

Abstract
Although the adaptive immune response almost invariably fails to completely eliminate retroviral infections, it can exert significant protection from disease and long-term control of viral replication. Friend virus (FV), a mouse retrovirus, causes persistent infection in all strains of mice and erythroleukaemia in susceptible strains, the course of which can be strongly influenced by both genetic and extrinsic factors. Here we examine the impact of coinfection on the requirements for immune control of FV infection. We show that congenic C57BL/6 mice, in which the introduction of an allele of the Friend virus susceptibility 2 (Fv2) gene provides the potential for FV-induced leukaemia development, effectively resist FV infection and both T cell- and antibody-dependent mechanisms contribute to their resistance. However, we further demonstrate that coinfection with lactate dehydrogenase-elevating virus (LDV) renders these otherwise immunocompetent mice highly susceptible to FV infection and subsequent disease. The presence of LDV delays induction of FV-specific neutralizing antibodies and counteracts the protective contribution of adaptive immunity. Importantly, the disease-enhancing effect of LDV coinfection requires the presence of a polyclonal B cell repertoire and is reproduced by direct polyclonal B cell activation. Thus, immune activation by coinfecting pathogens or their products can contribute to the pathogenicity of retroviral infection.
Keywords: Viral infection, T Cells, B cells, Immunodeficiency Diseases
Introduction
The immune system has evolved in the face of simultaneous exposure to multiple microorganisms with variable pathogenicity and strategies to evade or divert the immune response. At the same time, the host's response needs to be regulated at levels proportional to the infection, such that a balance between efficient pathogen clearance and excessive immune pathology is achieved. A heightened state of immune activation induced by one infecting pathogen can be beneficial for the control of another and coinfection with certain persistent herpesviruses has been recently shown to confer resistance to bacterial infection (1). In contrast, resistance to other infections may be compromised by concurrent infection with heterologous pathogens. Such examples include resistance to retroviral infection, and HIV in particular, in which activation of the immune system in response to viral, bacterial or parasitic coinfections has been implicated in the increase of both the risk of transmission and rate of disease progression in HIV-infected individuals (2-8). HIV infection is characterized by systemic immune hyperactivation, the extent of which correlates with more rapid disease progression (2, 5, 9), while low levels of T cell activation are associated with reduced susceptibility to infection (10). Although experimental models for infection with a single infectious agent have been and will continue to be an invaluable tool in the study of immunity to infection, infectious pathogens, in a natural setting, are rarely encountered in isolation and almost all humans are chronically infected by one or more persistent viruses. It is therefore also important to establish and study animal models for coinfection in order to define the mechanisms of immunological control during complex infections.
Infection of mice with Friend virus (FV)3, a mouse retrovirus, results in viral persistence, the outcome of which is influenced by a number of genetic and extrinsic factors (11, 12). FV is a retroviral complex, consisting of a replication-competent Friend helper murine leukemia virus (F-MuLV) and a replication-defective spleen focus-forming virus (SFFV). SFFV encodes a truncated form of a retroviral envelope protein, gp55, which can bind to and activate the erythropoietin receptor (EpoR) (13). As a result of constitutive EpoR activation by gp55, the initial phase of FV infection in susceptible mice is characterized by massive splenic enlargement due to polyclonal proliferation of splenic erythroblasts (14). Susceptibility to FV-induced splenomegaly is genetically determined and controlled by the Friend virus susceptibility 2 (Fv2) locus (14, 15). Fv2 encodes the Stk receptor tyrosine kinase, and a naturally-expressed truncated form of Stk (Sf-stk) determines the potential of FV-infected erythroblasts to proliferate in response to viral gp55, in a cell-autonomous manner (16). Sf-stk is the most abundant form of Stk in Fv2s mice and is not expressed in Fv2r mice and thus susceptibility is dominant. Most inbred mouse strains are Fv2s, except C57BL/6 (B6) and related strains, in which Sf-stk expression is decreased or absent and are thus Fv2r (16). In Fv2s strains, recovery from acute FV-induced splenomegaly is largely immune mediated and depends on the host's MHC haplotype (12, 17). Recovery is associated with an H2b haplotype, while mice of an H2a or H2d haplotype, such as A or BALB/c (C), respectively, readily succumb to the disease (12, 17).
It has long been observed that resistance to FV-induced splenomegaly can be modified by concurrent infections with unrelated viruses or microbial products. Although coinfection with some viruses, such as Sendai virus, results in interferon-mediated suppression of FV infection and subsequent disease (18, 19), preinfection or coinfection with other viruses, such as Newcastle disease virus, Guaroa virus or lactate dehydrogenase-elevating virus (LDV), enhances the pathogenicity of FV infection (20-22). Historical considerations suggest that LDV has, in fact, been present in many FV stocks for more than 30 years (21, 23). As there is no selective pressure to maintain the replication-defective SFFV component of FV in vitro, stocks of FV have been traditionally prepared by in vivo passages in mice of susceptible strains. Recent findings suggest that LDV has also been propagated during these passages and has been maintained as an unsuspected passenger in most FV stocks to date (23). It has also been demonstrated that the presence of LDV in FV stocks delays the FV-specific cytotoxic T cell response, thus increasing FV replication and subsequent disease (23).
Despite the important influence of coinfecting viruses on the course of retroviral infection, our knowledge of the precise mechanisms by which coinfections mediate their effects on retrovirus infection and the specific immune response to it, remains incomplete. We have used the FV/LDV system in an effort to understand the impact of coinfection on the requirements for immune control of a retroviral infection. LDV is a fast-replicating cytopathic virus which nevertheless establishes persistence in immunocompetent mice by infecting a small subset of macrophages with limited renewal capacity specialized in scavenging metabolic enzymes such as lactate dehydrogenase (24). Although ineffective in controlling the virus, a relatively weak LDV-specific T cell response is induced during infection (25, 26). In contrast, LDV infection triggers a potent polyclonal activation of B cells leading to hypergammaglobulinaemia, particularly of the IgG2 subtypes (27-33). However, only a small proportion of these antibodies are LDV-specific. Instead, many of these antibodies are directed against self-antigens and can form immune complexes in the absence of LDV-specificity (28, 29, 33).
Here we have used a newly-generated erythroleukaemia-susceptible B6 congenic strain to reveal a strong dependency on LDV for efficient infection with FV. We show that, when these mice are infected with FV alone, a highly effective immune response contains acute FV replication and prevents the development of splenomegaly. In contrast, LDV coinfection renders these otherwise immunocompetent mice unable to mount an FV-specific response and thus highly susceptible to acute splenomegaly. Importantly, this effect of LDV on FV pathogenicity appears crucially dependent on its ability to activate B cells in a polyclonal manner since it is negated in mice with a monoclonal B cell population of an unrelated specificity or in mice lacking B cells. Furthermore, the disease-enhancing effect of LDV co-infection can be mimicked by specific immune stimulation of B cells at the time of FV infection. Thus, a heightened activation state of the immune system is detrimental to the control of acute retroviral infection.
