

Abstract
N-CoR and SMRT are corepressor paralogs that partner with and mediate transcriptional repression by a wide variety of metazoan transcription factors, including nuclear hormone receptors. Although encoded by distinct genetic loci, N-CoR and SMRT share substantial sequence interrelatedness, form analogous assemblies with histone deacetylases and auxiliary factors, can interact with overlapping sets of transcription factor partners, and exert overlapping functions in cells. SMRT is subject to negative regulation by MAPK signaling pathways operating downstream of growth factor and stress signaling pathways. We report here that whereas activation of MEKK1 leads to phosphorylation of SMRT, its dissociation from its transcription factor partners in vivo and in vitro, and its redistribution from the cell nucleus to a cytoplasmic compartment, N-CoR is refractory to all these forms of regulation. In contrast to this MAPK cascade, other signal transduction pathways operating downstream of growth factor/cytokine receptors appear able to affect both corepressor paralogs. Our results indicate that SMRT and N-CoR are embedded in distinct regulatory networks and that the two corepressors interpret growth factor, cytokine, differentiation, and prosurvival signals differently.
Many transcription factors display bimodal regulatory properties and can confer both repression and activation on their target genes. This functional dualism reflects the ability of these transcription factors to recruit two alternative classes of auxiliary proteins, denoted corepressors and coactivators, that determine the polarity of the transcriptional response (1-9). Nuclear receptors, for example, are a family of ligand-regulated transcription factors that regulate key aspects of metazoan development, differentiation, and homeostasis (10-13). In the absence of hormone ligand, nuclear receptors can recruit a corepressor complex containing the SMRT protein, leading to repression of target gene expression (14-19). Conversely, binding of hormone agonist causes the release of the SMRT corepressor complex and the recruitment of coactivator complexes that enhance target gene expression (20, 21). Analogous corepressor and coactivator complexes partner with a broad assortment of other transcriptional regulators, including NF-κB, serum response factor, AP-1 proteins, Smad proteins, CCAAT binding factor, c-Myb, PLZF, Bcl-6, Pbx/Hox proteins, ETO-1 and ETO-2, aryl hydrocarbon receptor, and MyoD, among others (reviewed in Ref. 6).
Corepressors and coactivators modulate gene expression by modifying the chromatin template and by making inhibitory or stimulatory contacts with the general transcriptional machinery (22-34). Many coactivators possess histone acetyltransferase activity, whereas the SMRT corepressor recruits histone deacetylases, such as HDAC3 (1, 3-6, 9). Acetylation and deacetylation of nucleosomal histones by these coactivator and corepressor complexes, operating together with other covalent histone modifications, create a code that influences the interaction of the chromatin with additional factors and its accessibility to the general transcriptional machinery (22-26, 28-31, 33, 34). Besides histone deacetylases, the SMRT complex contains additional protein components, such as TBL1/TBLR1 and GPS2, that help stabilize its overall structure and that may contribute to the release of the corepressor complex in response to hormone agonist (35-39); other polypeptides, such as mSin3 and an assortment of additional histone deacetylases, can also interact with SMRT, but the association of these latter polypeptides with the SMRT complex in vivo and their contribution to SMRT-mediated repression remain incompletely elucidated (reviewed in Ref. 6). SMRT therefore acts as a molecular platform on which the remainder of the corepressor complex assembles and serves as the principal contact between the corepressor complex and its transcription factor partners. Regulatory events that cause a dissociation of SMRT also cause the release of the remainder of the corepressor complex and a loss of repression (20, 21).
Notably, a second corepressor protein, denoted N-CoR, is widely distributed in vertebrates and performs similar or identical functions compared with SMRT (40, 41). Although encoded by a distinct genetic locus, N-CoR shares the same overall molecular architecture and significant amino acid identity with SMRT (see Fig. 1A); interacts with many of the same transcription factors partners (although, in some cases, with different affinities); and assembles into similar or identical complexes with TBL1, TBLR1, and GPS2 and with other known or suspected corepressor components (reviewed in Ref. 6). Despite these many parallels between SMRT and N-CoR, these corepressor paralogs were established and subsequently maintained as distinct gene products from the beginning of the vertebrate evolutionary radiation and perform distinct functions in cells (reviewed in Ref. 6). What differences do N-CoR and SMRT therefore manifest at the molecular level to account for their distinct biological and evolutionary properties?
Fig. 1. Interaction of SMRT, but not of N-CoR, with nuclear receptors is inhibited by MEKK1.
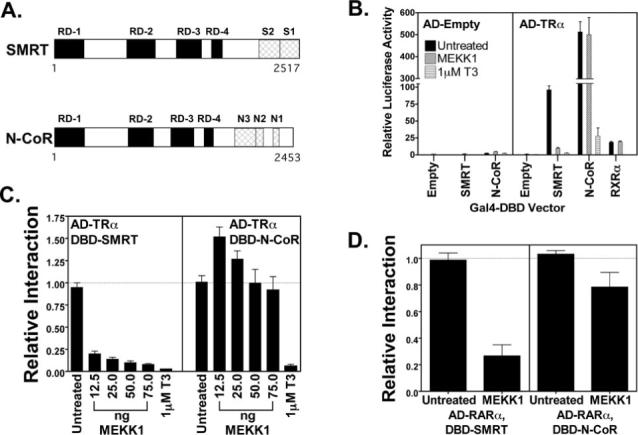
A, schematic of SMRT and N-CoR. The primary sequences of SMRT and N-CoR are represented from the N to the C terminus. The positions of domains involved in repression (RD-1 to RD-4) or involved in interaction with nuclear receptors (S1 and S2 in SMRT and N1, N2, and N3 in N-CoR) are depicted. B, both SMRT and N-CoR interact with T3Rα (TRα) in a mammalian two-hybrid assay. Either an empty Gal4AD construct or a Gal4AD-T3Rα construct was tested for the ability to interact with the Gal4DBD constructs noted below the panel. A positive interaction resulted in elevated expression of a Gal4−17-mer luciferase reporter. Relative luciferase activity is presented, representing the absolute luciferase normalized to the expression of a β-galactosidase reporter used as an internal transfection control. The effect of T3 on the interaction of Gal4AD-T3Rα with Gal4DBD-SMRT or Gal4DBD-N-CoR and the effect of a constitutively active MEKK1 allele on the interaction of Gal4-T3Rα with Gal4DBD-SMRT, Gal4DBD-N-CoR, or Gal4DBD-retinoid X receptor-α (RXRα) are also indicated. The means ± S.D. of three or more experiments are shown. C, MEKK1 inhibits the interaction of SMRT, but not of N-CoR, with T3Rα. The effect of co-introducing increasing amounts of a constitutively active MEKK1 allele on the mammalian two-hybrid interactions described for B was tested. The means ± S.D. of three or more experiments are shown. The ability of T3 to disrupt the corepressor/receptor interaction was also tested as a control. D, MEKK1 strongly inhibits the interaction of SMRT, but only weakly interferes with the interaction of N-CoR with RARα. The same type of assay as described for C was performed using Gal4AD-RARα in place of Gal4AD-T3Rα. The means ± S.D. of three or more experiments are shown.
We have shown that growth factor receptors are important regulators of SMRT function and operate through a MAPK1 cascade (42, 43). Activation of the epidermal growth factor (EGF) receptor or its downstream mediator, MEKK1, leads to inhibition of the ability of SMRT to interact with its transcription factor partners and a redistribution of SMRT from the nucleus to the cytoplasm (42, 43). These effects of MEKK1 on SMRT represent an important nexus between growth factor signaling and nuclear receptor function and contribute to the differentiation-promoting effects of arsenic trioxide treatment in acute promyelocytic leukemia (44). We report here that direct phosphorylation of SMRT by MEKK1 is sufficient to inhibit the SMRT/thyroid hormone receptor (T3R) interaction in vitro and that the relocalization of SMRT to the cytoplasm in cells expressing MEKK1 occurs unaccompanied by the T3R partner (which is retained in the nucleus). More important, we also report that N-CoR is unexpectedly resistant to these inhibitory effects of MEKK1 under conditions in which SMRT function is strongly suppressed. Unlike SMRT, N-CoR is refractory to MEKK1 phosphorylation, does not release from nuclear receptor partners in vitro or in vivo, and does not detectably change in its subcellular distribution in response to MEKK1 signaling. Taken together with the observations by other investigators, these results indicate that the SMRT and N-CoR corepressor paralogs are subject to distinct forms of regulation. We suggest that these divergent forms of control help account for the establishment and retention of these two distinct forms of corepressor during vertebrate evolution.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
The construction of the mammalian expression plasmids pSG5-Gal4AD, pSG5-Gal4AD-T3Rα, pSG5-Gal4DBD, pSG5-Gal4DBD-SMRTτ-(1773−2471), pSG5-Gal4DBD-N-CoR-(1946−2435), pSG5-Gal4AD-RARα, and pSG5-Gal4DBD-RARα was described previously (32, 43, 45, 46). The pSG5-Myc vector was created by inserting a synthetic oligonucleotide (MWG Biotech, High Point, NC) encoding a Myc epitope tag into an expanded multiple cloning site in pSG5. The pSG5-Myc-T3Rα, pSG5-Myc-SMRTτ-(1−2423), and pSG5-Myc-N-CoR-(1−2453) vectors were created using PCR to introduce approximate restriction sites on the ends of the corresponding open reading frames and by ligating the DNA products into the pSG5-Myc vector. The pCMV-GFP-SMRTτ-(1−2423) and pCMV-GFP-N-CoR-(1−2453) expression vectors were created by inserting PCR-generated DNAs containing the corresponding open reading frames into the pCMV-GFP vector (43). PCR-generated DNAs encoding the S1 domain of SMRTα (amino acids 2313−2517) or the S1 and S2 domains of SMRTα (amino acids 2077−2517) were cloned into pGEX-KG (47) to yield the pGEX-SMRTα-S1-(2313−2517) and pGEX-SMRTα-S1/S2-(2077−2517) constructs. The pGEX-N-CoR-N1-(2211−2453) and the pGEX-N-CoR-N1/N2/N3-(1817−2453) vectors were created by inserting the HindIII-SalI or ApaI-SalI fragment of N-CoR into a pGEX-KG vector bearing an expanded multiple cloning site. All clones were confirmed by DNA sequence analysis.
