

Abstract
Hydrogen exchange (HX) rates for backbone amide protons of oxidized Pseudomonas aeruginosa cytochrome c551 (Pa-cyt c) have been measured in the presence of low concentrations of the denaturant guanidine hydrochloride. Analysis of the data has allowed identification of submolecular unfolding units known as foldons. The highest energy foldon bears similarity to the proposed folding intermediate for Pa-cyt c. Parallels are seen to the foldons of the structurally homologous horse cyt c, although the heme axial methionine-bearing loop has greater local stability in Pa-cyt c, in accord with previous folding studies. Regions of low local stability are observed to correspond with regions that interact with redox partners, providing a link between foldon properties and function.
Keywords: Cytochrome, Denaturant, Electron Transfer, Heme, Nuclear magnetic resonance, Protein folding
Introduction
Cytochromes c (cyts c) serve as important model systems in the field of protein folding [1, 2]. Analyses of mitochondrial and bacterial monoheme cyt c folding have revealed common intermediates and transition states, leading to the proposal of a common folding mechanism for the cyt c family consistent with a conserved topology (Fig. 1) [2, 3]. At the same time, intriguing differences in the folding properties of monoheme cyts c have been noted. In particular, the ability to displace the heme axial methionine ligand, located on a loop (loop 3) toward the C-terminus of the protein (Fig. 1), is dependent on cyt c species. In horse cytochrome c (h-cyt c), the heme axial Met interaction with Fe(III) is disrupted under mildly denaturing conditions [4, 5] or slightly alkaline pH values [6, 7]. In contrast, disruption of the Met-Fe(III) bond in some small bacterial cyts c requires near complete unfolding of the protein [8, 9]. In line with these results, NMR analysis of loop 3 has revealed that this loop has greater mobility in mitochondrial cyts c (Saccharomyces cerevisiae iso-1-cyt c [10, 11] and h-cyt c [12]) relative to small bacterial cyts c (Pseudomonas aeruginosa cytochrome c551 [13] and Bacillus pasteurii cytochrome c [8, 14, 15]). The differences in unfolding properties and local stability of loop 3 among cyts c will be expressed in differences in their folding energy landscapes [16-18]. On the funnel-shaped energy landscape, high-energy unfolded conformations are found at the top, the native state at the bottom, and intermediates and transition states are represented by local energy minima and maxima.
Fig. 1.
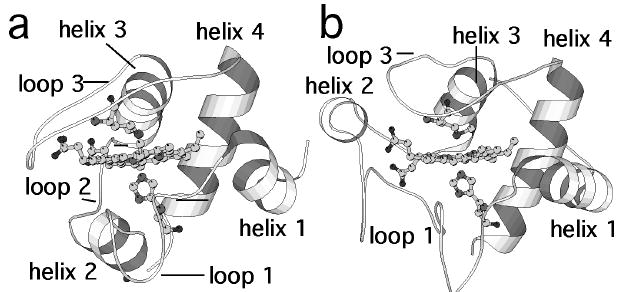
Structures of (a) Pa-cyt c [19] and (b) h-cyt c [20], with substructures indicated as referred to in this work. The axial His and Met ligands are shown in ball-and-stick mode. Figure prepared using Molscript [21].
Hydrogen exchange (HX) is a valuable tool in the elucidation of fleeting partially unfolded conformations of proteins on their energy landscapes [22, 23]. Although there are many protons within a protein that exchange rapidly with solvent, the amide protons of some residues are protected from exchange by participating in hydrogen bonding interactions and/or by being buried inside the protein’s hydrophobic core. Exchange of these protected protons takes place when a conformational fluctuation occurs, exposing the proton to solvent [24]. These opening events range from local fluctuations, in which single hydrogen bonds are broken, to subglobal unfolding events, in which groups of residues unfold, to global unfolding events, in which most or all of the protein unfolds [12, 25]. In the presence of denaturant, the Boltzmann distribution shifts to favor higher energy (more unfolded) conformations; therefore, performing HX on a protein in the presence of denaturant allows for detection of unfolding events that are rare under native conditions. By monitoring HX as a function of denaturant concentration, it is possible to map residues that unfold as a concerted unit, known as a foldon [12]. The foldons of h-cyt c have been identified, and the Met-bearing loop 3 constitutes a low-energy foldon [12, 26], consistent with the ease with which the axial Met-Fe(III) bond is broken in this protein [4, 5].
In this study, foldons of cytochrome c551 from Pseudomonas aeruginosa (Pa-cyt c) are identified through analysis of backbone amide proton HX rates and compared to foldons of h-cyt c (structures are compared in Fig. 1). While the Pa-cyt c foldons show overall similarity to those of h-cyt c, there also are significant differences in the foldon structures and stabilities. Finally, a relationship between foldon properties and the physiological electron transfer functions of h-cyt c and Pa-cyt c is proposed. These results yield detailed information of use in the interpretation of folding data on cyts c, and also provide a link between foldon properties and protein function.
Materials and Methods
Sample preparation
Pa-cyt c was expressed in E. coli and purified as previously described [13, 27]. Preparation of uniformly 15N-labeled Pa-cyt c (U-[15N]-Pa-cyt c) was by expression on minimal medium containing [15N, 99%]NH4Cl (Cambridge Isotope Laboratories, Inc.) as the sole nitrogen source as described [13, 28]. For preparation of NMR samples for HX studies, a 500-uL sample of 2.5 – 3 mM Pa-cyt c or U-[15N]-Pa-cyt c in 50 mM NaPi, pH 6.0, and 5x molar excess K3[Fe(CN)6] in H2O was lyophilized. All experiments were performed on ferric protein.
