

Abstract
The grain of the self-pollinating diploid barley species offers two modes of producing recombinant enzymes or other proteins. One uses the promoters of genes with aleurone-specific expression during germination and the signal peptide code for export of the protein into the endosperm. The other uses promoters of the structural genes for storage proteins deposited in the developing endosperm. Production of a protein-engineered thermotolerant (1, 3–1, 4)-β-glucanase with the D hordein gene (Hor3–1) promoter during endosperm development was analyzed in transgenic plants with four different constructs. High expression of the enzyme and its activity in the endosperm of the mature grain required codon optimization to a C+G content of 63% and synthesis as a precursor with a signal peptide for transport through the endoplasmic reticulum and targeting into the storage vacuoles. Synthesis of the recombinant enzyme in the aleurone of germinating transgenic grain with an α-amylase promoter and the code for the export signal peptide yielded ≈1 μg⋅mg−1 soluble protein, whereas 54 μg⋅mg−1 soluble protein was produced on average in the maturing grain of 10 transgenic lines with the vector containing the gene for the (1, 3–1, 4)-β-glucanase under the control of the Hor3–1 promoter.
Barley cultivars have been and are being bred to store large amounts of carbohydrates, oil, and protein during grain development. In germination, the stored amino nitrogen and carbohydrates are mobilized to sustain the development of the embryo and seedling. The stored carbohydrates, oils, and proteins in the kernels are exploited for feed, food, and beverages, both in the form of mature grain and as malt produced in large scale by steeping, germination, and kiln drying (1, 2). Advances in the molecular genetics of barley storage proteins (3, 4), elucidation of mechanisms by which storage proteins are synthesized, targeted, and sequestered for long-term storage in the endosperm cells (5, 6), and establishment of genetic transformation procedures (7–10) provide the basis for studying the production of recombinant proteins in the developing barley grain. The unique capacity of the scutellum and aleurone cells to synthesize hydrolytic enzymes during germination/malting and secrete them into the starchy endosperm (11, 12) provides an additional avenue for the production of recombinant proteins in this cereal (9, 10).
Barley grains of cultivars with a 1,000-grain weight of 30–45 g contain 8–16% protein on a dry-weight basis (1). A thousand grains of barley contain on the average 4.5 g of protein of which ≈2.3 g is distributed to the endosperm and the remainder to the embryo and aleurone tissue. The promoters and signal peptide codes of the genes encoding the barley prolamins (hordeins) are suitable candidates for deposition of recombinant proteins in the protein bodies of the developing endosperm cells. These isopropanol-soluble proteins account for approximately 50% of the total endosperm protein, comprising the high molecular weight D, the sulfur-poor C, and the sulfur-rich B and γ hordein polypeptides (3, 4). The different hordein proteins are coordinately expressed during endosperm development (13). As diagrammed in Fig. 1 (14), electron microscopic analyses of wild types and mutants deficient in groups or individual hordein polypeptides in combination with biochemical analyses have revealed the following picture of the synthesis and deposition of the hordein polypeptides: the mRNAs of the Hor1, Hor2, Hor3, and Hor5 loci on chromosome 5 encoding the hordein C, B, D, and γ precursor polypeptides (3, 5) are translated on the polysomes of the endoplasmic reticulum (ER) (15). The N-terminal signal peptides direct the nascent polypeptide chains into the lumen of the ER, where they are cleaved off during or immediately after translocation (16). Monoclonal and polyclonal antibodies recognizing individual hordein polypeptides or polypeptide families indicate that they are transported in small vesicles via the Golgi cisternae into the storage vacuoles of the endosperm cells (5). Surrounded by the tonoplast, the protein bodies consist of globules (H in Fig. 1) containing C, B, and γ hordein (5, 17, 18) and of a reticulate component, identified as D hordein (19). In certain genotypes and under not clearly defined conditions, hordein polypeptides also accumulate in the cisternae of the ER (Fig. 1 and ref. 5).
Figure 1.

The developing endosperm cell of barley. C, chromatin; E, ER; G, Golgi apparatus; H, hordein in protein body; M, mitochondrion; P, plasmodesma; S, starch grain; V, vacuole. (Reproduced with permission from the Carlsberg Foundation.)
This paper describes the expression of a protein-engineered thermostable (1, 3–1, 4)-β-glucanase with the Hor3–1 D hordein gene promoter in transgenic barley plants obtained by Agrobacterium-mediated transformation. The characteristics of and requirements for the synthesis of the recombinant protein with the endosperm-specific promoter are compared with results obtained with transgenic barley plants expressing the same recombinant protein with an aleurone-specific α-amylase promoter.
Materials and Methods
Plasmid Constructions.
Methods used for PCR and DNA manipulations were as described (20). Plasmids pJH 270 (14.943 kb), pJH 271 (15.003 kb), pJH 276 (14.943 kb) and pJH277 (15.003 kb) were constructed in the binary cloning vector pJH2600, which is derived from pBIN 19 (21). Plasmid pJH2600 was assembled by cloning a HindIII-SmaI fragment containing the bar gene 3′ to the ubiquitin promoter and a SmaI-EcoRI fragment with the nos terminator from pUBARN (10) into HindIII-EcoRI-digested derivative of the pBIN 19 plasmid by a three-way ligation.