The origins of pCMV5-FLAG-ΔMEKK1-(817−1493), pCMV-HA-MEK1(R4F), pMT3-ERK1, pSG5-v-ErbB, pLNC-v-Raf1, and pCMV-v-Ras plasmids were described previously (42, 43). A constitutively active clone of Akt, pCMV-Akt1(S473D), was the generous gift of Marty Mayo (University of Virginia). Baculovirus constructs for T3Rα and His6-ΔMEKK1 were described previously (43, 48).
Cell Culture
CV-1 cells were propagated in Dulbecco's modified Eagle's medium containing high glucose, l-glutamine, and pyridoxine hydrochloride (Invitrogen) and supplemented with 10% heat-inactivated fetal bovine serum (Hyclone Laboratories, Logan, UT). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. For expression of His6-ΔMEKK1 in the baculovirus expression system, Sf9 cells were maintained and infected in Ex-cell 420 medium (JRH Biosciences, Lenexa, KS) supplemented with 10% heat-inactivated fetal bovine serum; cells were incubated at 28 °C in a humidified atmosphere.
Mammalian Two-hybrid Analysis
CV-1 cells (3.0 × 104 cells/well in a 24-well plate) were transiently transfected, 24 h after plating, using Effectene transfection reagent (QIAGEN Inc.) following the manufacturer's recommended protocol. Transfection mixtures included 50 ng of the appropriate pSG5-Gal4AD vector, 12.5 ng of the appropriate pSG5-Gal4DBD vector, 50 ng of the pADH-Gal4−17-mer luciferase reporter, either 50 ng of pCH110 or 10 ng of pCMV-LacZ as an internal transfection control, appropriate expression vectors for the indicated signal transducers, and/or an empty vector, as appropriate. Twenty-four hours after transfection, the medium was replaced with fresh medium with or without 1 μm triiodothyronine (T3), 1 ng/ml interleukin-1β (IL-1β), 10 ng/ml anisomycin, and/or 1 μm U0126, as indicated. Cells were collected 48 h after transfection and lysed in lysis buffer (100 μl/well) containing 0.2% Triton X-100, 91 mm K2HPO4, and 9.2 mm KH2PO4. Luciferase and β-galactosidase activities were determined as described previously (43, 45).
Co-immunoprecipitation Assays
CV-1 cells (1.5 × 105 cells/well in a 6-well plate) were transfected with various combinations of Myc-T3Rα, Myc-SMRTτ, Myc-N-CoR, a constitutively active MEKK1 construct, or appropriate amounts of equivalent empty vectors using the Effectene protocol described above. Cells were collected 48 h after transfection and lysed by a 30-min incubation at 4 °C in 300 μl of immunoprecipitation buffer consisting of phosphate-buffered saline (PBS; 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, and 1.5 mm KH2PO4) plus 1 mm EDTA, 1.5 mg/ml iodoacetamide, 100 μm Na3VO4, 0.5% Triton X-100, 20 mm β-glycerophosphate, 1 mm NaF, 0.2 mm phenylmethylsulfonyl fluoride, 1× Complete phosphatase inhibitor mixture I (EMD Biosciences, Inc., La Jolla, CA), and 1× Complete protease inhibitor mixture (Roche Applied Science, Mannheim, Germany). The cell lysates were cleared by centrifugation at 14,000 rpm at 4 °C. A 15-μl aliquot of each cell lysate was saved, and the remaining lysate was incubated at 4 °C for 1 h with rabbit anti-v-ErbA polyclonal antiserum (diluted 1:100) (49). Next, 40 μl of protein G-Sepharose beads (50% slurry) were added, and the samples were incubated overnight at 4 °C on a rotator. The Sepharose beads and any proteins bound to them were collected by centrifugation at 3000 rpm in a microcentrifuge at 4 °C for 2 min. The beads were washed four times with 300 μl of immunoprecipitation buffer, and any proteins remaining bound to the beads were then eluted by boiling in SDS sample buffer; resolved by SDS-PAGE using a NuPAGE Novex Tris acetate 3−8% gradient gel system (Invitrogen); and visualized by immunoblotting using mouse anti-Myc monoclonal antibody (diluted 1:200; Gamma One Laboratories, Lexington, KY), horseradish peroxidase-conjugated goat anti-mouse IgG antibody (diluted 1:1500; Bio-Rad), and the ECL Plus Western blot detection system (Amersham Biosciences). The resulting chemiluminescent signal was detected and quantified using a Fluorchem 8900 digital detection system (Alpha Innotech, San Leandro, CA).
In Vitro Kinase Assays
GST-SMRT and GST-N-CoR fusion proteins were expressed in Escherichia coli BL21 cells and purified by binding to glutathione-agarose beads as described previously (32). Purified GST-corepressor proteins were eluted in buffer containing 20 mm glutathione, 5% glycerol, 10 mg/ml bovine serum albumin (BSA), and 1× Complete protease inhibitor mixture in 100 mm Tris-Cl (pH 8.0). The His6-tagged ΔMEKK1 proteins were expressed by baculoviral infection of Sf9 cells. Approximately 7 × 106 Sf9 cells were infected with recombinant baculovirus encoding His6-ΔMEKK1. Infected cells were harvested 72 h after infection, washed with PBS, resuspended in 3 ml of sonication buffer (20 mm Tris-Cl (pH 8.0), 100 mm NaCl, 0.5 mm β-mercaptoethanol, and 1 μg/ml leupeptin (Sigma)), and lysed by sonication. Triton X-100 was then added to a final concentration of 0.1%; the samples were vortexed briefly; and the lysate was cleared by centrifugation at 14,000 rpm at 4 °C. The His6-ΔMEKK1 protein was then purified by adding 200 μl of prewashed Talon Superflow metal affinity resin (Clontech), mixing the samples for 20 min at room temperature, and collecting the resin beads by centrifugation at 3000 rpm for 2 min at 4 °C. The resin was washed four times with sonication buffer, and the protein was eluted in 200 μl of buffer containing 20 mm Tris-Cl (pH 8.0), 100 mm NaCl, 400 mm imidazole, and 1× Complete protease inhibitor mixture. The eluate was dialyzed overnight in 50 mm Tris-Cl (pH 7.5), 50 mm NaCl, 0.1% β-mercaptoethanol, and 5% glycerol. 1× Complete protease inhibitor mixture was added, and the samples were flash-frozen in liquid nitrogen and stored as aliquots at −80 °C.
For phosphorylation in vitro, 10 μl of the GST-SMRT or GST-N-CoR protein were incubated overnight at 30 °C with 2 μl of His6-ΔMEKK1 and 1 mm ATP (Sigma) in MEKK1 assay dilution buffer (20 mm MOPS (pH 7.2), 25 mm β-glycerophosphate, 5 mm EGTA, 1 mm Na3VO4, 16.7 mm MgCl2, 1 mm sodium fluoride, 1 mm dithiothreitol, and 1× Complete phosphatase inhibitor mixture I) in a total reaction volume of 20 μl. The reactions were collected the following day for use in the appropriate electrophoretic mobility shift assays.
Electrophoretic Mobility Shift Assays
An annealed oligonucleotide probe representing a direct repeat of AGGTCA with a 4-base spacer (termed DR-4) was radiolabeled with 32P by fill-in synthesis with Klenow DNA polymerase. T3Rα was isolated from recombinant baculovirus-infected Sf9 cells (50). GST-SMRT-S1, GST-SMRT-S1/S2, GST-NCoR-N1, and GST-N-CoR-N1/N2/N3 protein constructs were isolated from E. coli and incubated with or without recombinant ΔMEKK1 as described above. Electrophoretic mobility shift assays (EMSAs) were initiated by mixing the T3Rα preparation with the radiolabeled DNA probe (50,000 cpm) in binding buffer containing 10 mm Tris-Cl (pH 7.5), 2mm MgCl2, 50 mm KCl, 2.5 mg/ml BSA, 20 μg/ml poly(dI-dC), and 1 mm dithiothreitol in a total volume of 14.5 μl. For supershift experiments, the above reactions were subsequently incubated for 15 min on ice with 5 μl of the indicated dilution of the GST-corepressor protein (either treated with ΔMEKK1 or not). The resulting DNA·protein complexes were resolved using a 5% polyacrylamide (29:1 acrylamide/bisacrylamide) gel and 0.5× 44 mm Tris base, 44 mm boric acid, and 1 mm EDTA electrophoresis system. The gels were dried, and radioactivity was visualized and quantified by PhosphorImager analysis.
Fluorescence Microscopy
CV-1 cells (1.0 × 105 cells/well in a 6-well plate) were allowed to attach to 22 × 22-mm coverslips and transfected using the Effectene protocol described above. Cells were fixed 48 h after transfection in a chilled (−20 °C) mixture of 50% acetone and 50% methanol for 10 m at 4 °C. After aspiration of the fixing agent, cells were washed three times with PBS and incubated for1hat room temperature in PBS containing 2% BSA. The primary mouse anti-Myc monoclonal antibody (diluted 1:500) or a pre-absorbed control mixed with Myc-neutralizing peptide (Affinity Bioreagents, Golden, CO) was added to the coverslips in PBS containing 2% BSA and incubated for 60 min at room temperature. The coverslips were then washed three times with PBS containing 2% BSA and incubated for1hat room temperature with Texas Red-conjugated horse anti-mouse IgG antibody (diluted 1:1000; Vector Laboratories, Burlingame, CA) in PBS containing 2% BSA. The coverslips were washed three times with PBS containing 2% BSA and three times with PBS alone and incubated for5mat room temperature in PBS containing 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were again washed three times with PBS and once with distilled water, and the excess moisture was removed by aspiration. The coverslips were mounted on slides using 25 μl of Vectashield (Vector Laboratories) and sealed with fingernail polish. The slides were visualized using a Nikon Microphot epifluorescence microscope. Digital images were captured with a Nikon Cool Pix 4500 digital camera. For quantification of the fluorescence microscopic data, 100 transfected cells were counted at random from each slide and scored for the following GFP-SMRT or GFP-N-CoR subcellular localization: nuclear, cytoplasmic, nuclear equal to cytoplasmic, or undeterminable.