Materials and methods
Mice
Inbred C57BL/6 (B6), BALB/c (C) and B6-backcrossed Rag1-deficient mice (B6.129S7-Rag1tm1Mom/J or B6-Rag1−/−) were originally obtained from the Jackson Laboratory (Bar Harbor, Maine, USA) and were subsequently maintained at NIMR. Hen-egg lysozyme (HEL)-specific BCR-transgenic MD4 (34) and TCR-transgenic TCR7 mice (35) have been described previously. Fv2s-congenic B6 mice (B6.A-Fv2s) were generated by 10 consecutive backcrosses of A strain (Fv2s) mice onto the B6 background, selecting for the Fv2s gene in each generation by PCR based on published sequence (16). Mice were then rendered homozygous for the Fv2s allele by intercrossing. All animal experiments were conducted according to UK Home Office regulations and local guidelines.
Viruses and infections
The Friend virus (FV) used in this study is a retroviral complex of a replication-competent B-tropic helper murine leukemia virus (F-MuLV-B) and a replication-defective polycythemia-inducing spleen focus-forming virus (SFFVp), referred to as FV. The FV stock was free of lactate dehydrogenase-elevating virus (LDV) and was obtained as previously described (23). FV stocks were propagated in vivo and prepared as 10% w/v homogenate from the spleens of LDV-free BALB/c mice infected with FV 12 days previously. A stock of FV, which historically contained LDV (23), was also used, and is referred to as FV/LDV. FV/LDV stocks were also propagated in vivo and prepared as 10% w/v homogenate from the spleen of LDV-free BALB/c mice coinfected with FV/LDV 12 days previously. A pool of 20 LDV-free BALB/c mice was used for the preparation of FV or FV/LDV stocks. Infectious stocks of FV-free LDV were prepared as previously described (23). Mice received an inoculum of FV complex containing ∼2,000 infectious units (iu) of F-MuLV (assayed in vitro) and between 1,000-2,000 spleen focus-forming units (SFFU) (assayed in vivo). In addition to FV, the FV/LDV inoculum also contained 10-100 iu of LDV, based on RT-PCR assays for viral RNA (23) and published data (24). Alternatively, mice received 10-100 iu of LDV alone. All viruses were injected via the tail vein in 0.1 ml of phosphate-buffered saline. Cell-associated FV in infected mice was estimated by flow cytometric detection of FV infected cells using surface staining for the glycosylated product of the viral gag gene (glyco-Gag), using the matrix (MA)-specific monoclonal antibody 34 (mAb 34). Splenomegaly is expressed as spleen index (SI), which is calculated as the weight of the spleen (in mg) divided by the weight of the rest of the body (in g), to control for the effect of the size of the animal on spleen size. LDV infection was confirmed by serum lactate dehydrogenase (LDH) assays (QuantiChrom Lactate Dehydrogenase kit, BioAssay Systems, Hayward, CA, USA) according the manufacturer's instructions.
Flow cytometric analysis
Single-cell suspensions were prepared from the spleen of donor mice following mechanical disruption of the organs on nylon mesh and were treated with ammonium chloride for erythrocyte lysis. For analysis of B cell activation cells were stained with directly-conjugated antibodies to B220, CD19, IgD, CD38 and GL7 (eBiosciences, San Diego, CA, USA). FV-infected cells erythroid cells were detected by direct staining with PE-conjugated antibodies to Ter119 (eBiosciences) in conjunction with the anti-MA mAb 34 (mouse IgG2b). Anti-MA mAb 34 was prepared from the supernatant of hybridoma cultures with ammonium sulfate precipitation. Binding of mAb 34 to infected cells was visualized by an anti-mouse IgG2b FITC-conjugated secondary reagent (BD, San Jose, CA, USA). Four- and 5-color cytometry were performed on FACSCalibur (BD Biosciences) and CyAn (Dako, Fort Collins, CO) flow cytometers, respectively, and analyzed with FlowJo v8.7 (Tree Star Inc., Ashland, OR, USA) or Summit v4.3 (Dako) analysis software, respectively.
Neutralizing antibody assay
In vitro FV-neutralizing antibodies in the sera of infected mice were measured using a modification of a previously described viral titer assay (36). Mus dunni cells (37) were transduced with the XG7 replication-defective retroviral vector, expressing green fluorescent protein (GFP) from a human cytomegalovirus (hCMV) promoter and a neomycin-resistance gene under the control of the LTR (36). Maintenance of GFP expression was ensured by constant selection with 1 mg/ml G418. Mus dunni-XG7 cells were then infected with F-MuLV-B and supernatant, which contained the pseudotyped XG7 vector was harvested. Serial dilutions of sera from infected mice were mixed with ∼1,500 iu/ml pseudotyped XG7 vector and allowed to incubate for 30 min at 37°C in IMDM cell culture medium containing 5% FCS. Mixtures were then added to untransduced Mus dunni cells and incubated for 3 days. The percentage of GFP+ Mus dunni cells at the end of the incubation period was assessed by flow cytometry, using a FACSCalibur (BD, San Jose, CA, USA), and the dilution of serum which resulted in 75% neutralization (i.e. 75% reduction in the percentage of GFP+ Mus dunni cells) was taken as the neutralizing titer.
Treatment of mice with FV-neutralizing antibodies
FV and FV/LDV infected were administered with the anti-gp70 of F-MuLV monoclonal antibody 48 (38), at indicated time points post infection. The anti-gp70 mAb 48 was prepared from the supernatant of hybridoma cultures with ammonium sulfate precipitation and was diluted in phosphate-buffered saline at a titer, which had in vitro FV-neutralizing activity equivalent to sera obtained from FV infected mice at day 21 of infection. Mice received 0.3 ml of mA48 dilution intraperitoneally.
Histology
Spleens from infected mice were removed, fixed with Bouin's fixative and embedded in paraffin. Sections were then stained with hematoxylin and eosin and photomicrographs were taken with a stereo-microscope (Carl Zeiss) equipped with a digital camera.
In vivo lymphocyte activation
For in vivo polyclonal lymphocyte activation mice were first infected with FV and then received stimuli 30 min later. A single dose of 10 μg LPS (from Salmonella minnesota R595, Axxora, CA, USA) or 130 μg activating anti-mouse IgM (polyclonal goat anti-mouse IgM, Jackson ImmunoResearch Europe, UK) were injected i.v. in 0.1 ml PBS. These doses of LPS or anti-IgM were found to induce no splenomegaly when given alone to FV-uninfected control mice.