HX data collection
HX was initiated (time = 0) by adding 500 μl of 0 – 1.4 M NaCl and 0 – 1.4 M GuHCl in D2O to lyophilized Pa-cyt c or U-[15N]-Pa-cyt c, yielding a final protein concentration of 2.5 – 3 mM in 50 mM NaPi. The experiment was performed on protein samples in the presence of 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, and 1.4 M GuHCl; the concentration of NaCl was decreased correspondingly from 1.4 M to 0 M in increments of 0.2 M to maintain a constant ionic strength. The concentration of GuHCl in stock solutions and in samples was determined using refractive index measurement [29]. The pH* (uncorrected pH) of samples immediately after addition of D2O was between 5.5 and 6.0. For samples with pH* < 6.0, microliter amounts of NaOD were used to adjust the pH* to 6.0. One sample of U-[15N]-Pa-cyt c was prepared in 1.4 M GuHCl at pH* = 5.0 to determine whether exchange is well approximated by the EX2 limit.
NMR data were collected on a Varian INOVA 500-MHz spectrometer operating at 499.839 MHz. After HX initiation for the unlabeled protein samples (0 – 0.8 M GuHCl only) consecutive 2-D TOCSY spectra (32 scans, (4096 × 256 points, 1.1-second recycle time, 4.5 hours per spectrum) at 299 K without solvent suppression were collected. Data collection commenced approximately 7 minutes after exchange initiation. When NMR spectra were not being collected, samples were kept at 299 K in a water bath. For U-[15N]-Pa-cyt c, HX was initiated as described above, after which 3 hours of consecutive HSQC spectra (2048 × 32 points, recycle time of 1.5 seconds, total collection time of 5 minutes and 15 seconds) were collected at 299 K. Additional HSQC spectra were taken at approximately 4, 7, 14, and 20 to 24 hours after HX initiation.
HX Data Analysis
NMR spectra were processed using Felix 97 (Accelrys). Resonance assignments were determined using standard methods guided by extensive assignments available in the literature [13, 30]. Peak intensities in TOCSY spectra were normalized using peaks of non-exchangeable protons as references. Cross-peak intensities at various delay times (I(t)) were plotted against delay time (t) and fit using Kaleidagraph (Synergy software) to a three-parameter single-exponential equation to obtain exchange rates (kobs):
| (1) |
In the HX experiment, equilibrium “opening” (allowing exchange) and “closing” events dictate the observed exchange rates:
| (2) |
The protein exchanges within the EX2 limit when the closing rate is much faster than the intrinsic exchange rate (kcl ≫ kint) and the folded protein is stable (kcl ≫ kop) [31]; under these conditions, kobs is directly related to the equilibrium constant for the opening reaction leading to exchange (Kop) according to
| (3) |
Commonly, this relationship is expressed in terms of a protection factor (P), where
| (4) |
The free energy for the opening reaction leading to exchange is given by
| (5) |
where R is the gas constant and T is the absolute temperature. If exchange of a single proton can occur via multiple unfolding events, the unfolding event yielding the most efficient exchange will dominate. The program SPHERE [32, 33] was used to determine values of kint.
For each residue with a measurable protection factor, ΔGHX was plotted against concentration of GuHCl. Some of the plots show curvature, with dependence of ΔGHX on denaturant increasing with concentration. The plots for residues within a common foldon merge into a single “isotherm” at higher denaturant concentrations characterized by a common extrapolated ΔGHX at zero denaturant (ΔGHX,0) and m-value in the linear region of the plot [12]:
| (6) |
The ΔGHX,0 value corresponds to the difference in free energy between the native state and the partially unfolded higher energy conformation in which the foldon of interest is open to allow exchange. The m-values are related to the change in exposed surface area that occurs upon the unfolding event, with larger m-values corresponding to larger changes in exposed surface area [12, 34].
Denaturation monitored by circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy was performed on an Aviv Instruments CD model 202 spectropolarimeter equipped with a jacketed cell holder connected to a circulating water bath. For GuHCl denaturation experiments, protein samples contained 10 μM oxidized Pa-cyt c and varied (0 - 8 M) concentrations of GuHCl in 50 mM sodium phosphate buffer, pH 6.0. A quartz cell with a 0.100-cm path length was used. The pH of each sample was adjusted to 6.0 by the addition of a small amounts of sodium hydroxide or hydrochloric acid solutions to each sample. The denaturant concentration of each sample was determined by refractive index measurement. CD spectra of protein samples (200 - 240 nm) at 298 K as a function of [GuHCl] were recorded with an averaging time of 5 sec and a bandwidth of 1.00 nm. CD spectra of reference samples (buffer with 0 - 8 M GuHCl) were recorded under the same conditions as the protein samples to provide a baseline for subtraction from the spectra of the corresponding protein samples. Protein denaturation was followed by the change of the CD signal at 222 nm. Analysis of the CD denaturation data to determine the free energy of unfolding in the absence of denaturant (ΔGU(H2O)) was by fitting to a 2-state model, as described in detail elsewhere [35]. The resulting denaturation curve and results from the fit are shown in Supporting Fig. S1.
Results
Determination of protection factors in the EX2 limit
Protection factors were determined for 68% of the backbone amide protons of the non-proline residues in Pa-cyt c (Supplementary Material, Table S1). The backbone amide protons of all other residues exchanged too quickly with solvent to measure kobs using the methods employed here. The residues with rapidly exchanging protons were generally found within loops and at the ends of helices, as expected.
The HX experiments were performed at pH 6.0 in the presence of GuHCl concentrations of up to 1.4 M GuHCl. Under these conditions, > 95% of the protein is folded as determined by circular dichroism spectroscopy (Fig. S1), and Pa-cyt c is expected to exchange within the EX2 limit. To verify that exchange indeed is according to the EX2 mechanism, an additional HX experiment was performed at pH 5.0 and 1.4 M GuHCl. As expected for the EX2 limit, observed exchange rates were approximately 10-fold lower than the rates measured at pH 6.0, due to the pH dependence of the intrinsic rates [31] (Supplementary Material: Table S2).