Phosphorylation/Dephosphorylation Assays
CV-1 cells (1.5 × 105 cells/well in a 6-well plate) were transfected with the appropriate mammalian expression vectors using the Effectene protocol described above. Cells were collected 48 h after transfection by mechanical scraping and lysed by a 30-min incubation at 4 °C in 250 μl of cell extraction buffer containing 25 mm HEPES (pH 7.8), 300 mm NaCl, 1.5 mm MgCl2, 1% Triton X-100, 0.1 mm dithiothreitol, 0.2 mm phenylmethylsulfonyl fluoride, and 1× Complete protease inhibitor mixture. Lysates were clarified by centrifugation at 14,000 rpm for 30 min at 4 °C, and the lysates were divided into two equal aliquots. One aliquot was treated for 30 min at 37 °C with 10 units of shrimp alkaline phosphatase (Promega Corp., Madison, WI), and one aliquot was mock-treated. The samples were then resolved by SDS-PAGE using the NuPAGE Novex Tris acetate 3−8% gradient gel system. The electrophoretograms were visualized by immunoblotting using rabbit polyclonal antibody directed against either SMRT (diluted 1:2000; Affinity Bioreagents) or N-CoR (diluted 1:500; Upstate Biotechnology, Inc., Lake Placid, NY), horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (diluted 1:3000, Bio-Rad), and the ECL Plus Western blot detection system. The chemiluminescent signals were captured and quantified using the Fluorchem 8900 digital detection system.
RESULTS
MEKK1 Signaling Disrupts the Interaction of Nuclear Receptors with SMRT, but Not with N-CoR
We have reported that SMRT is negatively regulated by growth factor signals operating through a MEKK1 cascade (43), whereas other investigators have reported that N-CoR function can be inhibited by cytokines, such as ciliary neurotrophic factor, through an Akt-mediated phosphorylation pathway (51). To better understand these phenomena, we compared the actions of MEKK1 on N-CoR and SMRT. As reported previously (43), SMRT and a transcription factor partner, T3Rα, exhibited a strong interaction in a mammalian two-hybrid assay, whereas no two-hybrid signal was observed in negative control studies using either an empty Gal4 DNA-binding domain (Gal4DBD) construct or an empty Gal4 activation domain (Gal4AD) construct in place of the corresponding T3R-corepressor fusions (Fig. 1B). A strong two-hybrid interaction was also observed between N-CoR and T3Rα, and both the SMRT/T3Rα and N-CoR/T3Rα interactions were disrupted by T3 (Fig. 1B). The two-hybrid interaction between SMRT and T3Rα was disrupted by introduction of an activated MEKK1 allele (ΔMEKK1, representing codons 817−1493) in a dose-dependent manner over a wide range of MEKK1 expression vector concentrations (Fig. 1C, left panel). In contrast, the two-hybrid interaction between N-CoR and T3Rα was not inhibited by the introduction of MEKK1, but was actually slightly enhanced at low-to-intermediate MEKK1 transfection levels (12.5−25 ng) (Fig. 1C, right panel). Higher levels of MEKK1 vector (50−75 ng), although not enhancing, nonetheless did not inhibit the N-CoR/T3Rα interaction, with still higher levels of MEKK1 vector producing cytotoxic effects (Fig. 1C, right panel) (data not shown). Extension of the two-hybrid assay to retinoic acid receptor-α (RARα) demonstrated that MEKK1 similarly strongly interfered with the interaction of SMRT and RARα, but had very little effect on the interaction of N-CoR and RARα (Fig. 1D; also see below).
A control two-hybrid interaction between T3Rα and its heterodimeric partner retinoid X receptor-α was not affected by introduction of the MEKK1 construct, nor was the basal level of the Gal4−17-mer reporter activity significantly altered by MEKK1 coexpression (Fig. 1B). The ability of MEKK1 to inhibit the interaction between SMRT and T3R, but not between N-CoR and T3R, was observed over a range of Gal4DBD-corepressor and Gal4AD-receptor inputs. Immunoblotting confirmed that MEKK1 had little or no effect on the abundance of the Gal4DBD-corepressor and Gal4AD-T3Rα protein chimeras; however, a small decrease in the levels of Gal4AD-RARα was noted in response to MEKK1 signaling, which likely accounts for the slight inhibition of the Gal4DBD-N-CoR/Gal4AD-RARα two-hybrid assay in Fig. 1D. Taken together, these results indicate that it is the interaction between SMRT and its nuclear receptor partners that is inhibited by MEKK1 signaling, rather than MEKK1 exerting an artifactual effect on the two-hybrid assay itself. In contrast, the interaction of N-CoR and nuclear receptors appears largely refractory to the inhibitory effects of MEKK1.
MEKK1 Inhibits the Association of T3Rα with SMRT, but Not with N-CoR, in Co-immunoprecipitation and Electrophoretic Mobility Shift Assays
We next employed a co-immunoprecipitation protocol to examine the effects of MEKK1 on the physical interaction between full-length corepressors and nuclear receptors. We introduced Myc-tagged SMRT or Myc-tagged N-CoR together with Myc-T3Rα into CV-1 cells, immunoprecipitated T3R with T3R-specific antiserum, and determined the amount of co-associated corepressor by an immunoblotting procedure. Both SMRT and N-CoR could be co-immunoprecipitated with T3Rα in this fashion in the absence of hormone, whereas the association with T3Rα was significantly reduced by the addition of T3 agonist (Fig. 2A) (data not shown). Neither corepressor was detected in the immunoprecipitate in the absence of T3Rα, confirming that the coprecipitation reflects a physical interaction between the nuclear receptor and either SMRT or N-CoR (Fig. 2A, compare the Myc-tagged corepressor coprecipitating with T3R in lanes 5 and 7 with that precipitating in the absence of receptor in lanes 3 and 4). Introduction of an activated MEKK1 allele into the transfected cells significantly reduced the co-immunoprecipitation of SMRT with T3Rα with little or no change in the total amount of SMRT (Fig. 2A, upper two panels, compare lanes 5 and 6). Notably, although the introduction of an activated MEKK1 allele reduced the overall abundance of N-CoR in these cells (down-regulation of N-CoR levels by a proteasome-mediated pathway has been described previously (52)), there was no additional effect of MEKK1 on the relative amount of N-CoR coprecipitating with T3Rα (Fig. 2A, upper two panels, compare lanes 7 and 8). We quantified our results by calculating the percentage of total SMRT or N-CoR co-immunoprecipitating with T3Rα minus or plus MEKK1 (Fig. 2B). These results for the full-length corepressors are consistent with those from the mammalian two-hybrid assays and confirm that MEKK1 inhibits the physical interaction of SMRT with T3Rα, but has little effect on the interaction of N-CoR with T3Rα. It should also be noted that expression of the ectopically introduced tagged N-CoR and SMRT in these experiments was comparable with or only modestly higher than that of the corresponding endogenous corepressors (data not shown).
Fig. 2. MEKK1 inhibits the co-immunoprecipitation of SMRT with T3Rα, but not that of N-CoR.
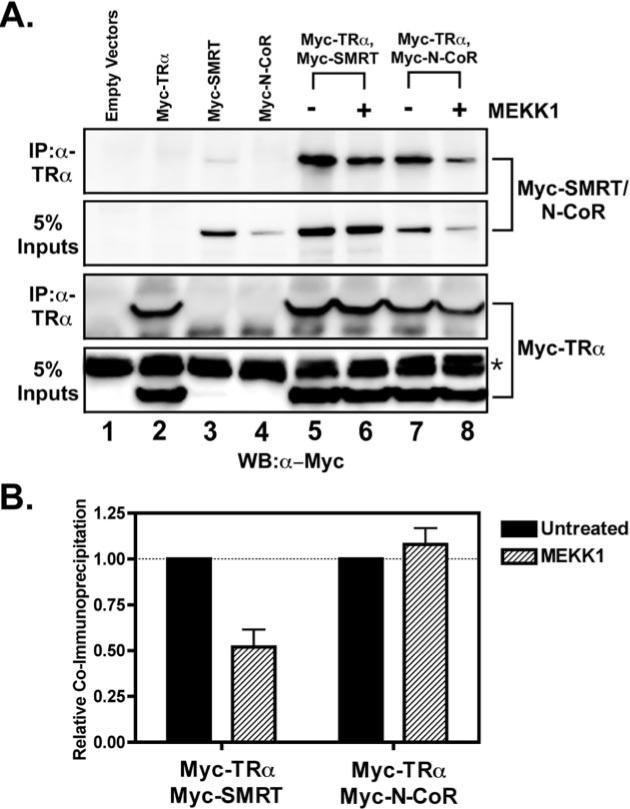
CV-1 cells were transfected with Myc epitope-tagged, full-length corepressor (either SMRT or N-CoR, as indicated), with Myc-tagged T3Rα (TRα), or with both, as indicated above the panels. Where indicated, the cells were also cotransfected with a constitutively active allele of MEKK1. Forty-eight hours after transfection, the cells were harvested and lysed. One aliquot of each lysate was analyzed directly by SDS-PAGE and immunoblotting with anti-Myc antiserum to visualize total expression levels of corepressor and T3Rα (5% Inputs). A second aliquot of each sample was first immunoprecipitated (IP) with anti-T3R antibody prior to analysis of the immunoprecipitate by SDS-PAGE and immunoblotting using the anti-Myc antiserum. The immunoblots were visualized using a chemiluminescence protocol. A, an Alpha Innotech Fluorchem scan of the resulting electrophoretogram is shown. The asterisk indicates a nonspecific immunoreactive band seen in all lysates. WB, Western blot. B, a quantification of the amount of each corepressor in the anti-T3Rα immunoprecipitate relative to the total amount of that corepressor in the cell (with or without MEKK1) is presented.