Results
Fv2s renders B6 mice susceptible to FV-induced splenomegaly
To overcome the Fv2r-mediated genetic resistance of B6 mice (H2b, Fv2r), which would allow the study of FV infection in a series of commonly used B6-backcrossed gene-targeted strains, we generated a congenic B6 strain (B6.A-Fv2s) carrying the Fv2s allele from the A strain. As the vast majority of previous studies of FV infection have been performed using LDV-containing FV stocks, we initially examined the susceptibility of B6.A-Fv2s mice to erythroleukaemia using an LDV-containing FV stock, such that comparisons with published data were possible. In contrast to B6 mice, B6.A-Fv2s mice exhibited significant splenic enlargement (∼18 times the normal spleen size) two weeks post infection (Fig. 1A), which was similar but not identical to fully susceptible C mice (H2d, Fv2s) (Fig. 1A). The splenomegaly induced by FV infection in B6.A-Fv2s mice displayed all the hallmarks of the early FV-induced disease in C mice, such as expansion of the splenic red pulp and disruption of normal splenic architecture (Fig. 1B, top row), and dramatic increase in nucleated Ter119+ erythroid precursors, most of which expressed on their membranes retroviral glyco-Gag (Fig. 1B, bottom row), an indicator for FV infection. In contrast to C mice, which subsequently succumbed to infection due to an inability to mount an effective cell-mediated antiviral response (12), B6.A-Fv2s mice recovered from severe splenomegaly within 3 weeks and remained splenomegaly-free throughout a 7 month observation period (Fig. 1C), as expected for a high-recovery strain (39). Thus, the congenic B6.A-Fv2s strain of mice combines the high-recovery phenotype of H2b mice with the susceptibility of Fv2s mice, as seen in hybrids, such as between C57BL/10 (B10) and A.BY mice (H2b/b, Fv2r/s), when FV/LDV coinfection is used (12, 39).
FIGURE 1.

An Fv2s allele restores susceptibility of B6 mice to splenomegaly induced by FV/LDV coinfection. (A) Spleen weights of B6, B6.A-Fv2s and C mice 14 days post FV/LDV coinfection. The mean values ± SEM of at least 10 mice per group, pooled from 3 experiments, are shown. (B) Hematoxylin and eosin staining of spleen sections (top row) and flow cytometric analysis of glyco-Gag+ Ter119+ erythroid precursors (bottom row) in the spleens of B6, B6.A-Fv2s and C mice, 9 or 14 days post FV/LDV coinfection, respectively. Results are representative of 5 mice per group from a single experiment. Numbers within the quadrants represent the percentage of cells in that quadrant in all splenocytes; numbers in parentheses represent the percentage of infected cells (glyco-Gag+) in erythroid precursors (Ter119+) only. (C) Course of FV/LDV coinfection, depicted as spleen index (SI), in FV/LDV coinfected B6.A-Fv2s and C mice. The mean values ± SEM of 4-5 mice per group per time point pooled from 3 experiments, are shown.
B6.A-Fv2s mice control FV infection but not FV/LDV coinfection
We next examined the impact of LDV coinfection on the course of FV infection, by comparing the original LDV-containing FV stock with an LDV-free FV stock. Both FV infection alone and FV/LDV coinfection resulted in rapid splenomegaly in fully-susceptible C mice (Fig. 2A, B), although the presence of LDV delayed its onset. In contrast, B6 mice remained resistant to both FV and FV/LDV (Fig. 2C, D), with only marginal splenic enlargement seen with FV/LDV (spleen index (SI)=8.2). Surprisingly, even though B6.A-Fv2s mice were susceptible to severe splenomegaly following FV/LDV infection (Fig. 2F), they were relatively resistant to FV alone (Fig. 2E), with a maximum SI of 11.7, on day 10 post infection, which represents an enlargement of ∼3 times the normal spleen size. Splenomegaly induction in FV/LDV coinfected B6.A-Fv2s mice was not an independent effect of infection with LDV, as it was dependent on the presence of the Fv2s allele (Fig. 2D, F) and infection of B6.A-Fv2s mice with LDV alone did not induce significant splenomegaly (Fig. 2F). Resistance of B6.A-Fv2s mice to FV-induced splenomegaly was not due to failure of FV to infect mice of this particular genetic background, as the percentage and absolute number of infected Ter119+ cells on day 7 of FV infection was either the same in resistant B6 mice (Fig. 3A) or even higher in susceptible B6.A-Fv2s mice (Fig. 3B) than those in FV/LDV coinfection. Instead, resistance correlated with rapid reduction of FV loads in the following 2 weeks of FV infection (particularly between day 7 and 14). In contrast, FV loads in FV/LDV coinfection continued to rise beyond day 7 and peaked on day 14 (Fig. 3A, B). This pattern is consistent with rapid immune-mediated control of FV infection in B6 and B6.A-Fv2s mice and delayed clearance in FV/LDV coinfection, in agreement with similar observations is B10 × A.BY F1 mice (12, 23).
FIGURE 2.

Effect of LDV coinfection on the immune requirements for containment of FV infection. Spleen size, depicted as SI, of C (A, B), B6 (C, D), B6.A-Fv2s (E, F), B6.A-Fv2s MD4 (G, H), B6.A-Fv2s TCR7 (I, J) and B6.A-Fv2s Rag1−/− mice (K, L) over the course of FV infection (top row) or LDV infection and FV/LDV coinfection (bottom row). Data are the mean values ± SEM of 5-12 mice per group per time point, pooled from 3-5 experiments.
FIGURE 3.
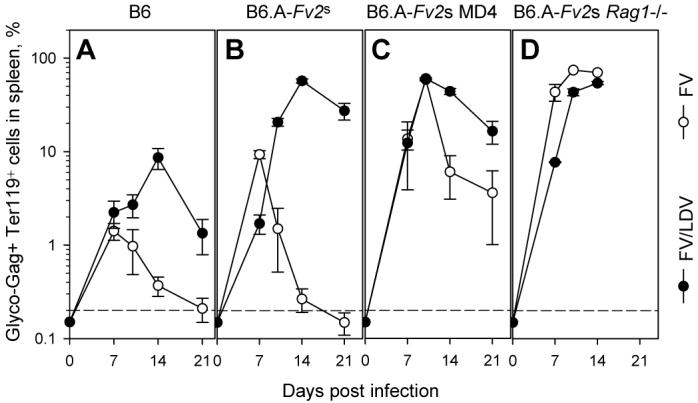
Effect of LDV coinfection on the kinetics of expansion and control of FV-infected erythroid precursors. The percentage of glyco-Gag+ Ter119+ cells, determined by flow cytometry, in splenocytes from B6 (A), B6.A-Fv2s (B), B6.A-Fv2s MD4 (C) and B6.A-Fv2s Rag1−/− mice (D) infected with FV alone or coinfected with FV and LDV is shown. Data are the mean ± SEM of 7-12 mice per group per time point, from 2 experiments. The dashed line represents the limit of detection (0.2%).