(1) pJH270.
Plasmid pHor-H(A12-M)ΔY13-GC-NOS contains a translational fusion between the 434-bp D hordein gene promoter, the ATG initiation codon (19), and the heat-stable codon-optimized (1, 3–1, 4)-β-glucanase gene (9). The D hordein gene promoter fragment was PCR amplified from a Hor3–1 genomic clone (19) by using the universal forward primer (5′-GTAAAACGACGGCCAGTG-3′) and primer dhor3 (5′CGGTCTGCATCTCGGTGGACTGTC-3′). The heat-stable β-glucanase gene was PCR amplified from plasmid pEII-αH(A12-M)ΔY13-GC-NOS (10) by using primer hor/glu (5′-CGAGATGCAGACCGGCGGCAGCTTC-3′) and the M13 reverse primer (5′-GGTTTTCCCAGTCACGAC-3′). These two PCR fragments were spliced together by overlap extension (SOE) (9). The resultant fragment was digested with BamHI and EcoRI and cloned into pUC18 digested with the same two enzymes.
(2) pJH271.
Plasmid pHsig-H(A12-M)ΔY13-GC-NOS is similar to plasmid (1) but also contains the coding sequence for the D hordein signal peptide. The D hordein gene fragment including the promoter, 5′ untranslated leader region, and the signal peptide-coding region was amplified from the Hor3–1 genomic clone (19) by using the universal forward primer and primer B (5′-CGGTCTGAGCGGTGGTGAGAGCCAC-3′). The heat-stable β-glucanase gene was PCR amplified from plasmid pEII-αH(A12-M)ΔY13-GC-NOS by using primer C (5′-CACCGCTCAGACCGGCGGGCAGCTTC-3′) and the M13 reverse primer. The two PCR fragments were spliced together by SOE and cloned into pUC18.
(3) pJH276.
Plasmid pHor-H(A12-M)ΔY13-NOS contains a translational fusion between the 437-bp promoter + initiator codon fragment of the D hordein gene and the heat-stable codon-unmodified, (1, 3–1, 4)-β-glucanase gene. The fragment was amplified with primer hor3′d13 (5′-CTGTTTGCATCTCGGTGGACTGT CA-3′) and the universal forward primer. The heat-stable β-glucanase gene was PCR amplified from plasmid pEII-ATG-H(A12-M)ΔY13-NOS with primer hor/d13 (5′-CGAGATGCAAACAGGCGGATCGTT T-3′) and the M13 reverse primer. The two PCR fragments were spliced by SOE and cloned in pUC18.
(4) pJH 277.
Plasmid pHsig-H(A12-M)ΔY13-NOS is similar to pHor-H(A12-M)ΔY13-NOS, except that it contains the coding sequence for the D hordein signal peptide. The four resultant plasmids were sequenced for the entire coding and the D hordein promoter regions. The translational fusions were confirmed, and no nucleotide alteration was found.
The third component of plasmids pJH 270, 271, 276, and 277 is the synthetic mutant green fluorescent protein gene (sGFP) (ref. 22; plasmid no. 5) under the control of the CaMV 35S promoter and the nos terminator. Plasmid pUC19-35S-SGFP-TYG-NOS was constructed by cloning a BamHI-EcoRI fragment containing the sGFP-TYG gene and the nos terminator into BamHI and EcoRI digested pBI221 (Clontech), 3′ to the 35S promoter. In a three-way ligation, the EcoRI-HindIII fragment from pHor-H(A12-M)ΔY13-GC-NOS and the EcoRI-HindIII fragment from pUC19-35S-SGFP-TYG-NOS were inserted into HindIII-digested pJH2600 to give pJH270. pJH271 (Fig. 2) was correspondingly constructed by using the EcoRI-HindIII fragment from pHsig-H(A12-M)ΔY13-GC-NOS, pJH276 by using the EcoRI-HindIII fragment from plasmid pHor-H(A12-M)ΔY13-NOS and pJH277 by using the EcoRI-HindIII fragment from pHsig-H(A12-M)ΔY13-NOS.
Figure 2.