To determine whether the inhibition of the SMRT/nuclear receptor interaction was a result of the direct phosphorylation of this corepressor by MEKK1, we employed an EMSA in vitro. T3Rα bound to a radiolabeled DR-4 DNA probe in vitro as a protein dimer, forming a receptor·DNA complex that migrates at a slower mobility than that of the free DNA probe (Fig. 3A, lane 1) (53-56); no complex was observed with non-recombinant baculovirus/Sf9 preparations, nor did the T3Rα preparation bind an irrelevant DNA probe (data not shown). Addition to the EMSA of a SMRT construct representing the S1 receptor interaction domain resulted in a further retardation (a super-shift) of the T3R·DNA complex, indicative of an interaction between the corepressor and the receptor (Fig. 3A, lanes 11−19; quantified in Fig. 3C). Incubation of the SMRT S1 domain construct with MEKK1 and ATP significantly inhibited its ability to supershift the T3R·DNA complex (Fig. 3A, lanes 2−10; quantified in Fig. 3C), indicating that phosphorylation of SMRT by MEKK1 reduces the avidity of the corepressor for its nuclear receptor partner (p < 0.002); omitting the ATP prevented phosphorylation and prevented inhibition by MEKK1 (data not shown). A similar ability of MEKK1 and ATP to inhibit the SMRT interaction with T3R·DNA was observed using a SMRT construct containing both S1 and S2 receptor interaction domains (p < 0.01) (Fig. 3E). In contrast to SMRT, the relevant N-CoR constructs interacted equally well with the T3R·probe complex in either the absence or presence of MEKK1 and ATP, indicating that MEKK1 does not alter the avidity of N-CoR for T3Rs (Fig. 3B, compare lanes 11−19 and 2−10; quantified in Fig. 3, D and F). Of note are the following: (a) treatment of the T3R·DNA complex with MEKK1 in the absence of SMRT or N-CoR had no observable effect on the T3R·DNA complex, and neither SMRT nor N-CoR bound to the DNA probe in the absence of T3R; (b) the non-recombinant GST preparation did not supershift the T3R·DNA complex in either the presence or absence of MEKK1; and (c) the mobilities of the T3R·DNA, SMRT·T3R·DNA, and N-CoR·T3R·DNA complexes were all shifted to slower mobilities by incubation with anti-T3R antibodies, further confirming their identities (data not shown). We conclude that direct MEKK1 modification of SMRT, but not of N-CoR, is a potent inhibitor of the corepressor/nuclear receptor interaction.
Fig. 3. MEKK1 inhibits the binding of SMRT, but not of N-CoR, to a T3Rα·DNA complex in vitro.
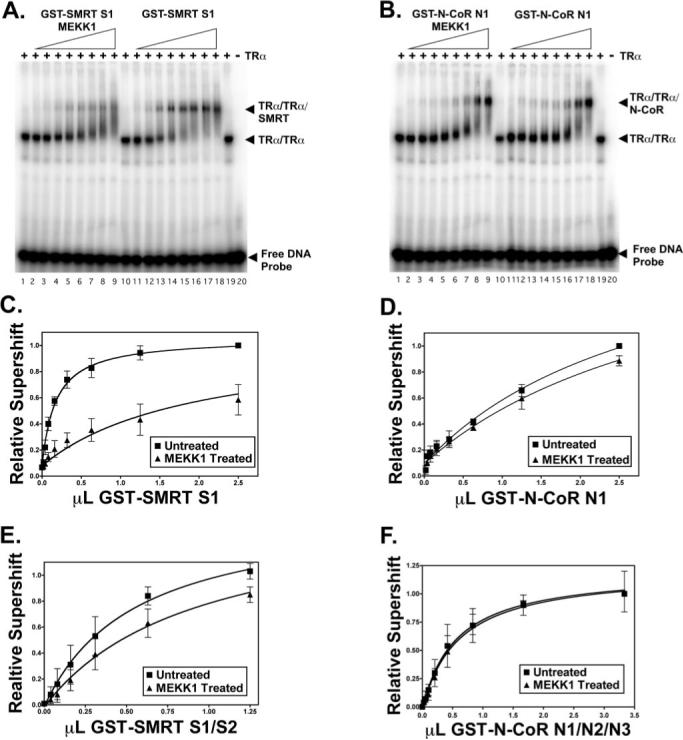
A, EMSA of the interaction of the SMRT S1 domain with T3Rα (TRα) in the absence and presence of MEKK1. T3Rα was incubated with a 32P-labeled DR-4 DNA response element in the presence of increasing levels of a SMRT S1 domain construct; the SMRT S1 domain was either previously phosphorylated by incubation with MEKK1 (lanes 2−9) or mock-treated (lanes 11−18), as indicated. SMRT was omitted in lanes 1, 10, and 19, and both SMRT and T3R were omitted in lane 20. The positions of the free DNA probe and the T3R·DNA and SMRT·T3R·DNA complexes are indicated. Radiolabel migrating between the latter two complexes most likely represents SMRT·T3R complexes that dissociated during electrophoresis. A representative experiment is shown. B, EMSA of the interaction of the N-CoR N1 domain with T3Rα in the absence and presence of MEKK1. The same type of experiment as described for A was performed using an N-CoR N1 domain construct. C, quantification of the interaction of the SMRT S1 domain with T3Rα in the absence and presence of MEKK1. The amount of T3Rα·DNA complex supershifted by SMRT when the corepressor was phosphorylated, or not, by MEKK1 was determined by PhosphorImager analysis of EMSAs performed as described for A. The means ± S.D. of three or more experiments are shown. D, quantification of the interaction of the N-CoR N1 domain with T3Rα in the absence and presence of MEKK1. E, quantification of the interaction of a SMRT S1/S2 domain construct with T3Rα in the absence and presence of MEKK1. F, quantification of the interaction of an N-CoR N1/N2/N3 domain construct with T3Rα in the absence and presence of MEKK1.
MEKK1 Signaling Alters the Subcellular Localization of SMRT, but Not of N-CoR
When expressed in CV-1 cells, GFP displayed a broad subcellular distribution extending over both nuclear and cytoplasmic compartments (data not shown). In contrast, a GFP fusion of full-length SMRT accumulated preferentially in the nucleus of transfected CV-1 cells, forming a pattern of small bright speckles superimposed over a more diffuse nucleoplasmic localization that was excluded from nucleoli (Fig. 4A). A comparable nuclear distribution was observed (a) in other cell types, such as 293T; (b) over a range of GFP-SMRT expression levels; (c) by immunofluorescence using Myc-directed antibodies to detect ectopically introduced, epitope-tagged SMRT; and (d) using SMRT-directed antibodies to detect endogenous SMRT (Fig. 4C) (data not shown). Although the vast majority of untreated cells displayed a nuclear SMRT localization, ∼6% of the SMRT-positive cells displayed a dual nuclear/cytoplasmic distribution, and 13% displayed a cytoplasmic localization (quantified in Fig. 4B). These “cytoplasmic” SMRT populations likely represent cells either in or recently transited through mitosis, as suggested by the absence of a discrete nuclear compartment, by the presence of DAPI-positive chromosomes arrayed on a mitotic plate, or by the presence of twinned, symmetrically arrayed cells that appeared to be the products of a recent cytokinesis (data not shown). Introduction of MEKK1 resulted in a redistribution of GFPSMRT into the cytoplasmic compartment in many, but not all, of the transfected cells (Fig. 4A; quantified in Fig. 4B). This cytoplasmic SMRT accumulation was observed in non-mitotic cells and therefore was not simply the result of an enhanced mitotic index in the MEKK1-treated population (data not shown). A similar cytoplasmic redistribution of SMRT in response to MEKK1 was also observed using GFP-SMRT in 293T cells, by immunofluorescence using Myc-tagged SMRT in CV-1 cells, or by immunofluorescence using endogenous SMRT in CV-1 cells and anisomycin to induce MEKK1 activity (Fig. 4C) (data not shown). No change in the subcellular localization of non-recombinant GFP was detected in response to MEKK1 (data not shown).
Fig. 4. MEKK1 induces nuclear export of SMRT, but not of N-CoR.
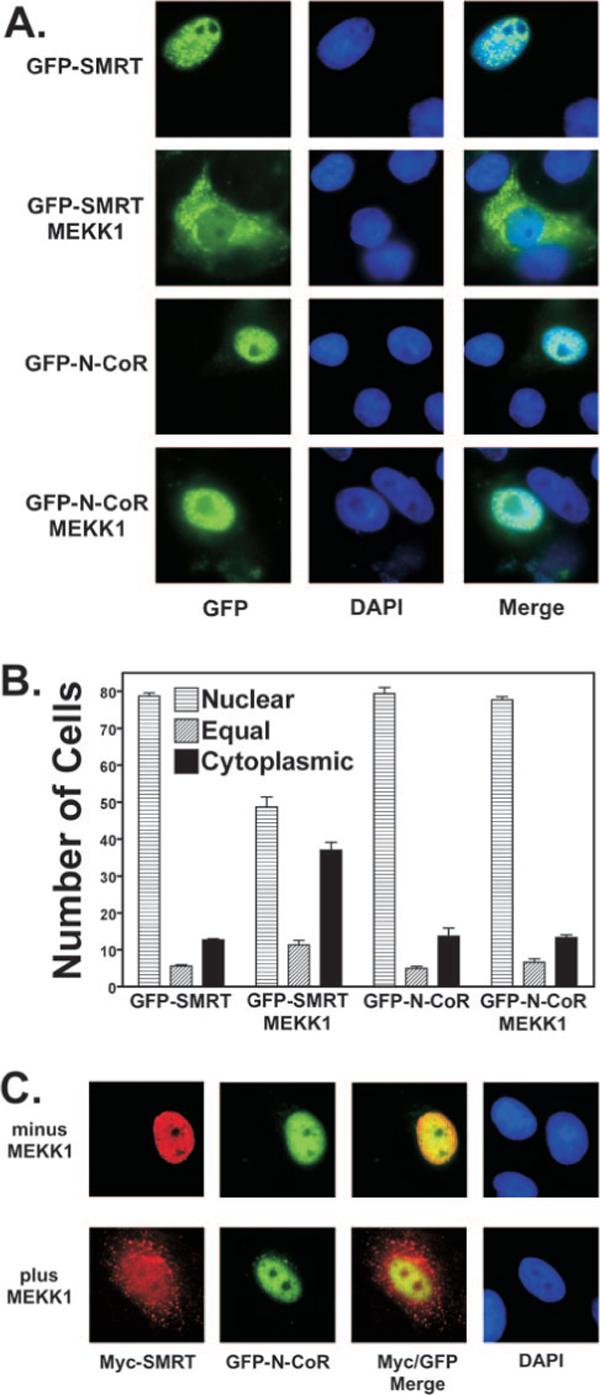
A, GFP-SMRT, but not GFP-N-CoR, is exported into the cytoplasm in response to MEKK1. GFP-SMRT and GFP-N-CoR constructs were transfected into CV-1 cells in the absence or presence of a constitutively active allele of MEKK1, as indicated. After 48 h, the cells were fixed, and the position of the GFP-corepressor construct was visualized by epifluorescence microscopy (left panels). Nuclei were visualized by DAPI staining (middle panels). A merge of the GFP-corepressor protein and DAPI signals is also shown (right panels). Representative microscopic fields are presented. B, quantification of changes in the subcellular localization of corepressors in response to MEKK1. The results from GFP-corepressor experiments such as in A were quantified. The means ± S.D. of three or more experiments are shown. When cotransfected, ∼90% of the cells expressing the GFP-corepressor construct also expressed the co-introduced MEKK1; the latter was detected exclusively in the cytoplasm (data not shown). C, SMRT is exported into the cytoplasm in response to MEKK1, whereas N-CoR in the same cells remains nuclear. CV-1 cells were transfected with Myc-tagged SMRT and GFP-tagged N-CoR in the presence or absence of a constitutively active MEKK1 allele. The cells were visualized 48 h later using immunofluorescence to detect Myc-SMRT (red channel) and GFP fluorescence to detect GFP-N-CoR (green channel). A merged image is also shown, as is a DAPI stain to visualize nuclei. Representative fields of fluorescent cells are presented.