Enhancement of FV-induced disease by LDV coinfection requires lymphocytes
As resistance of B6.A-Fv2s mice to FV infection was consistent with immune-mediated protection, we next investigated in more detail the immune response against FV infection and FV/LDV coinfection. To assess the contribution of humoral and cellular immunity to FV control, B6.A-Fv2s mice were rendered selectively immunodeficient in either virus-specific antibody or T cell responses, or both. B6.A-Fv2s mice were crossed to BCR transgenic mice, in which B cells were present in normal numbers, but express the MD4 BCR (34), instead of endogenous ones, and therefore can respond only to an unrelated epitope from HEL. In the absence of an FV-specific B cell response, FV infection caused significant early splenomegaly in B6.A-Fv2s MD4 mice (SI=32.2 at the peak on day 10 post infection) (Fig. 2G), compared with B6.A-Fv2s mice (p=0.0007, student's t-test on day 10). Splenomegaly in FV-infected B6.A-Fv2s MD4 mice was not, however, as severe as in fully susceptible C mice and was also followed by partial recovery until day 21 post infection, after which time, B6.A-Fv2s MD4 mice failed to control FV infection and displayed chronic and progressive splenomegaly (Fig. 2G). Compared with FV infected B6.A-Fv2s mice (Fig. 3B), percentages of infected Ter119+ cells in the spleen were significantly elevated in FV infected B6.A-Fv2s MD4 mice (Fig. 3C), in line with increased peak splenomegaly in the latter (Fig. 2E, G). Furthermore, FV infected B6.A-Fv2s MD4 mice harbored readily detectable numbers of infected Ter119+ cells throughout the course of infection (Fig. 3C), despite transient control of splenomegaly by day 21 of infection (Fig. 2G). Therefore, inability of B6.A-Fv2s MD4 mice to mount a neutralizing antibody response exacerbated FV infection. B6.A-Fv2s MD4 mice coinfected with FV/LDV progressed to splenomegaly with the same rate as FV/LDV coinfected B6.A-Fv2s mice (Fig. 2H). Surprisingly, the lack of neutralizing antibody response did not accelerate or exacerbate splenomegaly in FV/LDV coinfected B6.A-Fv2s MD4 mice (Fig. 2H). Indeed, its severity was significantly reduced in these mice compared with FV/LDV coinfected B6.A-Fv2s mice (p=0.012, student's t-test on day 14). Moreover, percentages of infected Ter119+ cells in the spleen of FV/LDV coinfected B6.A-Fv2s MD4 mice were not increased in comparison with those in the spleen of FV/LDV coinfected B6.A-Fv2s mice (Fig. 3B, C). Thus, a functional B cell response was beneficial in early control of FV infection but not in FV/LDV coinfection.
As the absence of a functional B cell response in B6.A-Fv2s MD4 mice only partially increased susceptibility to FV infection, we examined whether protection was mediated by virus-specific T cells. B6.A-Fv2s mice were crossed to TCR transgenic mice, in which T cells were present in normal numbers, but express the HEL-specific TCR7 TCR (35). B6.A-Fv2s TCR7 mice fail to mount a T cell response to FV due to lack of appropriate TCR, and would also be defective in T cell-dependent neutralizing antibody production due to lack of T cell help. As a control, B6.A-Fv2s mice were also crossed to lymphocyte-deficient Rag1−/− mice. The lack of virus-specific T cells in B6.A-Fv2s TCR7 mice rendered them fully susceptible to FV-induced splenomegaly, at levels and rate of progression comparable to C mice (Fig. 2I), from which they did not recover, demonstrating that resistance of B6.A-Fv2s mice to infection with FV alone is immune mediated. Progression to severe splenomegaly caused by infection with FV alone was also similar between B6.A-Fv2s TCR7 mice and lymphocyte-deficient B6.A-Fv2s Rag1−/− mice (Fig. 2K), indicating that B6.A-Fv2s TCR7 mice exhibit a complete lack of adaptive immunity to FV infection. However, neither B6.A-Fv2s TCR7 nor B6.A-Fv2s Rag1−/− mice, showed any enhancement or acceleration of splenomegaly, in comparison to fully immune competent B6.A-Fv2s mice, when coinfected with FV and LDV (Fig. 2J, L). Again, induction of splenomegaly in FV/LDV coinfected B6.A-Fv2s Rag1−/− mice was caused by FV, rather than LDV, as infection with LDV alone did not result in significant splenomegaly (Fig. 2L). Furthermore, following FV/LDV coinfection, percentages of infected Ter119+ cells in the spleen of immunodeficient B6.A-Fv2s Rag1−/− mice were comparable to those in the spleen of immunocompetent B6.A-Fv2s mice (Fig. 3B, D). Together, these results suggested that adaptive immunity does not contribute appreciably to the control of FV replication in the first 2 weeks of this coinfection.
Delayed induction of FV-neutralizing antibodies in FV/LDV coinfection
The use of B cell-deficient mice has previously demonstrated an indispensable role for B cells and neutralizing antibodies in the recovery from FV-induced splenomegaly during FV/LDV coinfection (40-43). However, no positive contribution of virus-neutralizing antibodies can be seen in FV/LDV coinfection of B6.A-Fv2s mice before day 21 of infection (Fig. 2F, H), suggesting either that the antibody response did not provide any protection against acute FV/LDV coinfection or that was not yet induced. In contrast, virus-specific antibodies appeared to play an essential role in controlling infection with FV alone already from day 7 post infection. Indeed, percentages of FV-infected Ter119+ cells in the spleen were increased in the absence of an FV-neutralizing antibody response when mice were infected with FV alone, but were unchanged or even decreased at later time points when mice were coinfected with FV and LDV (Fig. 3B, C). To confirm that enhanced infection with FV in B6.A-Fv2s MD4 mice, compared with B6.A-Fv2s mice, was due to lack of FV-specific antibody response, FV-infected B6.A-Fv2s MD4 mice were administered with FV-neutralizing antibodies. Injection of the anti-gp70 of F-MuLV monoclonal antibody 48 (38), completely prevented induction of splenomegaly (Fig. 4A) and dramatically reduced the percentages of FV-infected Ter119+ cells (Fig. 4B) in the spleen of B6.A-Fv2s MD4 mice infected with FV. Thus, administration of FV-neutralizing antibodies restored resistance of B6.A-Fv2s MD4 mice to FV infection. Similarly, injection of the monoclonal antibody 48 significantly reduced the degree of splenomegaly and percentages of FV-infected Ter119+ cells in the spleen of B6.A-Fv2s MD4 mice coinfected with FV and LDV (Fig. 4C, D). These results suggested that both FV infection and FV/LDV coinfection can be prevented or controlled by timely induction of FV neutralizing antibodies. We thus hypothesized that a possible explanation for the observed contribution of naturally-induced virus-specific antibodies in protection during FV infection, but not FV/LDV coinfection, was that these antibodies were only induced in the former. Contributing factors could be polyclonal B cell activation induced by LDV and the disruption of normal splenic architecture caused by FV/LDV-induced splenomegaly. Therefore, to examine the effect of LDV coinfection on induction of FV-specific antibodies we used B6 mice, which do not experience substantial splenomegaly or alterations in splenic structure with either FV alone or in combination with LDV (Fig. 1B; Fig, 2E, F). Sera from FV-infected B6 mice had detectable in vitro FV-neutralizing activity from day 14 post infection, which increased dramatically only by day 21 and approached a plateau by day 28 (Fig. 5). FV/LDV coinfection in B6 mice transiently induced serum FV-neutralizing activity in a proportion of mice on day 14 post infection (Fig. 5), which corresponds to an early IgM response (44). In comparison to FV infection, FV-neutralizing activity in sera from FV/LDV coinfected B6 mice was homogeneously below the detection limit (1:50) on day 21, suggestive of delayed class-switching (44), and only started rising between days 21 and 28 (Fig. 5), without however reaching the levels seen in FV-infected B6 mice. Thus, coinfection with LDV delayed the neutralizing antibody response to FV.