Mapping the foldons of Pseudomonas aeruginosa cytochrome c551
Most residues fall within three major categories of HX behavior in denaturant, determined by their extrapolated ΔGHX,0 and m-values: local fluctuations (small ΔGHX,0, m-value close to 0), subglobal unfolding events (moderate ΔGHX,0 and m-value), or large/global unfolding events (ΔGHX,0 and m-values similar to those for denaturation). Additionally, residues can exchange via local fluctuations or subglobal unfolding events at low concentrations of denaturant, and then via larger unfolding events with increased denaturant. The latter exchange behavior will determine to which foldon the residue belongs (i.e., which isotherm it joins).
Most of the residues in Pa-cyt c could be categorized within five foldons, as summarized in Fig. 2 and Table 1. To facilitate comparison to h-cyt c, the foldons of Pa-cyt c were assigned colors in order of ΔGHX,0 values (red < yellow < green < blue < violet) in analogy to published results on h-cyt c [12, 26]. Following are descriptions of foldon assignments grouped according to the Pa-cyt c substructures.
Fig. 2.
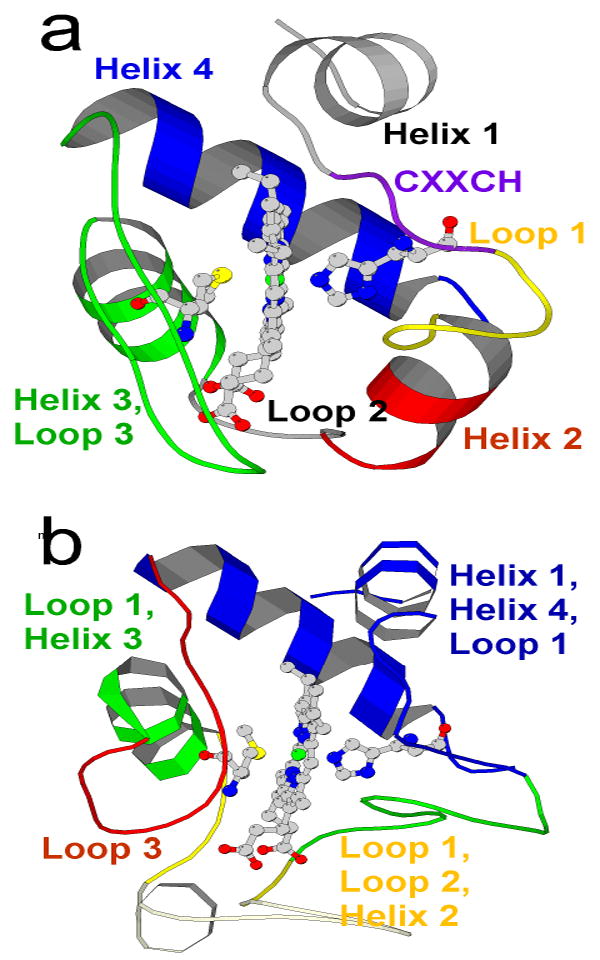
a The proposed foldons of Pa-cyt c; the extrapolated free energies ΔGHX,0 for the foldons are violet > blue > green > yellow > red. Helix 1 and loop 2 (grey) could not be placed into any of the foldons. b Foldons of h-cyt c [12, 26]. The lowest energy h-cyt c foldon is found within the omega loop at the bottom of the molecule as shown and is termed “nested yellow.”
Table 1.
Assignment and Properties of Pa-cyt c foldons
| Structural element | residues | ΔGHX,0 (kJ mol-1) |
m (kJ mol-1 M-1) |
Foldon |
|---|---|---|---|---|
| Helix 1 | 3 - 10 | < 15 | 0 – 2 | Local a |
| CXXCH | 11 - 16 | 36 – 39 | 11 – 13 | Violet |
| Loop 1 | 17 - 26 | 24 – 26 | 6 | Yellow |
| Helix 2 | 27 - 33 | 20 – 24 | 4 – 8 | Red |
| Loop 2 | 34 - 39 | -- b | -- b | Local a |
| Helix 3 | 40 - 49 | 26 – 34 | 11 – 15 | Green |
| Loop 3 | 50 - 67 | 26 – 34 | 11 – 15 | Green |
| Helix 4 | 68 - 80 | 30 – 36 | 12 – 16 | Blue |
Exchange is through local fluctuations; no foldon assigned
Not measured; fast exchange
Helix 1 (residues 3 – 10) and residues undergoing local fluctuations
Residues in helix 1 (Phe7, Lys8, Lys10), as well as Ala35, and Gly54 exchange via local fluctuations at low GuHCl concentrations, and display steady or increasing ΔGHX as denaturant concentration is increased, but exchange too quickly to observe an exchange rate at higher GuHCl concentrations. This behavior precludes the definitive placement of these residues within a foldon based on exchange behavior alone. There are 13 residues (13, 14, 26, 27, 28, 37, 42, 55, 57, 59, 66, 67, and 70) with ΔGHX values that are independent of GuHCl concentration (m < 2 kJ mol-1 M-1), indicating that they exchange via local fluctuations. For comparison to the h-cyt c foldons, the residues in Pa-cyt c that exchange via local fluctuations or those that exchange too quickly to measure an exchange rate were placed into foldons based on the HX behavior of their neighboring residues and the residues within their common secondary structure. This approach was taken in analysis of HX behavior of h-cyt c [12].
Loop 1
Loop 1 is placed in the yellow foldon (Fig. 3, ΔGHX,0 = 24 – 26 kJ mol-1, m = 6 kJ mol-1 M-1). Isotherms for representative residues are shown in Fig. 3a. Ala17 is hydrogen bonded to an internal water molecule, whereas Gly24 is hydrogen bonded to the carbonyl oxygen of Cys15. The relatively large ΔGHX,0 values exhibited by Ala17 and Gly24 may result from their interactions with the stable Cys-X-X-Cys-His heme-binding unit (residues 12-16).
