
Supplementary Material
Acknowledgements
We thank M.-C. Chan and N. Neuman for critical discussion and H. Surks and M. Ivan for critical reading of the manuscript and technical advice. We also thank S. Kato, E. Olson, R. Bassel-Duby, Y.Y. Mo, K. Miyazono, B. Cochran and G.-R. Wang for sharing reagents. This work was supported by grants from the National Institute of Health HD042149 and HL082854 to A.H. and HL086572 to G.L.
METHODS
RT–PCR primers
Human pri-miR-21: 5′-TTTTGTTTTGCTTGGGAGGA-3′ and 5′-AGCAGACAGTCAGGCAGGAT-3′. Human pre-miR-21: 5′-TGTCGGGTAGCTTATCAGAC-3′ and 5′-TGTCAGACAGCCCATCGACT-3′. Human GAPDH: 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. Human SMA: 5′-CCAGCTATGTGTGAAGAAGAGG-3′ and 5′-GTGATCTCCTTCTGCATTCGGT-3′. Human ID1: 5′-CCCATTCTGTTTCAGCCAGT-3′ and 5′-TGTCGTAGAGCAGCACGTTT-3′. Human ID3: 5′-ACTCAGCTTAGCCAGGTGGA-3′ and 5′-AAGCTCCTTTTGTCGTTGGA-3′. Human PDCD4: 5′-TATGATGTGGAGGAGGTGGATGTGA-3′ and 5′-TATGATGTGGAGGAGGTGGATGTGA-3′. Human p68: 5′-TATGATGTGGAGGAGGTGGATGTGA-3′ and 5′-TATGATGTGGAGGAGGTGGATGTGA-3′. Human calponin: 5′-GAGTGTGCAGACGGAACTTCAGCC-3′ and 5′-GTCTGTGCCCAGCTTGGGGTC-3′. Human SM22α: 5′-CGCGAAGTGCAGTCCAAAATCG-3′ and 5′-GGGCTGGTTCTTCTTCAATGGGC-3′.
siRNAs
Synthetic siRNAs targeting human SMAD1, SMAD4 or SMAD5 and p68 were validated Stealth DuoPak (Invitrogen) and Stealth Select RNAi (Invitrogen), respectively. For SMAD4: 5′-CCUGAGUAUUGGUGUUCCAUUGCUU-3’ and 5′-GCAAAGGUGUGCAGUUGGAAUGUAA-3’. For SMAD1: 5′-GCAACCGAGUAACUGUGUCACCAUU-3’ and 5′-GGUCUGCAUCAAUCCCUACCACUAU-3’. For SMAD5:5′-GCCACCUGAUGAUCAGAUGGGUCAA-3’ and 5′-GCUUGGGUUUGUUGUCAAAUGUUAA-3’. For p68: 5′-GGAAUCUUGAUGAGCUGCCUAAAUU-3′, 5′-ACAACUGCCCGAAGCCAGUUCUAAA-3′, and 5′-GGUGCAGCAAGUAGCUGCUGAAUAAA-3′. siRNA for human PDCD4 was described previously11 and synthesized by Dharmacon. As a negative control, Stealth RNAi Negative Control Duplex number 1−3 (Invitrogen) or scrambled siRNA (Dharmacon) was used.
RNA-ChIP primers
Human miR-21: 5′-TTTTGTTTTGCTTGGGAGGA-3′ and 5′-AGCAGACAGTCAGGCAGGAT-3′. Human miR-199a: 5′-GCCAACCCAGTGTTCAGACTA-3′ and 5′-GCCTAACCAATGTGCAGACTA-3′. Human miR-214: 5′-GCCTAACCAATGTGCAGACTA-3′ and 5′-CTATGGTGTGAGGGCTGCTT-3′. Human TM: 5′-GCAAGCACATAGTGGAGCAA-3′ and 5′-TCAAACATCCAGGACAACCA-3′.