A GFP fusion with full-length N-CoR displayed a subcellular distribution very similar to that of SMRT when expressed in unstimulated CV-1 cells; 80% of the untreated cells displayed a nuclear localization of GFP-N-CoR consisting of a microspecular pattern superimposed over a more diffuse nucleoplasm fluorescence that was excluded from nucleoli. This same type of pattern has been reported previously for endogenous N-CoR (57). The remaining cells exhibited a nuclear/cytoplasmic (5%) or cytoplasmic (14%) GFP-N-CoR fluorescence, with many of these cells mitotic or post-mitotic by the same criteria as noted for SMRT. In contrast to SMRT, however, GFP-N-CoR failed to detectably relocalize in response to co-introduced MEKK1 (Fig. 4B). Analogous results were observed in experiments using 293T cells over a range of GFP-N-CoR expression levels or using Myc-tagged N-CoR or endogenous N-CoR in an immunofluorescence protocol (data not shown). Notably, the divergent response of N-CoR and SMRT to MEKK1 signaling could be observed in individual cells by visualizing these corepressors simultaneously: in unstimulated cells, both GFP-N-CoR (green channel) and Myc-SMRT (red channel) immunoreactivities were primarily nuclear in the absence of MEKK1, whereas MEKK1 induced a cytoplasmic localization of Myc-SMRT in cells that retained the nuclear localization of GFP-N-CoR (Fig. 4C). We conclude that N-CoR, unlike SMRT, is refractory to MEKK1-mediated alterations in subcellular distribution under the conditions studied.
MEKK1 Signaling Disrupts the Co-distribution of T3R and SMRT, but Not of T3R and N-CoR
We next extended our subcellular visualization studies to examine the effects of MEKK1 on the association of SMRT and N-CoR with their transcription factor partners, such as T3R. We employed GFP-tagged corepressors in these studies and used an immunofluorescence procedure to detect co-introduced, Myc-tagged T3Rα. Myc-T3R introduced alone or together with GFP-SMRT or with GFP-N-CoR was primarily nuclear in these cells, displaying a diffuse, grainy nucleoplasmic distribution (Fig. 5); a very similar distribution has been reported for endogenous T3R (58, 59). Co-introduced GFP-SMRT or GFP-N-CoR displayed a distribution that largely overlapped that of the T3R signal (Fig. 5, note the merged images). Introduction of an activated MEKK1 allele had no detectable effect on the distribution of Myc-T3R, but resulted in a cytoplasmic redistribution of the GFP-SMRT signal in many of the cotransfected cells, resulting in a loss of co-localization between T3R and SMRT (Fig. 5). In contrast, GFP-N-CoR and Myc-T3Rα remained closely co-localized in both the absence and presence of the activated MEKK1 allele (Fig. 5). These results further support the proposal that MEKK1 signaling results in the release of the SMRT corepressor from its nuclear receptor partner, but that N-CoR is resistant to this form of regulation.
Fig. 5. Co-localization of T3Rα and SMRT is disrupted by MEKK1, whereas co-localization of T3Rα and N-CoR is main tained.
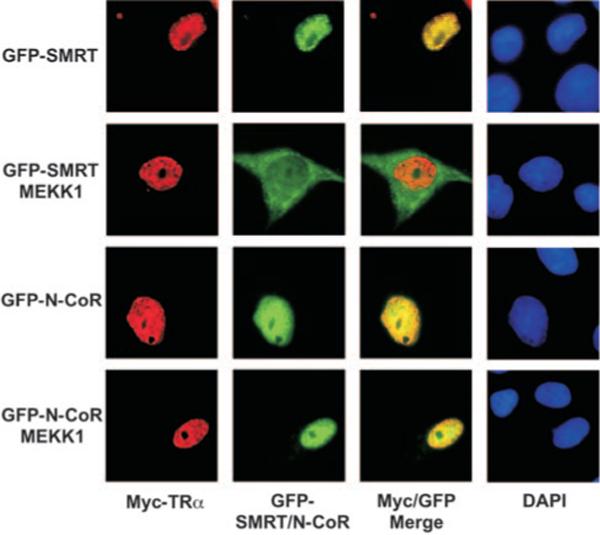
GFP-tagged SMRT or GFP-tagged N-CoR was transfected into CV-1 cells together with Myc-tagged T3Rα (TRα) in the absence or presence of MEKK1, as indicated. The subcellular localization of Myc-T3Rα was visualized by immunofluorescence (red channel) and that of GFP-SMRT (upper two panels) and GFP-N-CoR (lower two panels) by GFP fluorescence (green channel). An image merge is also shown, as is a DAPI stain to visualize nuclei. Representative fields of fluorescent cells are presented.
SMRT Phosphorylation Is Increased in Response to MEKK1 Signaling in Vivo
Protein phosphorylation can frequently be detected as an alteration in electrophoretic mobility on SDS-polyacrylamide gels/immunoblots (e.g. Refs. 43 and 44). We used this property to determine whether the phosphorylation pattern of SMRT and N-CoR differed in cells transfected with an activated MEKK1 construct. Full-length SMRT and N-CoR (>2400 amino acids long) were too large to accurately detect a change in electrophoretic mobility; therefore, we performed these experiments using the C-terminal corepressor constructs that were sufficient to confer inhibition of SMRT by MEKK1 in our two-hybrid assays. The mobility of the SMRT protein construct was measurably decreased by the co-introduction of an activated MEKK1 allele (Fig. 6, upper panel, compare lanes 3 and 4), and this reduced mobility was reversed by incubation with alkaline phosphatase (lane 5). In contrast, MEKK1 had little or no effect on an equivalent N-CoR construct (Fig. 6, lower panel, compare lanes 3 and 4). Intriguingly, alkaline phosphatase treatment increased the mobility of N-CoR whether isolated from unstimulated or stimulated cells (Fig. 6, lower panel, compare lanes 3 and 4 with lanes 2 and 5), indicating that N-CoR is constitutively phosphorylated at sites distinct from those that respond (in SMRT) to MEKK1 activation. These experiments suggest that SMRT is more extensively phosphorylated in response to MEKK1 signaling than is N-CoR, paralleling the greater susceptibility of SMRT to inhibition by MEKK1. It should be noted, however, that the effects of phosphorylation on electrophoretic mobility are difficult to predict. It is possible that both SMRT and N-CoR are phosphorylated in response to MEKK1, but that this modification takes place at different sites or in a different chemical environment in the two corepressors so as to have different consequences for their electrophoretic mobilities.
Fig. 6. MEKK1 detectably alters the electrophoretic mobility of a SMRT construct, but not that of an N-CoR construct.
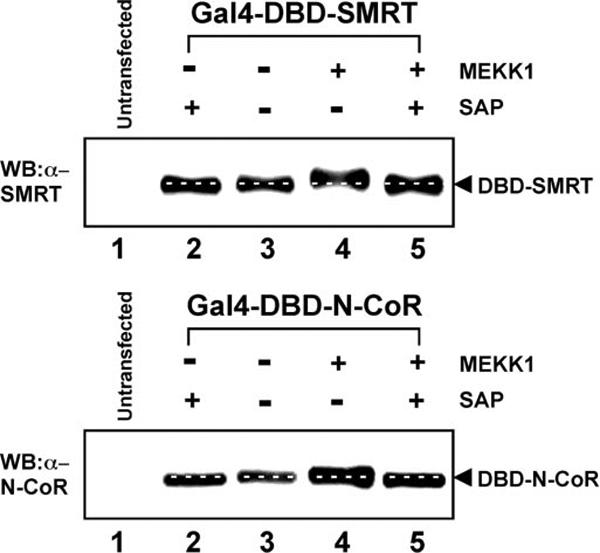
The same Gal4DBD-SMRT (upper panel) and Gal4DBD-N-CoR (lower panel) constructs employed in the two-hybrid assays were transfected into CV-1 cells in the absence and presence of the constitutively active MEKK1 allele, as indicated (lanes 2−5); control cells not expressing the corepressor constructs were analyzed in lane 1. The cells were lysed; aliquots of each lysate were incubated (lanes 2 and 5) or not (lanes 1, 3, and 4) with shrimp alkaline phosphatase (SAP); and the lysates were analyzed by SDS-PAGE and immunoblotting with either anti-SMRT or anti-N-CoR antiserum as described under “Experimental Procedures.” White dashed lines indicate the positions of the corepressor in the absence of MEKK1 and in the absence of shrimp alkaline phosphatase treatment. WB, Western blot.