Fig. 3.
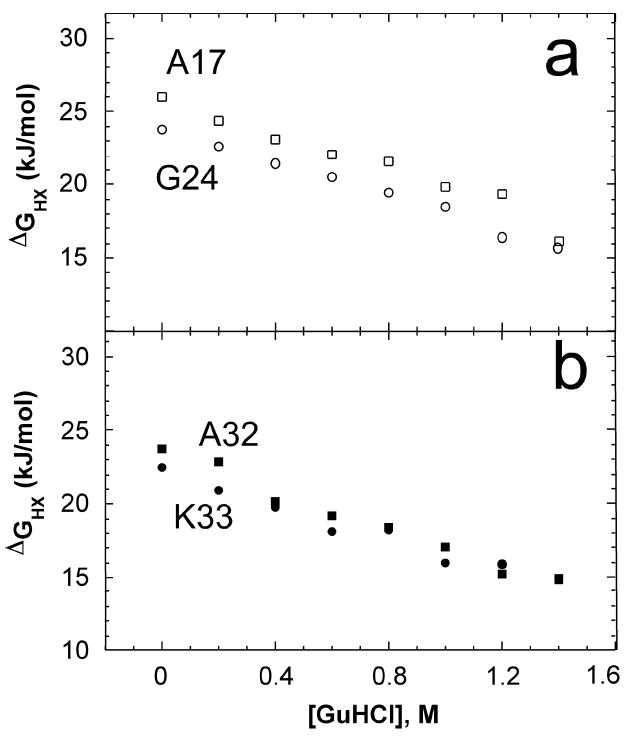
a Plot of ΔGHX vs. [GuHCl] for loop 1 residues Ala17 (□) and Gly24 (○), in the yellow foldon. b Plot of ΔGHX vs. [GuHCl] for helix 2 residues Ala32 ( ) and Lys33 ( ) in the red foldon.
Helix 2 and Loop 2
Helix 2 residues 29 – 33 along with loop 2 residue Phe34 exchange via a relatively low-energy unfolding event with a small m-value (ΔGHX,0 = 20 – 24 kJ mol-1 m = 4 – 8 kJ mol-1 M-1) and are placed in the red foldon (Fig. 3b). Most loop 2 residues exchange too quickly to measure exchange rates, suggesting that at equilibrium, loop 2 exhibits high local mobility. Note that the properties of residues in helix 2 are very similar to those in loop 1. It may be appropriate to consider loop 1, helix 2, and loop 2 to together form one foldon, although they are separated into two here. It also is reasonable to consider loop 2 to correspond to a lower energy foldon analogous to the “nested yellow” foldon of h-cyt c.
Helix 3 and Loop 3
Residues in helix 3 and loop 3 exchange via the same high energy unfolding event (Fig. 4a). Helix 3 residues 44 – 46 exchange via a lower energy unfolding event at low GuHCl concentrations (ΔGHX,0 = 20 – 23 kJ mol-1 m = 3 – 4 kJ mol-1 M-1), but join the isotherms of other residues in helix 3 and loop 3 (notably, axial ligand Met61) at higher concentrations of GuHCl (ΔGHX,0 = 26 – 34 kJ mol-1, m = 11 – 15 kJ mol-1 M-1).
Fig. 4.
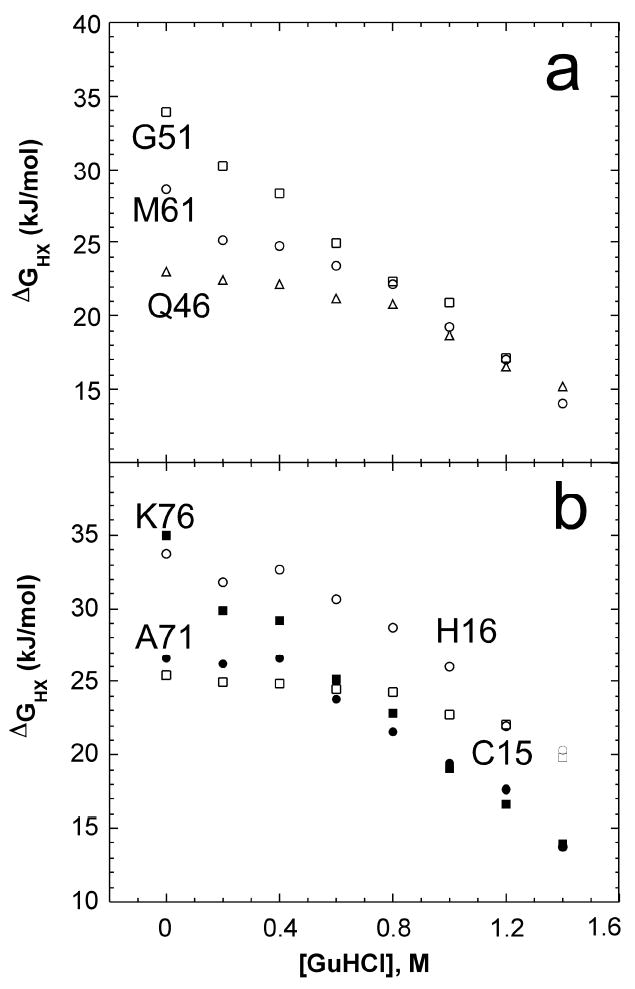
a Plot of ΔGHX vs. [GuHCl] for representative residues in the green foldon: helix 3 residue Gln46 (Δ) and loop 3 residues Gly51 (□) and Met61 (○). b Open symbols: Plot of ΔGHX vs. [GuHCl] for violet foldon residues Cys15 (□) and His16 (○). Filled symbols: Plot of ΔGHX vs. [GuHCl] for blue foldon residues Ala71 ( ) and Lys76 ( ).