Antibodies
Anti-Flag epitope tag (M2, Sigma), anti-p68 (clone PAb204, Upstate), anti-SMA (clone 1A4, Sigma), anti-calponin (clone hCP, Sigma), anti-GAPDH (2E3−2E10, Abnova), anti-SMAD2/SMAD3 (06−654, Upstate), anti-SMAD1/SMAD5/SMAD8 (Calbiochem), anti-SMAD4 (H-552, Santa Cruz), anti-Myc epitope tag (clone 9E10, Tufts Antibody Core Facility), anti-Lamin-A/C (2032, Cell Signalling) and anti-DROSHA (07−717, Upstate) antibodies were used.
In vitro pri-miRNA processing assays
In vitro pri-miRNA processing assay was performed as described previously26. In brief, the 480-nucleotide radiolabelled pri-miR-21 was prepared by standard in vitro transcription with T7 RNA polymerase in the presence of [α-32P]–UTP using human miR-21 gene cloned into pGEM-3 vector as a template. Nuclear extracts were prepared from ∼5 × 106 Cos7 cells treated with vehicle, 400 pM TGF-β or 3 nM BMP4 for 2 h. After dialysis into reaction buffer, nuclear extracts were incubated with pri-miR-21 substrates for 90 min at 37°C. Reaction mixtures were subjected to phenolchroloform extraction, precipitation and 10% (w/v) denaturing gel electrophodresis, followed by autoradiography. The amount of pri-miR-21 (input) and pre-miR-21 was quantified by the phosphoimager (Typhoon9410, GE Healthcare) using ImageQuant 350 software (GE Healthcare).
miRNA and cDNA expression constructs
The pCMV-miR-21 construct and recombinant adenovirus carrying miR-21 or miR-125b (Ad-miR-21 or Ad-miR-125b) were reported previously9,32. In brief, the pCMV-miR-21 construct contains 480-bp human miR-21 genomic fragments cloned into a modified pCMVMyc vector (Clontech). Ad-miR-21 and Ad-miR-125b contain 280-bp rat miR-21 and 366-bp miR-125b genomic fragments into CMV-driven adenoviral vector, respectively. To monitor the amount of pri-miR-21 and pre-miR-21 derived from the pCMV-miR-21 construct in mouse 10T1/2 cells, human-specific RT–PCR primers complementary to sequences in the miR-21 flanking region were used. Unlike pri-miR-21 or pre-miR-21, mature miR-21, which is identical in mouse and human, was detected as the sum of the endogenous and recombinant products. Human PDCD4 and p68 cDNA constructs were purchased from OriGene. In brief, a full-length human PDCD4 cDNA with a 1.9 kb 3′ UTR (NM_01445), which contains miR-21 target sequence, was cloned into pCMV6 vector. The human DROSHA cDNA construct was purchased from Addgene. The Flag–SMAD1(3SA) construct (a gift from Massague laboratory) contains human SMAD1 cDNA with Ser to Ala mutations at amino acids 462, 463 and 465 and was cloned into pCMV5 vector34.
Plasmid DNA and siRNA transfection
Cos7, 10T1/2 cells or PASMCs were transfected with FuGENE6 (Roche Applied Science) for plasmid DNAs and Oligofectamine (Invitrogen) for siRNAs as described before14.
Adenoviral infection
The recombinant adenoviruses were generated and purified by standard procedures. Infection of adenoviruses was performed at 100 multiplicity of infection. There was no detectable toxicity to the cells under these conditions.
qRT–PCR assays
For qRT–PCR assays, total RNA was extracted from cells by Trizol (Invitrogen). cDNA was synthesized from 1 μg of purified RNA by SuperScript II First-Strand cDNA synthesis system (Invitrogen) according to the manufacturer's instructions. qRT–PCR was performed with a real-time PCR machine (iQ5, BioRad). The results of qRT–PCR assays presented are an average of three independent RNA preparations. Each sample was analysed in triplicate. PCR cycling parameters were: 94 °C for 3 min, and 40 cycles of 94 °C for 15 s, 60 °C for 20 s, 72 °C for 40 s. For detection of mature miRNAs, TaqMan MicroRNA assay kit (Applied Biosystems) was used according to manufacturer's protocol. Data analysis was done by using the comparative CT method in software by BioRad.
Luciferase assay
After transfection of the reporter construct together with LacZ plasmid as an internal control, the cells were reseeded onto 12-well plates and treated with 3 nM BMP3 or 400 pM TGF-β1 for 16−20 h in DMEM/0.2% FCS. Luciferase assays were carried out using Promega's Luciferase assay system. Luciferase activity was normalized with LacZ activity.