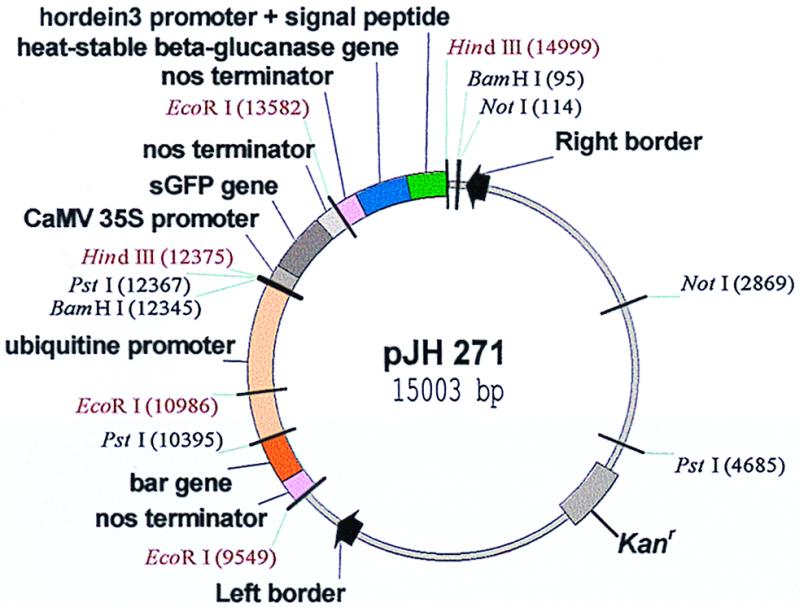
Agrobacterium vector pJH271 for expression of the protein-engineered heat-stable (1, 3–1, 4)-β-glucanase during development of the barley endosperm and its deposition in the protein bodies. The β-glucanase gene is codon optimized to a G+C content of 63%. The gene for the synthetic green fluorescent protein (sGFP) is under the control of the 35S promoter, and the bar gene is driven by the maize ubiquitin (pUbi1) promoter and the gene's first intron.
Barley Transformation.
Transgenic plants of the cultivar Golden Promise were produced by cocultivation of immature zygotic embryos with Agrobacterium tumefaciens strains AGL-1 carrying a disarmed Ti plasmid and one of the plasmids pJH270, 271, 276, or 277 essentially according to ref. 8 and described in detail in ref. 23. The transgenic barley plants obtained with the biolistic method have been described in refs. 9 and 10. Southern hybridizations and Western Blot analyses were carried out according to the procedures detailed in ref. 23.
Heat-Stable (1, 3–1, 4)-β-Glucanase Assays.
Identification of homozygous or heterozygous transgenic plants producing this enzyme are identified in the following way: grains are germinated for 3 days, heated for 2 hr at 65°C to inactivate the endogenous barley enzyme, cut in half, and placed endosperm-side down on Murashige–Skoog (24) medium containing 2 gL−1 lichenan [(1, 3–1, 4)-β-glucan]. The β-glucanase diffuses out of the endosperm into the medium and degrades the lichenan. The zymogram is developed after a few hours with Congo red, revealing degradation of the substrate as an unstained zone around the grain (Fig. 3). The amount of enzyme produced in transgenic grains is determined in extracts from mature or germinated grains with the method of McCleary (25) as described in refs. 9, 10.
Figure 3.
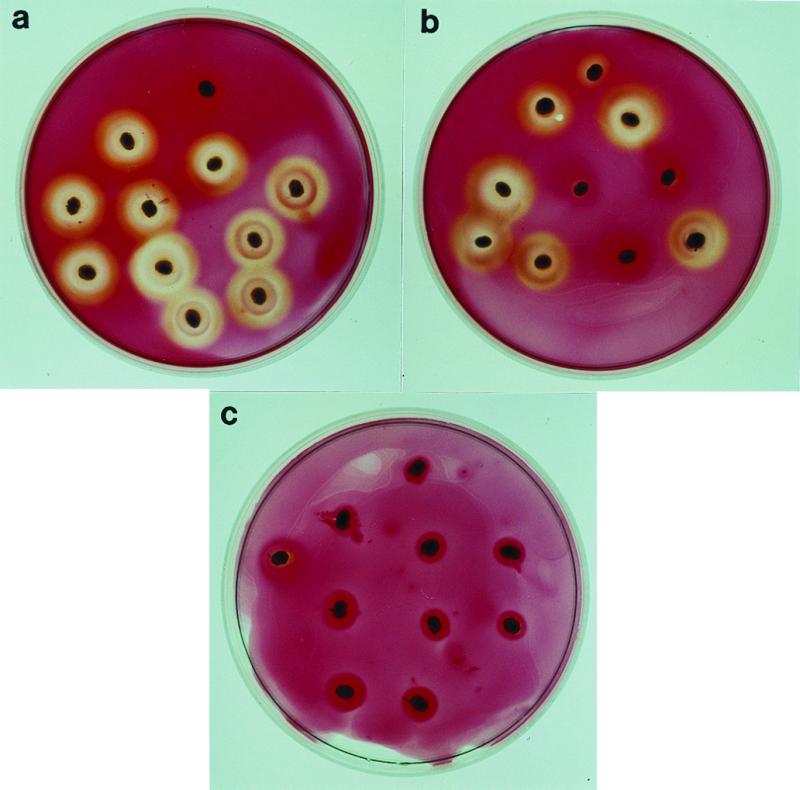
Zymogram assay for identification of homozygous and heterozygous transgenic plants producing heat-stable β-glucanase. Germinated and heated half grains are placed on lichenan-containing agar plates. Congo red binds to lichenan but not to its depolymerized saccharides. (a) Germlings of a homozygous transgenic plant, (b) of a heterozygous transgenic plant, and (c) of a nontransgenic segregant in the F2 of the cross transgenic line 5607 × Ca 803111(ant-499(Apex) × Alexis). An untransformed germling is placed at the top of each plate.