Helix 4
With the exception of residues on the ends, helix 4 exhibits high protection from exchange. At lower GuHCl concentrations, Ala71, Gln72, Thr73 and Ser80 exchange via an unfolding event with ΔGHX,0 = 25 – 29 kJ mol-1 and m = 5 – 11 kJ mol-1 M-1. As GuHCl concentration is increased, these residues join the isotherms of the other helix 4 residues Leu74 – 79 which exchange via an unfolding event with a free energy and m-value consistent with global unfolding (ΔGHX,0 = 30 – 36 kJ mol-1, m = 12 – 16 kJ mol-1 M-1). Both types of behavior are represented in Fig. 4b. Notably, the m-values are similar to that determined for GuHCl denaturation monitored by circular dichroism spectroscopy of 13.7 ± 2.7 kJ mol-1 M-1 [35].
The Cys-X-X-Cys-His motif
Cytochromes c are characterized by heme binding to a Cys-X-X-Cys-His motif in which the Cys form thioether bonds with heme and the His serves as a heme ligand (Fig. 5). Cys15 and His16, which hydrogen bond to Cys12 within the motif, exhibit unique HX behavior (Fig. 4b). At low and moderate GuHCl concentrations, both Cys15 and His16 exchange via high energy unfolding events (ΔGHX,0 = 26 and 34 kJ mol-1, respectively) that expose only small amounts of surface area upon unfolding (m = 2, 4 kJ mol-1 M-1, respectively). At high GuHCl concentrations, His16 exchanges via a high energy opening, exhibiting a large ΔGHX,0 (39 kJ mol-1) larger than is seen for helix 4 or for unfolding monitored by other methods. Notably, the isotherms of Cys15 and His16 cross those of blue foldon residues, indicating that even upon unfolding of the highly stable C-terminal helix, the hydrogen bonding interactions within the Cys-X-X-Cys-His motif remain intact. The isotherm of Cys15 joins that of His16 at the highest GuHCl concentrations, suggesting that their hydrogen bonding interactions with Cys12 are lost at high concentrations of GuHCl.
Fig. 5.
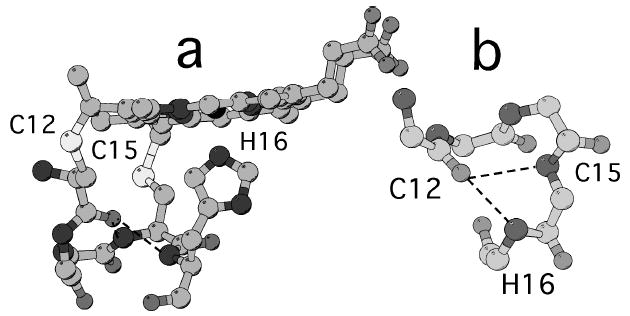
Three-dimensional structure of the Cys-X-X-Cys-His motif in Pa-cyt c [19]. a Heme and residues 12-16, showing His-Fe interaction and structure of the heme-binding peptide. b Backbone atoms of the heme-binding peptide, with intramotif hydrogen bonds from the backbone NH of Cys15 and His16 to Cys12 shown.
Discussion
Comparison of Pa-cyt c and h-cyt c foldons
The foldons of Pa-cyt c and h-cyt c show a number of similarities, in particular for helices (Fig. 2). In both proteins, the C-terminal helix, containing many of the proteins’ hydrophobic core residues, is in the highest energy blue foldon, and helix 3 (residues 60 – 70 in h-cyt c) is in the green foldon. Helix 2 (residues 49 – 54 in h-cyt c) has low local stability and is assigned to the red foldon in Pa-cyt c, and to the lowest energy “nested yellow” foldon in h-cyt c (placed below the red foldon). Helix 1 in h-cyt c is assigned to the blue foldon along with helix 4 which it contacts. Helix 1 is not assigned to a foldon in Pa-cyt c because its exchange rates are too fast to obtain measurements in the presence of denaturant. Notably, helix 1 also displays relatively fast exchange in h-cyt c. It is possible that the shorter length of helix 1 in Pa-cyt c reduces its local stability enough to preclude its identification as part of the blue foldon. Thus it is reasonable to expect that helix 1 unfolds cooperatively with helix 4 in Pa-cyt c as it does in h-cyt c.
The foldons of these two proteins show a number of notable differences. One is that the ΔGHX,0 values in h-cyt c cover a larger range than do the ΔGHX,0 values in Pa-cyt c (Fig. 6). It is possible that the compression of the ΔGHX,0 values in Pa-cyt c results from its being smaller in size than h-cyt c (82 vs. 104 residues) as well as exhibiting a lower global stability [36]. The second major difference concerns the foldon assignment of loop 3, the loop bearing the heme axial Met ligand. In h-cyt c, loop 3 forms the low energy red foldon. Consistent with its low-energy foldon assignment, NMR studies of equilibrium unfolding intermediates of h-cyt c revealed that loop 3 undergoes low energy fluctuations in which the heme axial Met80 is exchanged for one or more Lys residues on loop 3 (positions 72, 73, and 79) under mildly denaturing conditions [4, 5]. In Pa-cyt c, in contrast, displacement of the heme axial Met requires near complete unfolding of the protein [9]. In accord with this observation, the axial Met-bearing loop is in a locally stable region of the protein in Pa-cyt c, falling within the green foldon along with helix 3. The structural basis for this difference may be an increase in the number of interactions between loop 3 and other protein substructures in Pa-cyt c relative to h-cyt c. A similar proposal has been made to explain the local stability of the homologous loop in another small bacterial cyt c [14]. An analysis of hydrogen bonding interactions involving loop 3 reveals substantially more hydrogen bonds between loop 3 residues and other substructures in Pa-cyt c in contrast with h-cyt c loop 3 in which most hydrogen bonds are formed within the loop (Supporting Fig. S2). In addition, the high proline content of loop 3 in Pa-cyt c is expected to contribute to its entropic stabilization.