Anti-miRNAs
2′-O-methyl modified RNA oligonucleotides complementary to miRNA (anti-miR) or GFP (control) sequence were purchased from IDT. Anti-miRs were transfected to cells at a concentration of 106 nM using Oligofectamine (Invitrogen) according to the manufacturer's directions. Anti-miR-21: 5′-GUCAACAUCAGUCUGAUAAGCUA-3′. Anti-miR-199a: 5′-GAACAGGUAGUCUGAACACUGGG-3′. Anti-miR-125b: 5′-UCACAAGUUAGGGUCUCAGGGA-3′. Anti-miR-221: 5′-GAAACCCAGCAGACAAUGUAGCU-3′. Anti-miR-15b: 5′-UGUAAACCAUGAUGUGCUGCUA-3′. Anti-miR-100: 5′-CACAAGUUCGGAUCUACGGGUU-3′. Anti-GFP: 5′-AAGGCAAGCUGACCCUGAAGU-3′.
miRNA cloning
miRNA cloning from PASMCs was performed following the protocol from the Bartel laboratory (http://web.wi.mit.edu/bartel/pub/protocols_reagents.htm). In brief, miRNAs were prepared from PASMCs treated with 3 nM BMP4 for 24 h using Trizol (Invitrogen). After linker ligation and PCR amplification, miRNA sequences were concatemerized, cloned into Topo-TA vector, and sequenced by the DNA sequencing facility at Tufts University.
RNA-ChIP
RNA-ChIP was performed as described previously18. In brief, PASMCs or Cos7 cells were crosslinked for 15 min with 1% formaldehyde, the cell pellet was resuspended in Buffer A (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% Nonidet P-40). After 10 min on ice, the crude nuclei fraction was isolated by centrifugation, and then suspended in Buffer B (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1). After nuclei were disrupted by sonication, the lysates were cleared and subjected to immunoprecipitation with anti-Flag, anti-SMAD1/5/8, SMAD2/3 or anti-p68 antibody, followed by stringent washing, and elution. The RNA was isolated using Trizol (Invitrogen). Pellets were resuspended in TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA) and incubated with DNase I for 30 min at 37 °C to remove any remaining DNA. After extraction with phenol:chloroform (5:1), RNA was precipitated with ethanol and dissolved in 20 μl of water. 5 μl of RNA was used for a 20 μl cDNA synthesis reaction. Quantitative PCR reactions were then performed by real-time PCR machine (iQ5, BioRad).
GST pull-down assay
GST–SMAD fusion proteins were expressed in bacteria, followed by partial purification with GST–sepharose beads. Equal amounts of GST–SMAD fusion proteins conjugated to sepharose beads were added to nuclear extracts or total RNA. After washing the beads, proteins pulled-down with the beads were separated by SDS–PAGE, followed by immunoblotting or RT–PCR analysis. For RNase A treatment, 250 μg ml−1 RNase A (New England Biolab) were added to nuclear extracts 30 min before addition of GST–SMAD fusion proteins and throughout the pull-down assay.
Immunoprecipitation/immunoblot assay
Cells were lysed in TNE buffer (1% Nonidet P-40, 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 150 mM NaCl). Total cell lysates or proteins immunoprecipitated with antibodies were separated by SDS–PAGE, transferred to PVDF membranes (Millipore), immunoblotted with antibodies, and visualized using an enhanced chemiluminescence detection system (Amersham Biosciences).