Results
The Production of Recombinant (1, 3–1, 4)-β-Glucanase in Developing vs. Germinating Barley Grains.
Four different types of transformants were generated with plasmids pJH 271 (Fig. 2), pJH 270, pJH 277, and pJH 276. The ORF of the mature heat-stable hybrid (1, 3–1, 4)-β-glucanase gene in plasmid 271 is codon optimized to a G+C content of 63%, fused to the code for the D hordein signal peptide comprising 21 amino acids (MAKRLVLFVAVIVALVALTTA), and provided with the 437-bp Hor3–1 promoter. A CpG island in this promoter prevents the requirement of an endosperm-specific unmethylated state, which has to be established during endosperm development in the case of the B and C hordein gene promoters. The Hor3–1 promoter is 3- to 5-fold more active than the latter promoters (19). Ten transformants were regenerated after cocultivation with Agrobacterium and mature T1 grains analyzed for activity of heat-stable (1, 3–1, 4)-β-glucanase. Recombinant enzyme of up to 40 μg⋅mg−1 soluble protein was obtained in individual kernels (Fig. 4). When samples of five to six segregating T1 grains were germinated for 5 days, amounts of active recombinant enzyme ranging from 6.8 to 24.4 μg⋅mg−1 soluble protein were measured.
Figure 4.

Expression of recombinant heat-stable (1, 3–1, 4)-β-glucanase under the control of the D hordein gene (Hor3) promoter in individual T1 grains. For the codon optimized gene, two individual grains were analyzed, and values are given. The codon optimized gene and a signal peptide for targeting the recombinant protein into endosperm storage protein vacuoles are required for optimal production.
In the absence of the Hor3 signal peptide (pJH 270), localization of the recombinant enzyme is expected in the cytosol instead of the protein bodies. Much lower amounts of recombinant enzyme (2–5 μg⋅mg−1 soluble protein) were present in the mature grains of transformants with the gene lacking the signal peptide code. It remains to be determined whether this is caused by a decreased synthesis or by proteolytic degradation during programmed cell death in the last stages of grain maturation. The latter seems to be the case in transgenic barley grains expressing the uidA gene with the D or B1 hordein gene promoter without a signal peptide code, because it is reported that the GUS activities per milligram protein declined during grain filling from 3,800 pmol⋅min−1 at 10–20 days after fertilization to 280 pmol⋅min−1 at maturity (26). Barley transformed with the noncodon-optimized ORF produced small amounts of recombinant enzyme in the mature endosperm when it was synthesized as a precursor with the N-terminal signal peptide. In the absence of the signal peptide, no heat-stable glucanase accumulated (Fig. 4).
The segregation of the transgene in nine T1 lines comprising 185 plants was analyzed with the β-glucanase assay (Fig. 3) and the phosphinotricin acetyl transferase assay (27). All lines segregated 1:2:1 for homozygous transgenes, heterozygotes, and nontransgenes with χ2 values ranging from 0.74 to 6.3 (P≈0.6 to 0.05). This corresponds thus to single-locus mendelian ratios. The amount of recombinant enzyme measured in the grains of 34 homozygous transgenic plants is presented in Table 1. The 10 independent transgenic lines synthesized similar amounts (53.8 ± 2.8 μg⋅mg−1 soluble protein) of heat-stable (1, 3–1, 4)-β-glucanase. This is 40 times more than the maximum amount of recombinant enzyme synthesized during germination with the α-amylase promoter (Fig. 5). It has been previously estimated that 0.3 to 1 μg of recombinant (1, 3–1, 4)-β-glucanase could be obtained per grain or 20 to 40 mg per kilogram of malted grain. It thus seems not unreasonable that it will be possible to synthesize with the D hordein gene promoter 1–1.5 g recombinant protein per kilogram of barley grain. The recombinant enzyme synthesized during grain development was present in the mature grain and remained 100% active at day 3 and at day 5 of the hydrolytic depolymerization of the endosperm content during germination.
Table 1.
Expression of recombinant (1,3-1,4)-β-glucanase in T2-grains of homozygous plants from transgenic lines Hor,SP (A12-ΔY13)CG 271.0.1-10 obtained with vector pJH271
| Transgenic lines | Enzyme, μg⋅mg−1 soluble protein | x̄ ± error of the mean |
|---|---|---|
| 99-271.0.1 | 73, 60, 67, 17, 29 | 49.2 ± 11.1 |
| 0.2 | 99, 54 | 76.5 ± 22.5 |
| 0.3 | 25, 51, 51, 20 | 36.8 ± 8.3 |
| 0.4 | 79, 74, 62 | 71.7 ± 5.1 |
| 0.5 | 53, 51 | 52.0 ± 2.9 |
| 0.6 | 75, 41, 38, 68 | 55.5 ± 9.4 |
| 0.7 | 81, 56, 23, 26 | 46.5 ± 13.7 |
| 0.8 | 25, 36, 63, 59 | 45.8 ± 2.9 |
| 0.10 | 69, 57, 65, 58, 56, 69 | 62.3 ± 2.5 |
| 53.8 ± 2.8 |
Figure 5.