Fig. 6.
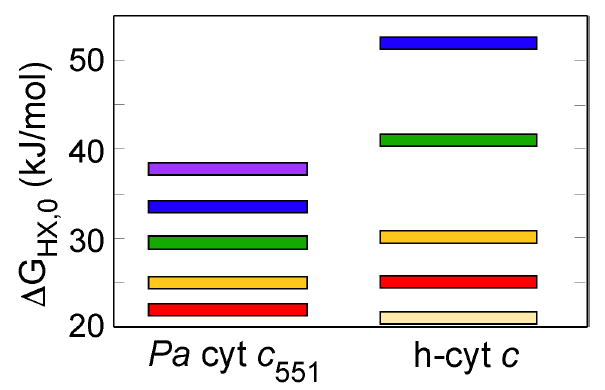
Average ΔGHX,0 values for the foldons in Pa-cyt c (left) and h-cyt c (right) [12, 26], demonstrating how the range of ΔGHX,0 values in h-cyt c is larger than the range of values in Pa-cyt c. The lowest energy foldon on h-cyt c is known as the “nested yellow” foldon.
Relationship to other folding studies: The IN-C state
The compression of the ΔGHX,0 values suggests that Pa-cyt c folds more cooperatively than does h-cyt c. Indeed, the equilibrium folding of Pa-cyt c more closely approximates a two-state transition than does the folding of h-cyt c [36]. In the proposed common folding mechanism for cyts c, helices 1 and 4 dock to form a folding intermediate, IN-C [2], corresponding generally to formation of the blue foldon. Results from kinetic folding experiments on Pa-cyt c suggest that IN-C is high in energy, such that Pa-cyt c does not populate the intermediate to a significant extent, in contrast with h-cyt c. Thus both equilibrium and kinetic folding of Pa-cyt c more closely approximates a 2-state process, whereas an intermediate is readily detected in equilibrium and kinetic studies of h-cyt c folding [37].
In Pa-cyt c, helix 1 residues display relatively rapid exchange in the presence of GuHCl and could not be assigned to a foldon. Despite the rapid exchange behavior of helix 1, it is possible that this relatively unstable helix in Pa-cyt c docks with helix 4 as part of the “blue foldon” corresponding to IN-C. Indeed, the local instability of helix 1 may contribute to the high energy of IN-C in Pa-cyt c relative to h-cyt c. In wild-type Pa-cyt c, helix 1 contains a kink at residue 5 (Val), which results in an unusually long hydrogen bond between Glu4 and Lys8 [19]. A Pa-cyt c mutation, F7A, has been shown to remove the helix 1 kink and to stabilize the native protein as well as the IN-C state, facilitating detection of IN-C [38]. It is expected that HX rates in helix 1 of this mutant would be significantly decreased. Thus foldon energy compression and local instability of helix 1 observed in this work are consistent with previous studies of Pa-cyt c folding, and provide further support for the assignment of the structure of IN-C.
Also of note, the presence of even low concentrations of GuHCl has been shown to affect the folding pathway of Pa-cyt c by masking the stabilizing effects of a salt bridge between two residues in helix 1 (Lys 10) and helix 4 (Glu 70) [39, 40]. This salt bridge has been shown to be an important factor in the formation of the IN-C intermediate [2]. Therefore, the use of non-denaturing concentrations of GuHCl, while not affecting the global stability of Pa-cyt c, may indeed have some effect on the stability of IN-C intermediate state.
Loop flexibility and the electron transfer partner binding site
The mitochondrial cyts c serve as electron donors to cytochrome c peroxidase and cytochrome c oxidase. Structural, kinetic, and mutational studies have implicated loop 3 of mitochondrial cyts c as the vital structural element in these interactions. More specifically, the Lys residues on loop 3 (72, 73, 86 and 87) make key electrostatic contacts with cyt c peroxidase in the crystal structure of the complex [41]; results of kinetic experiments confirm the functional importance of loop 3 in this interaction [42]. Analyses of the binding of h-cyt c to cyt c oxidase also implicate the positively charged patch of Lys residues in the binding interface [43, 44]. Whereas charged residues on loop 3 constitute the “hot spot” for protein-protein interactions involving the mitochondrial cyts c, Pa-cyt c interacts with its redox partners via a hydrophobic patch at the heme edge centering around Val23 (loop 1) and Ile59 (loop 3). Mutational studies implicated these two key residues in interactions with redox partners. Mutation of residues 23 and 59 has a large negative impact on the electron self-exchange rate, reactivity against Pa nitrite reductase (the physiological electron acceptor to Pa-cyt c), and the electron transfer rate to Pa azurin [45].
It is notable that the different foldon structures and distributions of low local ΔGHX in h-cyt c and Pa-cyt c correlate with their different “hot spots” for interactions with physiological partners. The electron transfer binding site for each protein is composed of residues with low ΔGHX values. Loop 1 in Pa-cyt c displays medium-to-low energy unfolding events (yellow foldon), and its residues found within the hydrophobic patch implicated in protein-protein interactions exchange by local fluctuations (Val13, Ala14) or show fast (not measured in these experiments) exchange (Gly11, Met22, Val23). Although loop 3 as a whole in Pa-cyt c is part of the higher-energy green foldon, the non-proline loop 3 residues that are within the interacting hydrophobic patch exchange by local fluctuations (Ile59) or show fast exchange (Ala65). In h-cyt c, the residues implicated in interactions with redox partners are primarily found within the low energy red foldon [4, 12]. As is seen for Pa-cyt c, the key interacting residues show low protection from exchange [10]. Thus, in both proteins, the residues central to protein-protein interactions are characterized by low-energy conformational fluctuations.