Immunofluorescence staining
PASMCs or 10T1/2 cells were fixed and permeabilized in a 50% acetone-50% methanol solution and subjected to staining using anti-SMA or anti-calponin antibody conjugated with fluorescein isothiocyanate (FITC) and nuclear staining with DAPI (Invitrogen).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
References
- 1.ten Dijke P, Arthur HM. Extracellular control of TGFβ signalling in vascular development and disease. Nature Rev. Mol. Cell Biol. 2007;8:857–868. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc. Am. Thorac. Soc. 2006;3:680–686. doi: 10.1513/pats.200605-118SF. [DOI] [PubMed] [Google Scholar]
- 3.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 4.Rensen SSM, Doevendans PAFM, van Eys GJJM. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Netherlands Heart J. 2007;15:100–108. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagna G, et al. Control of phenotypic plasticity of smooth muscle cells by BMP signaling through the myocardin-related transcription factors. J. Biol. Chem. 2007;282:37244–37255. doi: 10.1074/jbc.M708137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 7.Ji R, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ. Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 8.Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 9.van Rooij E, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl Acad. Sci. USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asangani IA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 11.Frankel LB, et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2007;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 12.Zhu S, et al. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
- 13.Hollnagel A, Oehlmann V, Heymer J, Ruther U, Nordheim A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J. Biol. Chem. 1999;274:19838–19845. doi: 10.1074/jbc.274.28.19838. [DOI] [PubMed] [Google Scholar]
- 14.Chan MC, et al. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol. Cell. Biol. 2007;27:5776–5789. doi: 10.1128/MCB.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee Y, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 16.Han J, et al. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. 2004;14:2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda T, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nature Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 19.Kim VN, Nam JW. Genomics of microRNA. Trends Genet. 2006;22:165–173. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nature Rev. Mol. Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem. Sci. 2007;32:189–197. doi: 10.1016/j.tibs.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Lee EJ, et al. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA. 2007;14:35–42. doi: 10.1261/rna.804508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson JM, et al. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wulczyn FG, et al. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 26.Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nature Struct. Mol. Biol. 2007;14:591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 27.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 28.Schmierer B, Hill CS. TGFβ–SMAD signal transduction: molecular specificity and functional flexibility. Nature Rev. Mol. Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 29.Schmittgen TD, et al. Real-time PCR quantification of precursor and mature microRNA. Methods. 2008;44:31–38. doi: 10.1016/j.ymeth.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 31.Loffler D, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 32.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J. Biol. Chem. 2007;282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 33.Warner DR, et al. Functional interaction between Smad, CREB binding protein, and p68 RNA helicase. Biochem. Biophys. Res. Commun. 2004;324:70–76. doi: 10.1016/j.bbrc.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 34.Kretzschmar M, Liu F, Hata A, Doody J, Massagué J. The TGF-β mediator Smad1 is directly phosphorylated and functionally activated by the BMP receptor kinase. Genes Dev. 1997;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- 35.Gomis RR, et al. A FoxO–Smad synexpression group in human keratinocytes. Proc. Natl Acad. Sci. USA. 2006;103:12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor β (TGF-β) target genes and distinguishes TGF-β-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giehl K, Imamichi Y, Menke A. Smad4-independent TGF-β signaling in tumor cell migration. Cells Tissues Organs. 2007;185:123–130. doi: 10.1159/000101313. [DOI] [PubMed] [Google Scholar]
- 38.Ijichi H, et al. Smad4-independent regulation of p21/WAF1 by transforming growth factor-β. Oncogene. 2004;23:1043–1051. doi: 10.1038/sj.onc.1207222. [DOI] [PubMed] [Google Scholar]
- 39.Si ML, et al. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 40.Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–6104. doi: 10.1158/0008-5472.CAN-06-0537. [DOI] [PubMed] [Google Scholar]
- 41.Volinia S, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl Acad. Sci. USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iorio MV, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 43.Wiemer EA. The role of microRNAs in cancer: no small matter. Eur. J. Cancer. 2007;43:1529–1544. doi: 10.1016/j.ejca.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nature Rev. Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 45.Arteaga CL. Inhibition of TGFβ signaling in cancer therapy. Curr. Opin. Genet. Dev. 2006;16:30–37. doi: 10.1016/j.gde.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Bachman KE, Park BH. Duel nature of TGF-β signaling: tumor suppressor vs. tumor promoter. Curr. Opin. Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 47.Glick AB. TGFβ1, back to the future: revisiting its role as a transforming growth factor. Cancer Biol. Ther. 2004;3:276–283. doi: 10.4161/cbt.3.3.849. [DOI] [PubMed] [Google Scholar]
- 48.Massague J, Gomis RR. The logic of TGFβ signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 49.Fujii M, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell. 1999;10:3801–3813. doi: 10.1091/mbc.10.11.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Y, et al. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA-binding in TGFβ signaling. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.