Expression of recombinant heat-stable (1, 3–1, 4)-β-glucanase under the control of a barley high pI α-amylase gene promoter in nine different transgenic lines grown in the field in 3 successive years. GP, Golden Promise.
Glycosylation of the Recombinant Enzyme.
Two of the three N-glycosylation sequons of the heat-stable hybrid (1, 3–1, 4)-β-glucanases H(A16-M) and H(A12-M)ΔY13 are glycosylated to different degrees when the protein is expressed in yeast (28, 29) or in barley aleurone protoplasts (9). It was therefore of interest to see whether the H(A12-M) ΔY13 protein is glycosylated when targeted via the ER and Golgi apparatus into the protein bodies of the endosperm. N-glycosylation is expected on the protein synthesized without the signal peptide. In Fig. 6 A and B, the molecular mass of the hybrid (1, 3–1, 4)-β-glucanase synthesized with the hordein gene promoter and signal peptide (lanes 2–4) is compared with that of the unglycosylated recombinant enzyme made in Escherichia coli (P, Mr = 24 kDa) and with the recombinant enzyme expressed with the α-amylase promoter in aleurone tissue (lane 1). The Western blot analysis with the heat-stable β-glucanase-specific antibody reveals the higher molecular mass isoforms (Mr 28 = kDa) of the glycosylated enzyme synthesized with the α-amylase promoter in the germinating transgenic barley grains (lane 1) as well as those produced with the D hordein gene promoter and signal peptide code in the developing endosperm (lanes 2–4). A slower migrating glycosylated isoform can be seen in the Coomassie blue-stained gel (Fig. 6A, arrow). The recombinant heat-stable (1, 3–1, 4)-β-glucanase synthesized without the signal peptide in the developing grains is not glycosylated because it migrates in the SDS gel like the purified enzyme expressed in E. coli (Fig. 6C, sharp stained narrow double band; D, single antibody decorated band). In contrast, the enzyme produced as a precursor with the signal peptide shows the slower migrating mature glycosylated isoforms (Fig. 6 E–F).
Figure 6.

Identification of heat-stable (1, 3–1, 4)-β-glucanase synthesized with the D hordein gene promoter in maturing grains and with an α-amylase promoter in germinating grains. GP, extract of untransformed Golden Promise; M, molecular marker; P, purified unglycosylated enzyme synthesized in E. coli; Mr, 24 kDa. (A) SDS/PAGE and Coomassie blue staining; (B) Western blot decorated with specific antibody and visualized by chemiluminescence. Lanes: 1, extract of germinating grains expressing the enzyme with the α-amylase promoter; 2–4, extracts of germinating grain containing the recombinant enzyme produced with the Hor3 promoter during grain maturation. The larger apparent molecular mass Mr 28 = kDa) of the barley-produced enzyme is because of N-glycosylation. (C—F) Comparison of the recombinant heat-stable (1, 3–1, 4)-β-glucanase synthesized with the D hordein gene promoter in developing grains with or without the signal peptide. (C and E): SDS/PAGE/Coomassie blue staining; and (D and F) Western blot of extracts containing recombinant enzyme synthesized in transformants with signal peptide (E and F, lanes 1–6) and in transformants without it (C and D, lanes 1–4). The recombinant enzyme synthesized without the signal peptide has an apparent molecular weight characteristic of the unglycosylated protein.
Southern Blot Analysis.
In Fig. 7, the hybridization patterns of DNA isolated from T1 seedlings of 10 transformants obtained with the Agrobacterium vector pJH271 (Fig. 2) are presented. The DNA was cut with the EcoRI restriction enzyme and probed with the 620-bp long coding region of the heat-stable (1, 3–1, 4)-β-glucanase lacking an EcoRI restriction site. The vector contains an EcoRI restriction site in the nos terminator of the transgene. The hybridizing restriction fragments of the blot are therefore bordered at one end by this EcoRI site and at the other end by a site in the chromosome near the place of insertion. The different sized bands thus indicate the number of insertions. In the present transformants, they range from one to four copies of the transgene. The EcoRI digest was also probed with the 569-bp coding region of the bar gene identifying in all transformants a single hybridizing fragment with the expected size of 1.43 kb (data not shown).
Figure 7.