FIGURE 4.

Effect of FV-neutralizing antibody administration on control of FV infection and FV/LDV coinfection. Spleen size, depicted as SI (A, C) and percentage of glyco-Gag+ Ter119+ cells (B, D) in the spleen of B6.A-Fv2s MD4 mice infected with FV alone (A, B) or coinfected with FV and LDV (C, D), with (mAb 48) or without (−) administration of monoclonal antibody 48. Antibodies were administered on day 3 and 6 and mice analyzed on day 10 of infection with FV alone, or on day 3, 6 and 9 and mice analyzed on day 10 of coinfection with FV and LDV. Data are the mean ± SEM of 3-4 mice per group from one experiment.
FIGURE 5.
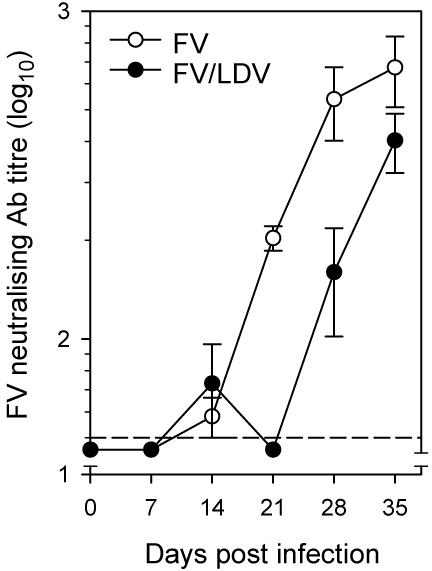
Effect of LDV coinfection on induction of FV-neutralizing antibodies. Titer of FV-neutralizing activity in the sera of FV infected or FV/LDV coinfected B6 mice. Data are the mean ± SEM of 4 mice per group per time point. One representative of 2 experiments in shown. The dashed line represents the limit of detection (1:50).
Polyclonal B cell stimulation overcomes resistance of B6.A-Fv2s mice to FV infection
LDV infection of immunocompetent mice causes polyclonal B cell activation and hypergammaglobulinaemia (27, 30, 31), which might prevent induction of antibody responses to new infections (45). Inhibition of the FV-neutralizing antibody response could be the mechanism underlying the enhancement of FV-induced splenomegaly by LDV coinfection. Alternatively, polyclonal activation of B cells per se could be directly responsible for the effect of LDV coinfection on FV-induced splenomegaly, independently of their ability produce FV-specific antibodies. We first addressed whether FV/LDV coinfection was associated with higher levels of B cell activation than infection with FV alone and whether or not LDV could cause activation of monoclonal B cells expressing the MD4 BCR transgene. Infection of B6.A-Fv2s mice with FV alone gave rise to significant numbers of B cells with the CD38lo GL7+ germinal center-phenotype in the spleen 14 days post infection (Fig. 6). In line with the B cell stimulatory effect of LDV, the spleens of B6.A-Fv2s mice coinfected with FV and LDV contained twice as many CD38lo GL7+ B cells at the same time-point (Fig. 6). Importantly, restriction of the BCR repertoire in B6.A-Fv2s MD4 mice severely restrained activation of B cells during either FV infection or FV/LDV coinfection (Fig. 6). The residual population of CD38lo GL7+ B cells observed in FV/LDV coinfected B6.A-Fv2s MD4 mice likely represent B cells expressing endogenous BCRs, which were still present at a frequency of ∼5% in these mice (34). These results indicated that BCR specificity is a crucial factor in B cell stimulation during these infections.
FIGURE 6.
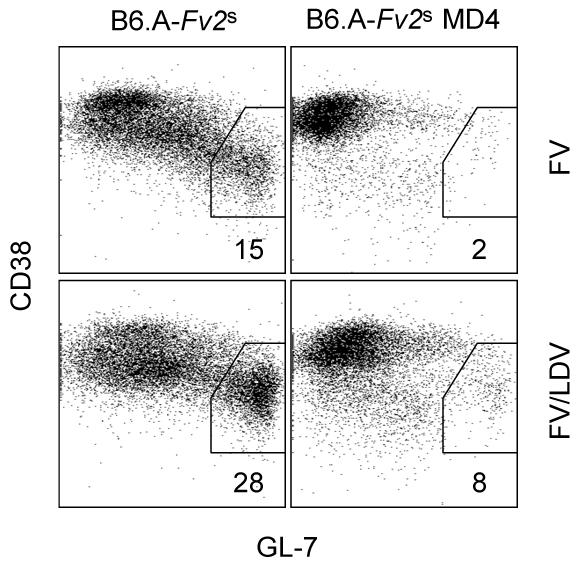
Activation of polyclonal or monoclonal B cells by FV infection or FV/LDV coinfection. Percentages of CD38lo GL7+ cells, in gated B220+ CD19+ IgD−B cells in the spleen of B6.A-Fv2s or B6.A-Fv2s MD4 mice 14 days post infection with FV alone or coinfection with FV and LDV. Numbers within the plots represent the mean percentage of CD38lo GL7+ cells of 2-3 mice per group. The absolute number of CD38lo GL7+ B cells was on average 5.0×106 and 0.1×106 in the spleen of FV-infected B6.A-Fv2s and B6.A-Fv2s MD4 mice, respectively; and 17.6×106 and 2.5×106 in the spleen of FV/LDV-coinfected B6.A-Fv2s and B6.A-Fv2s MD4 mice, respectively.