MEKK1 Can Reverse Repression by SMRT, but Not by N-CoR, in Transfected Cells
To examine the effect of MEKK1 on the ability of SMRT and N-CoR to function as corepressors in cells, we examined the ability of ectopic corepressors to mediate repression by RARα. We used a Gal4DBD-RARα construct and a Gal4−17-mer reporter in these studies to avoid interference from receptors endogenous to the CV-1 cells (45, 60). Ectopic expression of either SMRT or N-CoR repressed reporter gene expression in the presence of the Gal4DBD-RARα construct (Fig. 7). Co-introduction of activated MEKK1 counteracted this repression by SMRT, but had no detectable effect on repression mediated by N-CoR. These results are consistent with the results from our corepressor/receptor interaction assays and our subcellular localization experiments indicating that MEKK1 signaling interferes with SMRT (but not N-CoR) corepressor function.
Fig. 7. MEKK1 can counteract SMRT-mediated repression, but has no effect on N-CoR function.
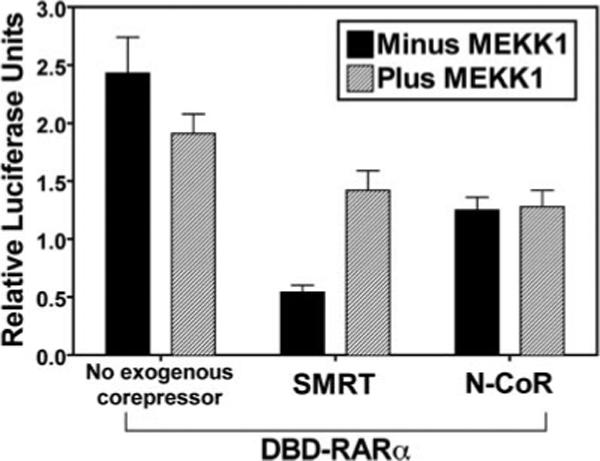
A Gal4DBD-RARα construct and a Gal4−17-mer reporter were transfected into CV-1 cells together with an empty expression vector, an expression vector for full-length SMRT, or an expression vector for full-length N-CoR, as indicated. Each experiment also included (hatched bars) or not (black bars) a constitutively active MEKK1 allele. After 48 h in hormone-stripped medium, the cells were harvested, and the luciferase activity of the Gal4−17-mer reporter relative to a β-galactosidase reporter used as an internal transfection control was determined. The means ± S.D. of three or more experiments are presented.
Both SMRT and N-CoR Respond to Growth Factor and Cytokine Receptors but Diverge in Their Response to Downstream Signal Transducers
Upstream activators of MEKK1, such as an activated allele of the EGF receptor, Ras, or anisomycin, also inhibited the two-hybrid interaction of SMRT with T3Rα and enhanced its cytoplasmic localization (p < 0.001) (Fig. 8, A and C); this inhibition is mediated both through MEKK1 and (to a lesser extent) through a distinct parallel pathway not fully defined (42, 43). Interestingly, the interaction of N-CoR with T3Rα and its nuclear localization, although refractory to MEKK1 and to MEKK1 activators such as anisomycin, were partially reduced by EGF receptor signaling, suggesting that both N-CoR and SMRT may be responsive to this second, MEKK1-independent pathway (Fig. 8A, right panel) (data not shown). Conversely, a MAPK kinase, MEK1, acting downstream of MEKK1, duplicated several of the actions of MEKK1 on the SMRT/nuclear receptor interaction and on GFP-SMRT localization (Fig. 8, A and B) (43). In contrast, MEK1 had little or no effect on the N-CoR/T3Rα interaction in our two-hybrid assay, nor did MEK1 alter the subcellular distribution of the GFP-N-CoR construct (Fig. 8, A, right panel; and B). Thus, although growth factors such as EGF can inhibit both N-CoR and SMRT function, the much stronger inhibitory effects of these growth factors on SMRT appear to be mediated through phosphorylation of SMRT by an MEKK1-MEK1 cascade to which N-CoR is non-responsive (modeled in Fig. 9).
Fig. 8. SMRT and N-CoR respond to distinct, although overlapping, growth factor and cytokine signaling pathways.
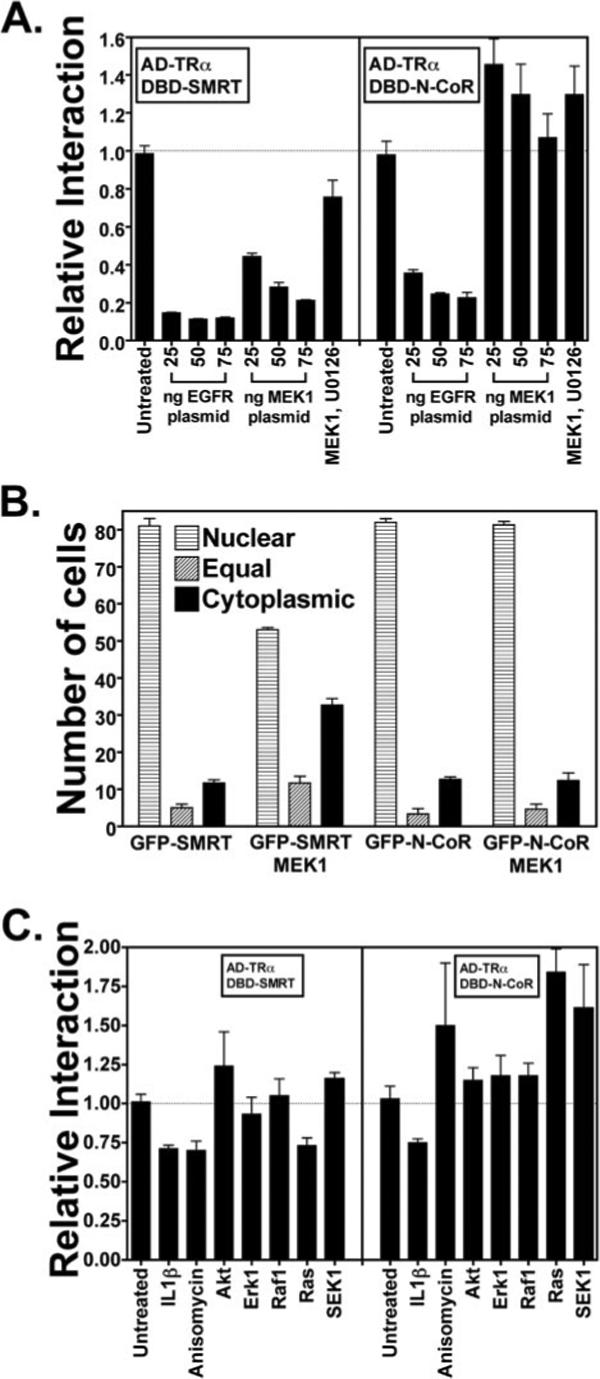
A, the interaction of both SMRT and N-CoR with T3Rα (TRα) is inhibited by an activated EGF receptor (EGFR), whereas only the SMRT interaction is inhibited by MEK1. The same mammalian two-hybrid interaction as described in the legend to Fig. 1 was used with increasing amounts of an expression vector for an activated allele of the EGF receptor (v-ErbB) or an expression vector for an activated allele of MEK1, as indicated. The experiments using 50 ng of MEK1 were also repeated in the presence of an MEK1 inhibitor, U0126, as indicated. The relative luciferase activity is shown. The means ± S.D. of three or more experiments are presented. B, GFP-SMRT, but not GFP-N-CoR, is exported from the nucleus in response to MEK1 signaling. The same type of experiment as described in the legend to Fig. 4B was repeated, but an expression vector for constitutively active MEK1 was used in place of the MEKK1 vector. The means ± S.D. of three or more experiments are presented. C, SMRT and N-CoR respond to a variety of different growth factor and cytokine signal transducers. The same type of experiment as described for A was repeated, but by exposing the cells to IL-1β or anisomycin or by cotransfecting activated alleles of Akt, ERK1, Raf1, Ras, or SEK1, as indicated. The means ± S.D. of three or more experiments are presented.
Fig. 9. Schematic model of kinase regulation of SMRT and N-CoR function.
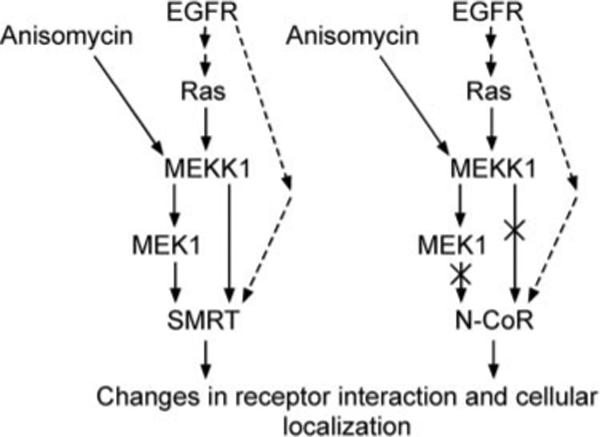
A model of the effects of growth factor receptors and downstream signal transducers on SMRT and N-CoR function is shown. The EGF receptor (EGFR) or anisomycin activates MEKK1 signaling, which, in turn, strongly inhibits SMRT function by causing release of SMRT from its nuclear receptor partners and export out of the nucleus. MEK1, activated by MEKK1, also has strong inhibitory effects on SMRT function (solid arrows). The EGF receptor exerts an additional, weaker inhibitory effect on SMRT through a second, poorly understood pathway (dashed arrows); N-CoR appears to be subject only to this secondary pathway.
IL-1β has been reported to inhibit the interaction of N-CoR with transcription factors and to cause its nuclear export (61). Consistent with these studies, IL-1β modestly but consistently inhibited the interaction of both SMRT and N-CoR with T3Rα in our two-hybrid assay, suggesting that both corepressors are responsive to this cytokine (p < 0.01) (Fig. 8C). Ciliary neurotrophic factor has also been reported to cause the dissociation of N-CoR from its transcription factor partners and nuclear export, due in this case to phosphorylation of N-CoR by an Akt pathway (51). Using CV-1 cells, we did not detect an effect of Akt on either SMRT or N-CoR in our two-hybrid and GFP localization assays. Raf1, SEK1, and ERK1 also had little or no effect on either N-CoR or SMRT in this assay (Fig. 8C). We conclude that SMRT and N-CoR are embedded in partially overlapping yet distinct kinase regulatory pathways that operate downstream from both growth factor and cytokine receptors. SMRT is strongly and directly inhibited by a MEKK1-MEK1 cascade, whereas N-CoR is refractory to this pathway in our studies. In contrast, both SMRT and N-CoR respond more weakly to additional signals that act downstream of the EGF and IL-1β receptors and that are distinct from the actions of this MAPK kinase kinase cascade (Fig. 8).