Southern blot analysis of T1 seedlings transgenic for the heat-stable (1, 3–1, 4)-β-glucanase (lanes 5–9 and 10–14). The transgenic plants were obtained by Agrobacterium-mediated transformation by using vector pJH271 with the transgene driven by the Hor3 promoter and translation effected on the ER with the D hordein signal peptide. Ten micrograms of genomic DNA was digested with EcoRI and probed with the digoxigenin-labeled coding region of the heat-stable (1, 3–1, 4)-β-glucanase (620 bp). Lanes: 1, 2, 3 = 20, 4, and 2 copies of the plasmid cut with EcoRI (10.2 kb); 4, Golden Promise; M, molecular marker.
Discussion
We used a protein-engineered heat-stable bacterial (1, 3–1, 4)-β-glucanase as a test protein and found that two conditions were required to obtain a high yield of the recombinant protein: (i) The gene had to be codon optimized to a C+G content of 63%, and (ii) the protein had to be synthesized as a precursor with a signal peptide that will likely deliver it to storage vacuoles in a manner similar to D hordein. The requirement for codon optimization is surprising, because genes for cereal prolamins are generally not especially C+G rich. The C+G content of the promoter of the Hor2–4 gene, a member of the gene family encoding B-hordeins, ranges from 35 to 45% (200-bp window) and that along the ORF from 35 to 52% (19). The promoters of the B and C hordein genes are hypermethylated in the leaf tissues but hypomethylated in the developing endosperm. The high-lysine mutation lys3a prevents the establishment of the hypomethylated state in the endosperm (19), and this results in very low transcription rates for the B and C hordein genes as well as several other genes. The nucleotide sequence of the D hordein gene promoter and the 5′ part of the coding region has characteristics of CpG dinucleotide islands and differs in this respect from the other hordein gene promoters. Analysis of the methylation pattern by genome sequencing revealed that the D hordein gene promoter remains hypomethylated in the leaf tissues as well as in the developing endosperm. Consequently, transcription and translation of the D hordein gene are not curtailed in the lys3a mutation. It will be of interest to determine whether codon optimization is a requirement for gene expression with the B, C, and γ hordein promoters.
Although direct demonstration of the localization of the recombinant (1, 3–1, 4)-β-glucanase in the protein bodies has to be undertaken, the requirement of the signal peptide for a high content of the recombinant protein in the mature grain and its retention in the endosperm of the germinating grain indicates deposition of value-adding proteins in the protein-storage bodies. Targeting the protein into storage vacuoles can provide protection against the extensive proteolytic action that eliminates a large part of the cellular components of the cereal endosperm during the programmed cell death at the final stages of grain maturation. The nucleotide sequence of the coding region for the D hordein gene is 71–79% identical to the high molecular weight (HMW) glutenin genes of wheat, whereas the homology is 85–89% in the promoter region. The close relationship among these genes and the similarity in their tissue-specific expression patterns in wheat and barley endosperms are underscored by the success of transgenic expression of the 1Ax1 and hybrid Dy10:Dx5 wheat genes for HMW glutenin subunits in the cultivar Bob White (30, 31). The transgenic wheat plants synthesized up to 2.3% of the total protein in the endosperm as 1Ax1 glutenin subunit, providing as much as a 71% increase in the glutenin class of prolamins, which are known to be beneficial for dough elasticity and bread-making quality.
In the absence of the signal peptide, only small amounts of the recombinant protein are present in the mature grain, and this may be caused by the synthesis of the recombinant protein on cytosolic ribosomes. Deposition in the cytosol might make it sensitive to proteolytic degradation during the final stages of grain maturation.
The B, C, and γ hordein polypeptides are stored in different parts of the protein bodies (5). These polypeptides are encoded by families of genes that are linked in gene clusters, with each member having its own promoter. These promoters provide additional opportunities for the expression of recombinant proteins in the developing endosperm. In rice, the gene promoters for the endosperm storage proteins glutelin, albumin, globulin, and prolamin direct the expression of β-glucuronidase in the developing endosperm of transgenic plants 12–15 days after flowering (32), and thus are good candidates for expressing recombinant proteins in this grain.
The Southern blots of the transformants here investigated reveal multiple integration sites with one to four copies of the transgene (Fig. 7). Multiple-copy integrants are observed commonly with cereal transgenic plants (7, 8, 23, 33). The multiple bands hybridizing in most Southern blots are caused by host DNA interspersed among the transgene copies at the integration locus (34, 35). The transgenic barley plants, which have been field tested, show reduced 1,000-grain weight and somewhat reduced growth rates (23). These defects can be overcome by relocating the transgenes into other genotypes. As recently pointed out by Srivastava, Anderson, and Ow (33), it is important to obtain single-copy transgenic cereal plants at integration sites that do not inactivate an essential gene in the crop plant. Furthermore, it is desirable to eliminate the selective marker to allow subsequent transformation of a successful transgenic plant. Srivastava and coworkers have used the site-specific 38-kDa Cre recombinase with two alleles of the 34 bp lox recognition sequence to resolve in wheat transgenic multicopy loci into single-copy transgenes while eliminating the bar selective marker. A combination of this transformation/selection strategy with the use of the efficient storage protein gene promoters and signal peptide codes will constitute a powerful expression system for recombinant proteins in cereal grains, irrespective of whether the protein is to be isolated and purified or used in the form of whole grains, flour, malt, or malt syrups.