To assess whether B cell activation by LDV at the time of FV/LDV coinfection contributes to the severity of splenomegaly, we substituted LDV with a polyclonal B cell stimulus. LPS is a potent polyclonal activator of murine B cells, which induces cellular proliferation and antibody secretion. Indeed, injection of a small dose of LPS in B6.A-Fv2s mice led to the emergence of CD38lo GL7+ germinal center B cells 10 days later (Fig. 7A). When LPS was injected at the time of infection with FV alone, B6.A-Fv2s mice, which are otherwise resistant, rapidly developed severe splenomegaly (Fig. 7B), with kinetics similar to immune-deficient B6.A-Fv2s TCR7 or B6.A-Fv2s Rag1−/− mice. Thus, LPS administration, which in itself did not cause any splenic enlargement (Fig. 7B), was sufficient to overcome the immune mediated resistance of B6.A-Fv2s mice. As observed in FV/LDV coinfected mice, the splenomegaly in FV-infected B6.A-Fv2s mice given LPS was predominantly due to an uncontrolled expansion of Ter119+ cells, most of which were expressing FV antigens (Fig. 7C). Although LPS activates B cells polyclonally, it also activates other immune and non-immune cell types, most notably macrophages, which can also become infected with FV (46). To confirm that polyclonal B cell activation was the primary cause of the failure to control acute FV infection, B6.A-Fv2s mice were injected with an activating anti-IgM antibody at the time of FV infection, which would activate B cells specifically. Administration of anti-IgM antibody was followed by the appearance of CD38lo GL7+ germinal center B cells 10 days later (Fig. 7D). Similar to LPS injection, polyclonal B cell activation induced by anti-IgM antibody injection, which alone had no effect of splenic size, overcame the resistance of B6.A-Fv2s mice to splenomegaly (Fig. 7E). Again, splenomegaly caused by anti-IgM antibody administration in FV-infected B6.A-Fv2s mice was due to expansion of infected Ter119+ cells (Fig. 7F). Thus, B cell activation by either LPS or anti-IgM antibody treatment prevents the control of acute FV infection by otherwise resistant mice.
FIGURE 7.
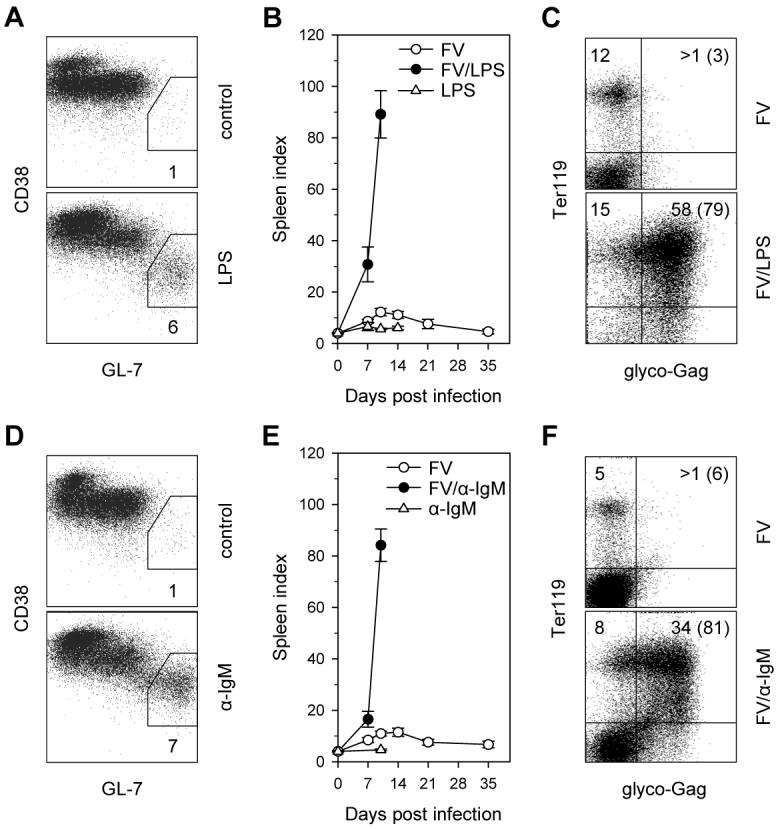
Effect of polyclonal B cell stimuli on immune control of FV infection. (A) Percentages of CD38lo GL7+ cells, in gated B220+ CD19+ IgD− B cells in the spleen of control B6.A-Fv2s mice or 10 days post administration of LPS. (B) Splenomegaly, depicted as SI, in B6.A-Fv2s mice infected with FV with or without concomitant administration of LPS or in mice given LPS alone. (C) Flow cytometric detection of FV-infected erythroid precursors in the spleen of FV-infected B6.A-Fv2s mice without (top) or with (bottom) LPS administration. (D) Percentages of CD38lo GL7+ cells, in gated B220+ CD19+ IgD− B cells in the spleen of control B6.A-Fv2s mice or 10 days post administration of anti-IgM antibody. (E) Splenomegaly in B6.A-Fv2s mice infected with FV with or without concomitant administration of anti-IgM antibody or in mice given anti-IgM antibody alone. (F) Flow cytometric detection of FV-infected erythroid precursors in the spleen of FV-infected B6.A-Fv2s mice without (top) or with (bottom) anti-IgM antibody administration. Data in (A) and (D) represent the mean percentage of CD38lo GL7+ cells of 2-3 mice per group. Data in (B) and (E) are the mean ± SEM of 4-11 mice per group per time point, pooled from 2-3 independent experiments. Numbers within the quadrants in (C) and (F), represent the percentage of cells in that quadrant in all splenocytes; numbers in parentheses represent the percentage of infected cells (glyco-Gag+) in erythroid precursors (Ter119+) only. Results in (C) and (F) are representative of 3-4 mice per group. No FV-infected cells were detected in mice given LPS or anti-IgM antibody alone (not shown).
Discussion
For this study we developed a congenic B6 strain, B6.A-Fv2s, which combines genetic susceptibility to FV-induced erythroid precursor expansion with immune-mediated resistance to FV infection. Experiments with this mouse strain allowed us to reveal a fundamental role for LDV coinfection-induced activation of the immune system in failure to control acute FV infection. B6.A-Fv2s mice effectively controlled acute splenomegaly when challenged with FV alone and their resistance was immune-mediated. In contrast, polyclonal B cell activation by concurrent infection with LDV or stimulation with B cell stimuli counteracted the immune response to FV and no evidence for immune-mediated protection could be demonstrated during the early acute phase of FV/LDV coinfection.
The introduction of an Fv2s allele onto the B6 genetic background fully imparted susceptibility to FV-induced splenomegaly. Disease progression and recovery upon FV/LDV coinfection of B6.A-Fv2s (H2b/b, Fv2s/s, Rfv3r/r) mice followed kinetics similar to what has been previously described for B10 × A.BY F1 mice (H2b/b, Fv2r/s, Rfv3r/s) mice (12, 39). In contrast to B10 × A.BY F1 mice, which still developed splenomegaly when infected with FV alone (23), our results suggest that B6.A-Fv2s mice are highly resistant to FV-induced disease in the absence of LDV coinfection. The composition of resistance and susceptibility alleles is similar in the two strains at all the described loci affecting FV infection, such as the H2, Fv1, Fv2 and Rfv3 loci (12). It could be that heterozygosity at one or more such loci or other alleles on the A strain genetic contribution in B10 × A.BY F1 mice contributes to their enhanced susceptibility to FV infection, in comparison with B6.A-Fv2s mice. Alternatively, the difference in susceptibility could be due to differences in the FV infecting dose used in different studies.