DISCUSSION
N-CoR and SMRT Differ in Their Response to MEKK1 Signaling
The SMRT corepressor is inhibited by a growth factor signaling pathway that operates through MEKK1 (42, 43). These MAPK cascade transducers result in inhibition of the SMRT interaction with its transcription factor partners and a change in the subcellular localization of SMRT from a nuclear to a cytoplasmic distribution. In this work, we have shown that SMRT function is regulated at multiple levels by MEKK1 signaling, whereas N-CoR function is refractory to these same forms of regulation. (a) MEKK1 signaling in vivo resulted in enhanced phosphorylation of the SMRT C terminus, but caused no detectable change in the phosphorylation of N-CoR under the same conditions. (b) Introduction of an activated version of MEKK1 into cells resulted in a nearly complete inhibition of the two-hybrid interaction of SMRT with nuclear receptors, whereas activated MEKK1 did not inhibit but instead appeared to slightly stabilize the interaction of N-CoR with its nuclear receptor partners, such as T3Rα. We do not understand the basis for this possible stabilization of the N-CoR/T3R interaction, which is absent at higher MEKK1 expression levels, but it is both reproducible and in sharp contrast to the strong inhibition seen for SMRT. (c) The co-immunoprecipitation of T3Rα with full-length SMRT, but not with N-CoR, was inhibited by activated MEKK1. (d) Incubation of a SMRT construct with MEKK1 in vitro significantly inhibited the ability of SMRT to interact with the T3R·DNA complex in an EMSA, whereas the interaction of N-CoR with the T3R·DNA complex was unaltered under the same conditions. (e) MEKK1 activation caused a relocalization of SMRT from a nuclear to a cytoplasmic compartment, although MEKK1 caused no observable change in the subcellular localization of N-CoR. (f) The ability of SMRT to function as a corepressor in a transfection analysis was abrogated by MEKK1, whereas that of N-CoR was not.
It is worth noting that whereas MEKK1 signaling resulted in loss of repression, it did not appear to result in a gain in target gene activation beyond basal reporter levels; the latter appears to require the presence of a hormone agonist. Notably, the experiments described here demonstrate that the MEKK1-induced SMRT translocation from the nucleus to the cytoplasm occurred independently of its T3Rα partner, which remained in the nucleus. This confirms that MEKK1 signaling causes a dissociation of SMRT and T3R in vivo and also suggests that MEKK1 signaling may permit T3Rα to remain bound to target promoters, but in a neutral state.
MEKK1 Is One of Several Signals Operating Downstream of Growth Factors and Cytokines That Can Inhibit Corepressor Function
MEKK1 and analogous MAPK kinase kinase cascades are only one of many signal transducers that operate downstream of growth factor and cytokine signaling. Consistent with the multiplex nature of growth factor signaling, we have observed that an activated version of the EGF receptor typically induces a stronger inhibition of SMRT function than does MEKK1 alone, and this EGF receptor-mediated inhibition of SMRT function appears to be blocked only partially by introduction of a dominant-negative MEKK1 construct (Ref. 43; diagrammed schematically in Fig. 9). Therefore, MEKK1 is the predominant (but not exclusive) mediator of the inhibitory actions of EGF receptor signaling on SMRT function. Consistent with this model, we found that N-CoR function, although fully refractory to MEKK1 inhibition in our studies, was nonetheless partially inhibited by EGF receptor signaling; we propose that N-CoR, in common with SMRT, is subject to this undefined but secondary pathway of EGF receptor signaling.
We have not yet identified the basis behind the secondary pathway of inhibition mediated by the EGF receptor independent of MEKK1. One plausible candidate appears to be Akt, which is activated by phosphatidylinositol 3-kinase and functions downstream of many growth factor and cytokine receptors. N-CoR has been reported to be phosphorylated by Akt at Ser401, leading to reversal of N-CoR-mediated repression and its nuclear export; this pathway was identified in neural stem cells, where it appears to mediate astroglial differentiation in response to ciliary neurotrophic factor (51). However, SMRT possesses an alanine at position 401 and is stated to be resistant to the actions of Akt (51). Furthermore, we could detect no inhibition of the two-hybrid interaction between either SMRT or N-CoR and T3R and no alteration in SMRT or N-CoR subcellular localization in response to introduction of phosphatidylinositol 3-kinase or of activated Akt, nor was the profound inhibition of SMRT by EGF receptor signaling or the weaker inhibition of N-CoR impaired by LY294002, a phosphatidylinositol 3-kinase inhibitor.2 We conclude that Akt does not contribute to inhibition of SMRT or N-CoR function under the conditions studied here.
Notably, the cytokine IL-1β also caused a moderate inhibition of both SMRT and N-CoR in our two-hybrid assay. IL-1β has been reported to inhibit N-CoR through an indirect pathway, resulting in MEKK1 phosphorylation of a TAB2 subunit present in a subset of N-CoR·HDAC3 complexes (61). In this prior study, SMRT was reported to be resistant to this TAB2 pathway, and the effects of TAB2 on N-CoR were restricted to NF-κB and estrogen receptor target genes. Although we do not exclude this TAB2-dependent mechanism functioning for a subpopulation of N-CoR target genes, such as those regulated by NF-κB, we detected no evidence of an inhibitory effect of MEKK1 on N-CoR in the context of our current study.
Regulation of Corepressor Function by Kinases, a Common Theme
Hormone ligands regulate the interaction of nuclear receptors with corepressors and coactivators by inducing allosteric changes in the nuclear receptor that mask or expose the corepressor-docking site on the receptor surface (reviewed in Refs. 20 and 21). However, recent studies have demonstrated that the corepressor/nuclear receptor interaction is also subject to regulation by a series of important kinase signaling pathways that modulate corepressor function in normal cells and that contribute to aberrant nuclear receptor function in disease (42-44, 51, 61-65). As noted here, SMRT, but not N-CoR, is negatively regulated by a MAPK kinase kinase cascade that operates downstream of EGF receptor signals. Inducers of cell stress, including arsenic trioxide and anisomycin, can also activate MEKK1 and are potent inhibitors of SMRT function; this MEKK1-dependent mechanism may contribute to the prodifferentiation effects of arsenic trioxide as used in the treatment of acute promyelocytic leukemia (44). The Drosophila EGF receptor has also been shown to regulate the function of the Drosophila SMRTER protein, although whether SMRTER is a true ortholog of mammalian SMRT remains unclear (66). Reciprocally, it has been reported that N-CoR, but not SMRT, can be inhibited in certain contexts in response to TAB2 and Akt pathways operating downstream of cytokine and ciliary neurotrophic factors (51, 61). N-CoR, but not SMRT, has also been reported to be down-regulated by a Siah2/proteasome-mediated pathway (52). Regulation of the corepressor interaction by modification of the transcription factor partner has also been noted; phosphorylation of c-Jun by c-Jun N-terminal kinase, for example, can lead to release of N-CoR complexes and exchange of c-Jun for c-Jun/c-Fos heterodimers (67). SMRT does not appear to participate in this process. Therefore, SMRT and N-CoR appear to be embedded in distinct regulatory networks, and these distinct regulatory properties may help account for the appearance and conservation of these two corepressors as distinct isotypes during the vertebrate evolutionary radiation. Notably, there are multiple phosphorylation sites in these corepressors that can be modified by growth factor and cytokine cascades and that appear to contribute combinatorially to the regulation processes described here.3 A more complete dissection of these different phosphorylation sites will be important for further understanding the differential impact of these different signaling cascades on the different corepressor isoforms.
In addition to SMRT and N-CoR, other components of the corepressor complex are also subject to regulation by phosphorylation. For example, calmodulin-dependent kinases have been reported to phosphorylate class II histone deacetylases in muscle cells, resulting in the tethering of these histone deacetylases to cytoplasmic 14−3−3 proteins and the derepression of their corresponding target genes (68-70). Phosphorylation of HDAC4 by ERK1 and ERK2 has been reported to result in the opposite response, resulting in an enhanced nuclear accumulation, whereas phosphorylation of HDAC1 and HDAC2 alters their interactions with one another and with other components of their corepressors complexes (71-73). Both nuclear receptors and coactivators are themselves also subject to an extensive series of regulatory phosphorylations (e.g. Refs. 74-80). These covalent modifications act together with ligand agonists and antagonists to integrate the multiplicity of signals impinging on the cell so as to produce the correct overall transcriptional and biological response for a given physiological context.
Acknowledgments
We thank Liming Liu for superb technical assistance and Fumihiko Hayakawa and Michael Goodson for many helpful discussions and reagents.
Footnotes
This work was supported in part by United States Public Health Service Grant DK53528 from NIDDK, National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: MAPK, mitogen-activated protein kinase; EGF, epidermal growth factor; MEKK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase; T3R, thyroid hormone receptor; T3, triiodothyronine; IL-1β, interleukin-1β; PBS, phosphate-buffered saline; GST, glutathione S-transferase; BSA, bovine serum albumin; MOPS, 4-morpholinepropanesulfonic acid; EMSA, electrophoretic mobility shift assay; DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; Gal4DBD, Gal4 DNA-binding domain; Gal4AD, Gal4 activation domain; RARα, retinoic acid receptor-α; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; ERK, extracellular signal-regulated kinase.