The barley (1, 3–1, 4)-β-glucanase (isoenzyme II) depolymerizes during germination the mixed-linked β-glucans of the endosperm cell walls. This has to take place before α-amylases and proteases can reach and convert the stored starch and proteins into sugars and amino acids to be used as nutrients by the growing germling. The high amount of (1, 3–1, 4)-β-glucans present in the endosperm walls is the reason why barley is considered a low-metabolic energy feed grain and therefore is an unacceptable component in chicken feed for poultry. The birds do not produce an enzyme in their gastrointestinal tract to depolymerize the (1, 3–1, 4)-β-glucans, and this leads to a high viscosity of the feed in the intestine, a limited uptake of nutrients, and sticky droppings adhering to the chickens and the floors of the production cages (36, 37). Inclusion of fungal enzyme preparations or purified (1, 3–1, 4)-β-glucanase from Bacillus amyloliquefaciens to barley diets can overcome the nutritional deficiencies of barley grain and prevent the occurrence of sticky droppings (38, 39). The increased thermostability of the hybrid enzyme expressed in the transgenic barley grains has a half-life at 70°C of more than 4 hr compared with the half-life of 6 min for the parent enzymes (40). This increased thermostability allows it to survive the heat generated by pressing barley into feed pellets and pasteurization of the feed required to prevent infection of the chicks by Salmonella typhimurium. In processing barley grain material, it was found that addition of 20 μg of the H(A12-M)Δ Y13 enzyme is sufficient for complete depolymerization of 5–10 g soluble barley (1, 3–1, 4)-β-glucan from 1 kg of malt containing high levels of the polymer (10). Thus enzyme in ≈1 g (40 grains) of transgenic malt of the high-enzyme producing lines in Fig. 5 is expected to depolymerize the soluble β-glucan of 1 kg (≈25,000 grains) of nontransgenic “normal” barley, whereas 200 μg enzyme or 10 g of transgenic malt will depolymerize the 50 g of water-soluble and insoluble (1, 3–1, 4)-β-glucan of 1 kg of normal barley cultivars. Chicken-feeding trials to verify these predictions have been initiated. When the transgenic lines described in this communication have been field propagated, mature grain containing 40 times more recombinant enzyme than is present in current transgenic malt will be available for use as a supplement in broiler chicken feed barley. This will convert barley into a high-metabolic feed grain.
Acknowledgments
Financial support by Cooperative State Research, Education and Extension Service/National Research Initiative Competitive Grants Program (grants nos. 9502390 and 9701248), Applied Phytologics, Sacramento, CA, and the Washington Technology Center (grant no. 99 B-1) is gratefully acknowledged. This is scientific paper CSS 0011-15 from the College of Agriculture and Home Economics Research Center, Washington State University, Pullman, WA.
Abbreviation
- ER
endoplasmic reticulum
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030527497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030527497
References
- 1.Newman C W, Newman R K. In: Barley: Genetics, Biochemistry, Molecular Biology and Biotechnology. Shewry P R, editor. Wallingford, Oxon, U.K.: C.A.B.; 1992. pp. 351–368. [Google Scholar]
- 2.Briggs D E. In: Barley: Genetics, Biochemistry, Molecular Biology and Biotechnology. Shewry P R, editor. Wallingford: C.A.B.; 1992. pp. 369–401. [Google Scholar]
- 3.Kreis M, Shewry P R. In: Barley: Genetics, Biochemistry, Molecular Biology and Biotechnology, Shewry P R, editor. Wallingford: C.A.B.; 1992. pp. 319–333. [Google Scholar]
- 4.Rechinger K B, Bougri O V, Cameron-Mills V. Theor Appl Genet. 1993;85:829–840. doi: 10.1007/BF00225026. [DOI] [PubMed] [Google Scholar]
- 5.Rechinger K B, Simpson D J, Svendsen I, Cameron-Mills V. Plant J. 1993;4:841–853. doi: 10.1046/j.1365-313x.1993.04050841.x. [DOI] [PubMed] [Google Scholar]
- 6.Møgelsvang S, Simpson D J. J Plant Physiol. 1998;153:1–15. [Google Scholar]