In electron transfer reactions between proteins, the protein-protein complex is transient in nature [46]. A conformationally flexible binding site may allow the electron transfer partners to rapidly sample a range of possible orientations of similar energies, a fraction of which are electron-transfer active, in a process known as “dynamic docking” [47]. In addition, a flexible binding site may facilitate rapid dissociation of the complex to allow for efficient turnover [48, 49]. Accordingly, analysis of the dynamics of the blue copper electron transfer protein azurin has revealed conformational exchange of amino acid residues that are implicated in interactions with redox partners [50]. The presence of conformational mobility of the portions of electron transfer proteins that bind redox partners may be a general phenomenon.
The properties of the Cys-X-X-Cys-His motif
The Cys-X-X-Cys-His motif that defines c-type cyts contains the His axial ligand and the two Cys residues that are responsible for covalently linking the heme to the rest of the protein (Fig. 5a). In h-cyt c, residues within the conserved motif display unique HX behavior, exchanging through high energy local fluctuations at all GuHCl concentrations, and exhibiting an isotherm that crosses the blue isotherm believed to be associated with global unfolding [12]. Similar behavior is seen here for Pa-cyt c. Cys15 and His16 hydrogen bond with Cys12 within the Cys-X-X-Cys-His motif (Fig. 5b), thus, exchange of these residues reports on fluctuations within this motif. The isotherm for residues 15 and 16 crosses the blue isotherm, indicating these interactions remain intact even under conditions promoting global unfolding (exchange of helix 4) (Fig. 4). These findings support the proposal that the Cys-X-X-Cys-His motif is a locally stable structure generally in cyts c [51]. Notably, a recent study of HX within this motif suggests that the degree of its flexibility may play a role in tuning heme redox potential by influencing the strength of the His-Fe interaction [52].
Conclusions
Submolecular unfolding units of oxidized Pa-cyt c have been characterized using HX NMR. Broad similarities are seen to h-cyt c, in particular in the behavior of the helices. The axial Met-bearing loop 3, however, shows markedly different behavior in these two proteins with this loop possessing greater local stability in Pa-cyt c relative to h-cyt c. The results reported here are in excellent agreement with previous studies of Pa-cyt c folding, including observation of higher cooperativity in Pa-cyt c folding and higher local stability of loop 3 in comparison with h-cyt c. Finally, the variations in distributions of local stability in these two proteins are proposed to relate to their different sites of interaction with redox partners, providing a link between foldon structure and protein function. The presence of conformational mobility in regions of electron transfer protein important for interactions with redox partners may be a general phenomenon.
Supplementary Material
Acknowledgments
This work is supported by NIH grant R01-GM63170.
Contributor Information
Lea V. Michel, Department of Biochemistry and Biophysics, University of Rochester, Rochester, NY 14642
Kara L. Bren, Department of Chemistry, University of Rochester, Rochester, NY 14627-0216, Tel: 585-275-4335, Fax: 585-276-0205, E-mail: bren@chem.rochester.edu
References
- 1.Winkler JR. Curr Opin Chem Biol. 2004;8:169–174. doi: 10.1016/j.cbpa.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Travaglini-Allocatelli C, Gianni S, Brunori M. Trends Biochem Sci. 2004;29:535–541. doi: 10.1016/j.tibs.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Ptitsyn OB. J Mol Biol. 1998;278:655–666. doi: 10.1006/jmbi.1997.1620. [DOI] [PubMed] [Google Scholar]
- 4.Russell BS, Melenkivitz R, Bren KL. Proc Natl Acad Sci USA. 2000;97:8312–8317. doi: 10.1073/pnas.150239397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell BS, Bren KL. J Biol Inorg Chem. 2002;7:909–916. doi: 10.1007/s00775-002-0381-z. [DOI] [PubMed] [Google Scholar]
- 6.Hong XL, Dixon DW. FEBS Lett. 1989;246:105–108. doi: 10.1016/0014-5793(89)80262-5. [DOI] [PubMed] [Google Scholar]
- 7.Rosell FI, Ferrer JC, Mauk AG. J Am Chem Soc. 1998;120:11234–11245. [Google Scholar]
- 8.Bartalesi L, Bertini I, Ghosh K, Rosato A, Turano P. J Mol Biol. 2002;321:693–701. doi: 10.1016/s0022-2836(02)00678-2. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto Y, Terui N, Tachiiri N, Minakawa K, Matsuo H, Kameda T, Hasegawa J, Sambongi Y, Uchiyama S, Kobayashi Y, Igarashi Y. J Am Chem Soc. 2002;124:11574–11575. doi: 10.1021/ja025597s. [DOI] [PubMed] [Google Scholar]
- 10.Baxter SM, Fetrow JS. Biochemistry. 1999;38:4493–4503. doi: 10.1021/bi982742z. [DOI] [PubMed] [Google Scholar]
- 11.Fetrow JS, Baxter SM. Biochemistry. 1999;38:4480–4492. doi: 10.1021/bi9827417. [DOI] [PubMed] [Google Scholar]
- 12.Bai YW, Sosnick TR, Mayne L, Englander SW. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russell BS, Zhong L, Bigotti MG, Cutruzzolà F, Bren KL. J Biol Inorg Chem. 2003;8:156–166. doi: 10.1007/s00775-002-0401-z. [DOI] [PubMed] [Google Scholar]