B. A. Jonas and M. L. Privalsky, unpublished data.
B. A. Jonas, F. Hayakawa, and M. L. Privalsky, unpublished data.
REFERENCES
- 1.Chen JD, Li H. Crit. Rev. Eukaryotic Gene Expression. 1998;8:169–190. doi: 10.1615/critreveukargeneexpr.v8.i2.40. [DOI] [PubMed] [Google Scholar]
- 2.Ito M, Roeder RG. Trends Endocrinol. Metab. 2001;12:127–134. doi: 10.1016/s1043-2760(00)00355-6. [DOI] [PubMed] [Google Scholar]
- 3.Lee JW, Lee YC, Na SY, Jung DJ, Lee SK. Cell. Mol. Life Sci. 2001;58:289–297. doi: 10.1007/PL00000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna NJ, O'Malley BW. Endocrinology. 2002;143:2461–2465. doi: 10.1210/endo.143.7.8892. [DOI] [PubMed] [Google Scholar]
- 5.Ordentlich P, Downes M, Evans RM. Curr. Top. Microbiol. Immunol. 2001;254:101–116. doi: 10.1007/978-3-662-10595-5_5. [DOI] [PubMed] [Google Scholar]
- 6.Privalsky ML. Annu. Rev. Physiol. 2004;66:315–360. doi: 10.1146/annurev.physiol.66.032802.155556. [DOI] [PubMed] [Google Scholar]
- 7.Rachez C, Freedman LP. Curr. Opin. Cell Biol. 2001;13:274–280. doi: 10.1016/s0955-0674(00)00209-x. [DOI] [PubMed] [Google Scholar]
- 8.Shibata H, Spencer TE, Onate SA, Jenster G, Tsai SY, Tsai MJ, O'Malley BW. Recent Prog. Horm. Res. 1997;52:141–164. Discussion 164−165. [PubMed] [Google Scholar]
- 9.Xu L, Glass CK, Rosenfeld MG. Curr. Opin. Genet. Dev. 1999;9:140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 10.Beato M, Klug J. Hum. Reprod. Update. 2000;6:225–236. doi: 10.1093/humupd/6.3.225. [DOI] [PubMed] [Google Scholar]
- 11.Hager GL. Prog. Nucleic Acid Res. Mol. Biol. 2001;66:279–305. doi: 10.1016/s0079-6603(00)66032-4. [DOI] [PubMed] [Google Scholar]
- 12.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Lazar MA. Annu. Rev. Physiol. 2000;62:439–466. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]
- 14.Chen JD, Evans RM. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 15.Chen JD, Umesono K, Evans RM. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7567–7571. doi: 10.1073/pnas.93.15.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ordentlich P, Downes M, Xie W, Genin A, Spinner NB, Evans RM. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2639–2644. doi: 10.1073/pnas.96.6.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park EJ, Schroen DJ, Yang M, Li H, Li L, Chen JD. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3519–3524. doi: 10.1073/pnas.96.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sande S, Privalsky ML. Mol. Endocrinol. 1996;10:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 19.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. Mol. Cell. Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glass CK, Rosenfeld MG. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 21.Privalsky ML. Curr. Top. Microbiol. Immunol. 2001;254:117–136. doi: 10.1007/978-3-662-10595-5_6. [DOI] [PubMed] [Google Scholar]
- 22.Ayer DE. Trends Cell Biol. 1999;9:193–198. doi: 10.1016/s0962-8924(99)01536-6. [DOI] [PubMed] [Google Scholar]
- 23.Berger SL. Science. 2001;292:64–65. [PubMed] [Google Scholar]
- 24.Chen H, Tini M, Evans RM. Curr. Opin. Cell Biology. 2001;13:218–224. doi: 10.1016/s0955-0674(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 25.Hassig CA, Schreiber SL. Curr. Opin. Chem. Biol. 1997;1:300–308. doi: 10.1016/s1367-5931(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 26.Jenuwein T, Allis CD. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 27.Muscat GE, Burke LJ, Downes M. Nucleic Acids Res. 1998;26:2899–2907. doi: 10.1093/nar/26.12.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pazin MJ, Kadonaga JT. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 29.Rice JC, Allis CD. Curr. Opin. Cell Biol. 2001;13:263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- 30.Strahl BD, Allis CD. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 31.Turner BM. BioEssays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 32.Wong CW, Privalsky ML. Mol. Cell. Biol. 1998;18:5500–5510. doi: 10.1128/mcb.18.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Workman JL, Kingston RE. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 34.Wu C. J. Biol. Chem. 1997;272:28171–28174. doi: 10.1074/jbc.272.45.28171. [DOI] [PubMed] [Google Scholar]
- 35.Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, Shiekhattar R. Genes Dev. 2000;14:1048–1057. [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- 38.Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. EMBO J. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Kalkum M, Chait BT, Roeder RG. Mol. Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- 40.Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, Rosenfeld GM. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 41.Seol W, Mahon MJ, Lee YK, Moore DD. Mol. Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- 42.Hong SH, Wong CW, Privalsky ML. Mol. Endocrinol. 1998;12:1161–1171. doi: 10.1210/mend.12.8.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong SH, Privalsky ML. Mol. Cell. Biol. 2000;20:6612–6625. doi: 10.1128/mcb.20.17.6612-6625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong SH, Yang Z, Privalsky ML. Mol. Cell. Biol. 2001;21:7172–7182. doi: 10.1128/MCB.21.21.7172-7182.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauksdottir H, Farboud B, Privalsky ML. Mol. Endocrinol. 2003;17:373–385. doi: 10.1210/me.2002-0340. [DOI] [PubMed] [Google Scholar]
- 46.Hong SH, David G, Wong CW, Dejean A, Privalsky ML. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guan KL, Dixon JE. Anal. Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 48.Chen HW, Privalsky ML. Mol. Cell. Biol. 1993;13:5970–5980. doi: 10.1128/mcb.13.10.5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonde BG, Privalsky ML. J. Virol. 1990;64:1314–1320. doi: 10.1128/jvi.64.3.1314-1320.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoh SM, Privalsky ML. J. Biol. Chem. 2001;276:16857–16867. doi: 10.1074/jbc.M010022200. [DOI] [PubMed] [Google Scholar]
- 51.Hermanson O, Jepsen K, Rosenfeld MG. Nature. 2002;419:934–939. doi: 10.1038/nature01156. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Guenther MG, Carthew RW, Lazar MA. Genes Dev. 1998;12:1775–1780. doi: 10.1101/gad.12.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forman BM, Casanova J, Raaka BM, Ghysdael J, Samuels HH. Mol. Endocrinol. 1992;6:429–442. doi: 10.1210/mend.6.3.1316541. [DOI] [PubMed] [Google Scholar]
- 54.Kurokawa R, Yu VC, Näär A, yakumoto S, Han Z, Silverman S, Rosenfeld MG, Glass CK. Genes Dev. 1993;7:1423–1435. doi: 10.1101/gad.7.7b.1423. [DOI] [PubMed] [Google Scholar]
- 55.Lazar MA, Berrodin TJ, Harding HP. Mol. Cell. Biol. 1991;11:5005–5015. doi: 10.1128/mcb.11.10.5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Näär AM, Boutin JM, Lipkin SM, Yu VC, Holloway JM, Glass CK, Rosenfeld MG. Cell. 1991;65:1267–1279. doi: 10.1016/0092-8674(91)90021-p. [DOI] [PubMed] [Google Scholar]
- 57.Soderstrom M, Vo A, Heinzel T, Lavinsky RM, Yang WM, Seto E, Peterson DA, Rosenfeld MG, Glass CK. Mol. Endocrinol. 1997;11:682–692. doi: 10.1210/mend.11.6.0018. [DOI] [PubMed] [Google Scholar]
- 58.Dace A, Zhao L, Park KS, Furuno T, Takamura N, Nakanishi M, West BL, Hanover JA, Cheng SY. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8985–8990. doi: 10.1073/pnas.160257997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strait KA, Schwartz HL, Seybold VS, Ling NC, Oppenheimer JH. Proc. Natl. Acad. Sci. U. S. A. 1991;88:3887–3891. doi: 10.1073/pnas.88.9.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farboud B, Hauksdottir H, Wu Y, Privalsky ML. Mol. Cell. Biol. 2003;23:2844–2858. doi: 10.1128/MCB.23.8.2844-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 62.Jang MK, Goo YH, Sohn YC, Kim YS, Lee SK, Kang H, Cheong J, Lee JW. J. Biol. Chem. 2001;276:20005–20010. doi: 10.1074/jbc.M010211200. [DOI] [PubMed] [Google Scholar]
- 63.Kurokawa H, Arteaga CL. Clin. Cancer Res. 2001;7:4436s–4442s. Discussion 4411s−4412s. [PubMed] [Google Scholar]
- 64.Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, Hilsenbeck SG, Osborne CK, Glass CK, Rosenfeld MG, Rose DW. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2920–2925. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, McDonnell DP. Mol. Cell. Biol. 2002;22:3663–3673. doi: 10.1128/MCB.22.11.3663-3673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuda L, Nagaraj R, Zipursky SL, Banerjee U. Cell. 2002;110:625–637. doi: 10.1016/s0092-8674(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 67.Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, Muscat GE. J. Biol. Chem. 2001;276:17007–17013. doi: 10.1074/jbc.M101508200. [DOI] [PubMed] [Google Scholar]
- 69.Grozinger CM, Schreiber SL. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKinsey TA, Zhang CL, Olson EN. Mol. Cell. Biol. 2001;21:6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galasinski SC, Resing KA, Goodrich JA, Ahn NG. J. Biol. Chem. 2002;277:19618–19626. doi: 10.1074/jbc.M201174200. [DOI] [PubMed] [Google Scholar]
- 72.Tsai SC, Seto E. J. Biol. Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 73.Zhou X, Richon VM, Wang AH, Yang XJ, Rifkind RA, Marks PA. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14329–14333. doi: 10.1073/pnas.250494697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, Robin P, Knibiehler M, Pritchard LL, Ducommun B, Trouche D, Harel-Bellan A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- 75.Hammer GD, Krylova I, Zhang Y, Darimont BD, Simpson K, Weigel NL, Ingraham HA. Mol. Cell. 1999;3:521–526. doi: 10.1016/s1097-2765(00)80480-3. [DOI] [PubMed] [Google Scholar]
- 76.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A, Chin WW, Burger AG, Meier CA. J. Biol. Chem. 1999;274:10505–10510. doi: 10.1074/jbc.274.15.10505. [DOI] [PubMed] [Google Scholar]
- 77.Lopez GN, Turck CW, Schaufele F, Stallcup MR, Kushner PJ. J. Biol. Chem. 2001;276:22177–22182. doi: 10.1074/jbc.M010718200. [DOI] [PubMed] [Google Scholar]
- 78.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Mol. Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 79.Rowan BG, Garrison N, Weigel NL, O'Malley BW. Mol. Cell. Biol. 2000;20:8720–8730. doi: 10.1128/mcb.20.23.8720-8730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O'Malley BW. Mol. Cell. Biol. 2002;22:3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]