- 7.Wan Y, Lemaux P G. Plant Physiol. 1994;104:37–48. doi: 10.1104/pp.104.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tingay S, McElroy D, Kalla R, Fieg S, Wang M, Thornton S, Bretell R. Plant J. 1997;11:1369–1376. [Google Scholar]
- 9.Jensen L G, Olsen O, Kops O, Wolf N, Thomsen K K, von Wettstein D. Proc Natl Acad Sci USA. 1996;93:3487–3491. doi: 10.1073/pnas.93.8.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jensen L G, Politz O, Olsen O, Thomsen K K, von Wettstein D. Hereditas. 1998;129:215–225. [Google Scholar]
- 11.Jones R L, Jacobsen J V. Int Rev Cytol. 1991;126:49–88. doi: 10.1016/s0074-7696(08)60682-8. [DOI] [PubMed] [Google Scholar]
- 12.Fincher G B. In: Barley: Genetics, Biochemistry, Molecular Biology and Biotechnology. Shewry P R, editor. Wallingford: C.A.B.; 1992. pp. 413–437. [Google Scholar]
- 13.Sørensen M B, Cameron-Mills V, Brandt A. Mol Gen Genet. 1989;217:195–201. [Google Scholar]
- 14.von Wettstein D. Proc. Europ. Brewery Convent. 17. Congr. Berlin 1979. 1979. pp. 587–629. [Google Scholar]
- 15.Cameron-Mills V, Ingversen J, Brandt A. Carlsberg Res Commun. 1978;43:91–102. [Google Scholar]
- 16.Cameron-Mills V, Madrid S M. Carlsberg Res Commun. 1989;54:181–192. [Google Scholar]
- 17.Cameron-Mills V. Carlsberg Res Commun. 1980;45:557–576. [Google Scholar]
- 18.Cameron-Mills V, von Wettstein D. Carlsberg Res Commun. 1980;45:577–594. [Google Scholar]
- 19.Sørensen M B, Müller M, Skerritt J, Simpson D. Mol Gen Genet. 1996;250:750–760. doi: 10.1007/BF02172987. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 21.Bevan M. Nucleic Acids Res. 1984;12:8711–8721. doi: 10.1093/nar/12.22.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu W L, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J. Curr Biol. 1996;6:325–330. doi: 10.1016/s0960-9822(02)00483-9. [DOI] [PubMed] [Google Scholar]
- 23.Horvath, H., Huang, J., Wong, O. T. & von Wettstein, D. (1999) J. Crop Prod., in press.
- 24.Murashige T, Skoog F. Physiol Plant. 1962;15:473–597. [Google Scholar]
- 25.McCleary B V. Methods Enzymol. 1988;160:74–86. [Google Scholar]
- 26.Cho M J, Choi H W, Buchanan B B, Lemaux P G. Theor Appl Genet. 1999;98:1253–1263. [Google Scholar]
- 27.Kramer C, DiMaio J, Carswell G K, Shillito R D. Planta. 1993;190:454–458. [Google Scholar]
- 28.Meldgaard M, Svendsen I. Microbiology. 1994;140:159–166. doi: 10.1099/13500872-140-1-159. [DOI] [PubMed] [Google Scholar]
- 29.Meldgaard M, Harthill J, Petersen B, Olsen O. Glycoconjugate J. 1995;12:380–390. doi: 10.1007/BF00731341. [DOI] [PubMed] [Google Scholar]
- 30.Altpeter F, Vasil V, Srivastava V, Vasil I K. Nat Biotechnol. 1996;14:1155–1159. doi: 10.1038/nbt0996-1155. [DOI] [PubMed] [Google Scholar]
- 31.Blechl A E, Anderson O D. Nat Biotechnol. 1996;14:875–879. doi: 10.1038/nbt0796-875. [DOI] [PubMed] [Google Scholar]
- 32.Wu C-Y, Adachi T, Hatano T, Washida H, Suzuki A, Takaiwa F. Plant Cell Physiol. 1998;39:885–889. doi: 10.1093/oxfordjournals.pcp.a029404. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava V, Anderson O D, Ow D W. Proc Natl Acad Sci USA. 1999;96:11117–11121. doi: 10.1073/pnas.96.20.11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pawlowski W P, Somers D A. Proc Natl Acad Sci USA. 1998;95:12106–12110. doi: 10.1073/pnas.95.21.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohli A, Leech M, Vain P, Laurie D A, Christou P. Proc Natl Acad Sci USA. 1998;95:7203–7208. doi: 10.1073/pnas.95.12.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White W B, Bird H R, Sunde M L, Prentice N, Burger W C, Marlett J A. Poult Sci. 1981;60:1043–1048. doi: 10.3382/ps.0601043. [DOI] [PubMed] [Google Scholar]
- 37.White W B, Bird H R, Sunde M L, Marlett J A, Prentice N A, Burger W C. Poult Sci. 1983;62:853–862. doi: 10.3382/ps.0601043. [DOI] [PubMed] [Google Scholar]
- 38.Jensen L, Fry R E, Allred J B, McGinnis J. Poult Sci. 1957;36:919–921. [Google Scholar]
- 39.Rickes E L, Ham E A, Moscatelli E A, Ott W H. Arch Biochem Biophys. 1962;96:371–375. doi: 10.1016/0003-9861(62)90422-8. [DOI] [PubMed] [Google Scholar]
- 40.Politz O, Simon O, Olsen O. Eur J Biochem. 1993;216:829–834. doi: 10.1111/j.1432-1033.1993.tb18204.x. [DOI] [PubMed] [Google Scholar]