- 14.Bartalesi I, Bertini I, Di Rocco G, Ranieri A, Rosato A, Vanarotti M, Vasos PR, Viezzoli MS. J Biol Inorg Chem. 2004;9:600–608. doi: 10.1007/s00775-004-0558-8. [DOI] [PubMed] [Google Scholar]
- 15.Bartalesi I, Rosato A, Zhang W. Biochemistry. 2003;42:10923–10930. doi: 10.1021/bi0348258. [DOI] [PubMed] [Google Scholar]
- 16.Dobson CM, Sali A, Karplus M. Angew Chem-Int Edit Eng. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 17.Dill KA, Chan HS. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 18.Dinner AR, Sali A, Smith LJ, Dobson CM, Karplus M. Trends Biochem Sci. 2000;25:331–339. doi: 10.1016/s0968-0004(00)01610-8. [DOI] [PubMed] [Google Scholar]
- 19.Matsuura Y, Takano T, Dickerson RE. J Mol Biol. 1982;156:389–409. doi: 10.1016/0022-2836(82)90335-7. [DOI] [PubMed] [Google Scholar]
- 20.Bushnell GW, Louie GV, Brayer GD. J Mol Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 21.Kraulis PJ. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 22.Krishna MMG, Hoang L, Lin Y, Englander SW. Methods. 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Weinkam P, Zong CH, Wolynes PG. Proc Natl Acad Sci USA. 2005;102:12401–12406. doi: 10.1073/pnas.0505274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Englander SW, Kallenbach NR. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 25.Maity H, Lim WK, Rumbley JN, Englander SW. Protein Sci. 2003;12:153–160. doi: 10.1110/ps.0225803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maity H, Maity M, Englander SW. J Mol Biol. 2004;343:223–233. doi: 10.1016/j.jmb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Wen X, Bren KL. Inorg Chem. 2005;44:8587–8593. doi: 10.1021/ic050976i. [DOI] [PubMed] [Google Scholar]
- 28.Morar AS, Kakouras D, Young GB, Boyd J, Pielak GJ. J Biol Inorg Chem. 1999;4:220–222. doi: 10.1007/s007750050307. [DOI] [PubMed] [Google Scholar]
- 29.Pace CN, Scholtz JM. In: Protein Structure: A Practical Approach. Creighton TE, editor. IRL; Oxford: 1997. pp. 299–321. [Google Scholar]
- 30.Timkovich R, Cai ML. Biochemistry. 1993;32:11516–11523. doi: 10.1021/bi00094a007. [DOI] [PubMed] [Google Scholar]
- 31.Hvidt A, Nielsen SO. Adv Prot Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 32.Bai YW, Milne JS, Mayne L, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y-Z. University of Pennsylvania 1995 [Google Scholar]
- 34.Myers JK, Pace CN, Scholtz JM. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen X, Patel KM, Russell BS, Bren KL. Biochemistry. 2007;46:2537–2544. doi: 10.1021/bi602380v. [DOI] [PubMed] [Google Scholar]
- 36.Bigotti MG, Allocatelli CT, Staniforth RA, Arese M, Cutruzzolà F, Brunori M. FEBS Lett. 1998;425:385–390. doi: 10.1016/s0014-5793(98)00256-7. [DOI] [PubMed] [Google Scholar]
- 37.Gianni S, Travaglini-Allocateli C, Cutruzzolà F, Brunori M, Shastry MCR, Roder H. J Mol Biol. 2003;330:1145–1152. doi: 10.1016/s0022-2836(03)00689-2. [DOI] [PubMed] [Google Scholar]
- 38.Borgia A, Bonivento D, Travaglini-Allocatelli C, Di Matteo A, Brunori M. J Biol Chem. 2006;281:9331–9336. doi: 10.1074/jbc.M512127200. [DOI] [PubMed] [Google Scholar]
- 39.Gianni S, Brunori M, Travaglini-Allocatelli C. Protein Sci. 2001;10:1685–1688. doi: 10.1110/ps.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gianni S, Travaglini-Allocatelli C, Cutruzzolà F, Bigotti MG, Brunori M. J Mol Biol. 2001;309:1177–1187. doi: 10.1006/jmbi.2001.4728. [DOI] [PubMed] [Google Scholar]
- 41.Pelletier H, Kraut J. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 42.Pearl NM, Jacobson T, Arisa M, Vitello LB, Erman JE. Biochemistry. 2007;46:8263–8272. doi: 10.1021/bi700623u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antalis TM, Palmer G. J Biol Chem. 1982;257:6194–6206. [PubMed] [Google Scholar]
- 44.Lappalainen P, Watmough NJ, Greenwood C, Saraste M. Biochemistry. 1995;34:5824–5830. doi: 10.1021/bi00017a014. [DOI] [PubMed] [Google Scholar]
- 45.Cutruzzola F, Arese M, Ranghino G, van Pouderoyen G, Canters G, Brunori M. J Inorg Biochem. 2002;88:353–361. doi: 10.1016/s0162-0134(01)00390-7. [DOI] [PubMed] [Google Scholar]
- 46.Crowley PB, Ubbink M. Acc Chem Res. 2003;36:723–730. doi: 10.1021/ar0200955. [DOI] [PubMed] [Google Scholar]
- 47.Liang ZX, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. J Am Chem Soc. 2002;124:6849–6859. doi: 10.1021/ja0127032. [DOI] [PubMed] [Google Scholar]
- 48.Crowley PB, Carrondo MA. Proteins. 2004;55:603–612. doi: 10.1002/prot.20043. [DOI] [PubMed] [Google Scholar]
- 49.Prudencio M, Ubbink M. Journal of Molecular Recognition. 2004;17:524–539. doi: 10.1002/jmr.686. [DOI] [PubMed] [Google Scholar]
- 50.Zhuravleva AV, Korzhnev DM, Kupce E, Arseniev AS, Billeter M, Orekhov VY. J Mol Biol. 2004;342:1599–1611. doi: 10.1016/j.jmb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 51.Low DW, Gray HB, Duus JØ. J Am Chem Soc. 1997;119:1–5. [Google Scholar]
- 52.Michel LV, Ye T, Bowman SEJ, Levin BD, Hahn MA, Russell BS, Elliott SJ, Bren KL. Biochemistry. 2007;46:11753–11760. doi: 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.