Our analysis of FV infection in B6.A-Fv2s mice with selective immunodeficiencies demonstrates that both T cell- and antibody-dependent mechanisms contributed to their resistance. The absence of an FV-specific B cell response in B6.A-Fv2s MD4 mice, revealed a biphasic effect in the control of FV-induced splenomegaly. FV-infected B6.A-Fv2s MD4 mice developed early (∼day 10) splenic enlargement, from which they partially recovered until day 21. Recovery was mediated by an FV-specific T cell response as B6.A-Fv2s TCR7 mice failed to recover. Nevertheless, T cell-mediated protection was incomplete as B6.A-Fv2s MD4 mice failed to clear FV-infected cells and progressed to splenomegaly at later time points. Enhanced susceptibility of B6.A-Fv2s MD4 mice to early FV-induced splenomegaly due to lack of an FV-specific antibody response is consistent with restoration of resistance in these mice, upon administration of FV-neutralizing monoclonal antibodies. However, an early requirement for the FV-specific antibody response revealed in B6.A-Fv2s MD4 mice seems to be at odds with relatively late induction of FV-neutralizing antibodies, even in infection with FV alone. Indeed, neither the assay employed in this study, nor previously used assays (23, 47, 48), detected significant FV-neutralizing activity before day 21 of infection. However, non-neutralizing antibodies may also contribute to protection and it has been previously shown that administration of an FV p15 Gag-specific non-neutralizing monoclonal antibody had a protective, albeit weak, effect against FV infection (43). Furthermore, the importance of antibody activity against infected cells, in addition to free virions, has been recently documented in a macaque model for HIV infection, by removal of the Fc-mediated effector function of the antibodies (49). Such activity would not be detected by a classical neutralization assay. Alternatively, an early effect of antibodies on FV infection, before measurable FV-neutralizing antibodies are induced, could reflect the action of natural, as opposed to induced, antibodies. The presence and protective function against infection of virus-specific antibodies naturally occurring in the sera of virus-naïve animals has been previously documented (50). These natural antibodies are present at low levels and although they may be virus-specific they do not detectably neutralize virus infectivity in vitro (50).
In contrast to their protective effect during infection with FV alone, neither T cell- nor antibody-mediated immunity appeared to have any impact on disease progression during the first 2 weeks of FV/LDV coinfection. Indeed, no enhancement of FV-induced disease could be demonstrated by partial or full adaptive immunodeficiency during this time. Although the role of both T cell- and antibody-mediated protection in recovery from FV-induced splenomegaly has been established (12, 17, 46), our findings are in line with previous studies with genetic or antibody-mediated lymphocyte depletion experiments, which also failed to reveal a role for either CD4+ or CD8+ T cells in protection from early splenomegaly in susceptible strains of mice following infection with, presumably, LDV-contaminated FV stocks (39). Although concurrent infection with LDV could potentially interfere with subsequent effector mechanisms of FV-specific immunity, recent data showed that the most likely explanation for the apparent lack of immune protection during early FV/LDV coinfection is that FV-specific immune response is prevented or delayed. Indeed, induction of FV-specific CD8+ T cells was recently found to be delayed by LDV coinfection (23). Similarly, our results showed that induction of FV-neutralizing antibodies was significantly delayed in B6 mice by LDV coinfection and that lack of FV-specific antibodies in B6.A-Fv2s MD4 mice had no detrimental effect in the first 3 weeks of the disease course when LDV was present. Thus, concurrent infection with LDV prevents early induction of both cytotoxic (23) and, in the present study, antibody responses to FV, suggesting that LDV may act by inhibiting the provision of a common requirement for both these arms of adaptive immunity, such as virus-specific CD4+ T cell help.
Coinfection of partially or fully immunodeficient B6.A-Fv2s mice with FV and LDV demonstrated that not only did the adaptive immune response fail to afford any noticeable protection during acute coinfection, surprisingly it contributed to the disease-enhancing effect of LDV. Mice with a monoclonal B cell population of unrelated specificity or mice lacking B cells had significantly milder disease, compared with fully immunocompetent mice coinfected with FV/LDV. Furthermore, the presence of LDV enhanced FV-induced splenomegaly in fully immunocompetent mice, but had no effect or even inhibited FV-induced splenomegaly in immunodeficient mice. Therefore, coinfection with LDV reverses the role of the adaptive immune response in FV infection from disease-inhibiting to disease-promoting. Importantly, this effect of LDV correlated with its ability to cause polyclonal B cell activation and could be mimicked by generic B cell stimuli, such as LPS and anti-IgM antibody treatment. It has long been observed that microbial products, such as LPS and CpG oligonucleotides (21, 51-53), or stimulation of haematopoiesis by erythropoietin injection or hypoxia (21), can exacerbate FV-induced splenomegaly. This enhancement has previously been thought to be the result of target cell expansion (21). Although expansion of erythroid precursor cells by these treatments would certainly contribute to enhancement of FV replication, this would have been in addition to the immune-hyperactivation effect of LDV, most likely present in these previous studies (21). Our results suggest that enhancement of FV pathogenicity induced by LDV coinfection is largely due to immune dysfunction. Firstly, resistance to infection with FV alone is immune-mediated and coinfection with LDV has a marked inhibitory effect on the induction FV-specific immune response. Secondly, the disease-enhancing effect of LDV coinfection depended on the presence of a polyclonal B cell repertoire and it could be reproduced by direct polyclonal B cell activation with anti-IgM antibodies. It must be noted, however, that delay of the FV-neutralizing antibody response may not be the only mechanism underlying the impact of B cell activation on FV infection. Treatment of B6.A-Fv2s mice with anti-IgM antibodies at the time of FV infection resulted in more severe splenomegaly than lack of FV-specific antibodies in B6.A-Fv2s MD4 mice. Moreover, B6.A-Fv2s mice experience more severe acute disease than B6.A-Fv2s MD4 mice following FV/LDV coinfection, despite the lack of an antibody response common in both strains. Although our results clearly implicate B cell activation in LDV-mediated disease enhancement, it is still unclear what proportion or subset of B cells is involved. Although LPS and anti-IgM antibody administration had similar effects on splenomegaly and induced comparable numbers of germinal center-phenotype B cells, anti-IgM antibodies target fewer B cells than LPS, owing to the wide-ranging expression of IgM on B cells. Furthermore, polyclonal B cell activation by LDV is selective to B cells with autoreactive BCRs (28, 29, 33). It is therefore likely that activation of only a fraction of B cells is responsible for the disease enhancement. Thus, it will be important to determine the precise nature of B cell activation during this coinfection and how it may additionally contribute to FV-induced disease enhancement, in order to establish any potential link with delay in induction of T cell-mediated immunity.
Although the most remarkable effect of LDV coinfection was the enhancement of FV pathogenicity specifically in immunocompetent mice, it also affected, to some extent, the kinetics of FV infection in all strains. The presence of LDV in stocks of FV delayed the progression to splenomegaly in susceptible mice by 3-4 days, compared with LDV-free stocks of FV, which was particularly evident in immunodeficient B6.A-Fv2s Rag1−/− mice. The precise reasons for the delay in FV-induced splenomegaly are not clear but they could be mediated by an antiviral response, through the action of type I IFN, induced by LDV, similarly to coinfection with other viruses (19). In fact, it has recently been shown that plasma type I IFN measured during the acute phase of FV/LDV coinfection was induced by LDV, while no measurable type I IFN was induced by FV alone (54). Alternatively, LDV could be delaying the progression of FV-induced splenomegaly at the level of FV-infected cell expansion and survival. Although LDV has a strict cell-tropism for functionally-specialized macrophages in vivo, it has been observed that retroviral infection renders non-permissive cells susceptible to LDV infection in vitro (55, 56) and endogenous retroviral activity is required for successful infection of neural tissue by LDV and development of neurological disease in vivo (57). Thus, it could be envisaged that FV-infected erythroid precursors, the expansion of which would normally lead to splenomegaly following infection with FV alone, become targets for infection and subsequent lysis by the cytopathic LDV in FV/LDV coinfection. Such a mechanism would be compatible with our observation that the delay in accumulation of FV-infected erythroid cells caused by LDV is influenced by the presence of an Fv2s allele and is noticeable in B6.A-Fv2s, but not in B6 mice (Fv2r). In contrast, any effect of type I IFN would be expected to be the same in both strains, regardless of the Fv2 status.
In conclusion, our results indicate that under certain conditions the immune response to retroviral infection can be highly effective in the prevention of acute disease and subsequent containment of infection. However, the effectiveness of the antiviral immunity can be severely undermined by concomitant immune activation in response to coinfecting pathogens or their products. Thus, although strong activation of the immune system is beneficial when focused on the infecting retrovirus, it can also be detrimental when diverted to other targets, especially in the case of retroviruses, which frequently target the immune system for their replication. Polyclonal activation of B cells might therefore represent an immune-evasion strategy, employed both by retroviruses, such as HIV (58) and other persistent viruses, such as LCMV (59). As coinfecting pathogens modify both susceptibility to and immune requirement for control of retroviral infection, our data also highlight the need to extend our understanding of the factors determining the outcome of retroviral infection to include the influence of coinfection and any host factors, which in turn modulate immune reactivity in general.
Acknowledgments
The authors would like to thank Drs Anne O'Garra for the TCR7 mice, Facundo Batista for the MD4 mice, and Elena Grigorieva for histology.
Footnotes
This work was supported by the UK's Medical Research Council (MRC) and the Portuguese Foundation of Science and Technology (SFRH/BD/15208/2004 to IA).
Abbreviations used in this paper: B6, C57BL/6; C, BALB/c; EpoR, erythropoietin receptor; FV, Friend virus; Fv2, Friend virus susceptibility 2; F-MuLV, Friend helper murine leukemia virus; HEL, hen-egg lysozyme; LDV, lactate dehydrogenase-elevating virus; SFFV, spleen focus-forming virus; SI, spleen index.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the United States National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 2.Bentwich Z, Kalinkovich A, Weisman Z. Immune activation is a dominant factor in the pathogenesis of African AIDS. Immunol. Today. 1995;16:187–191. doi: 10.1016/0167-5699(95)80119-7. [DOI] [PubMed] [Google Scholar]
- 3.Lusso P, Gallo RC. Human herpesvirus 6 in AIDS. Immunol. Today. 1995;16:67–71. doi: 10.1016/0167-5699(95)80090-5. [DOI] [PubMed] [Google Scholar]
- 4.Blanchard A, Montagnier L, Gougeon ML. Influence of microbial infections on the progression of HIV disease. Trends Microbiol. 1997;5:326–331. doi: 10.1016/S0966-842X(97)01089-5. [DOI] [PubMed] [Google Scholar]
- 5.Rizzardini G, Trabattoni D, Saresella M, Piconi S, Lukwiya M, Declich S, Fabiani M, Ferrante P, Clerici M. Immune activation in HIV-infected African individuals. Italian-Ugandan AIDS cooperation program. AIDS. 1998;12:2387–2396. doi: 10.1097/00002030-199818000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Wald A, Link K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J. Infect. Dis. 2002;185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- 7.Bafica A, Scanga CA, Schito M, Chaussabel D, Sher A. Influence of Coinfecting Pathogens on HIV Expression: Evidence for a Role of Toll-Like Receptors. J. Immunol. 2004;172:7229–7234. doi: 10.4049/jimmunol.172.12.7229. [DOI] [PubMed] [Google Scholar]
- 8.Abu-Raddad LJ, Patnaik P, Kublin JG. Dual Infection with HIV and Malaria Fuels the Spread of Both Diseases in Sub-Saharan Africa. Science. 2006;314:1603–1606. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- 9.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat. Med. 2006;12:289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 10.Koning FA, Otto SA, Hazenberg MD, Dekker L, Prins M, Miedema F, Schuitemaker H. Low-Level CD4+ T Cell Activation Is Associated with Low Susceptibility to HIV-1 Infection. J. Immunol. 2005;175:6117–6122. doi: 10.4049/jimmunol.175.9.6117. [DOI] [PubMed] [Google Scholar]
- 11.Friend C. Cell-free transmission in adult Swiss mice of a disease having the character of a leukemia. J. Exp. Med. 1957;105:307–318. doi: 10.1084/jem.105.4.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasenkrug KJ, Chesebro B. Immunity to retroviral infection: The Friend virus model. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7811–7816. doi: 10.1073/pnas.94.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JP, D'Andrea AD, Lodish HF, Baltimore D. Activation of cell growth by binding of Friend spleen focus-forming virus gp55 glycoprotein to the erythropoietin receptor. Nature. 1990;343:762–764. doi: 10.1038/343762a0. [DOI] [PubMed] [Google Scholar]
- 14.Ney PA, D'Andrea AD. Friend erythroleukemia revisited. Blood. 2000;96:3675–3680. [PubMed] [Google Scholar]
- 15.Lilly F. Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J. Natl. Cancer Inst. 1970;45:163–169. [PubMed] [Google Scholar]
- 16.Persons DA, Paulson RF, Loyd MR, Herley MT, Bodner SM, Bernstein A, Correll PH, Ney PA. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat. Genet. 1999;23:159–165. doi: 10.1038/13787. [DOI] [PubMed] [Google Scholar]
- 17.Hasenkrug KJ, Dittmer U. The Role of CD4 and CD8 T Cells in Recovery and Protection from Retroviral Infection: Lessons from the Friend Virus Model. Virology. 2000;272:244–249. doi: 10.1006/viro.2000.0387. [DOI] [PubMed] [Google Scholar]
- 18.Wheelock EF. The effects of nontumor viruses on virus-induced leukemia in mice: reciprocal interference between Sendai virus and Friend leukemia virus in DBA-2 mice. Proc. Natl. Acad. Sci. U. S. A. 1966;55:774–780. doi: 10.1073/pnas.55.4.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wheelock EF. Inhibitory effects of Sendai virus on Friend virus leukemia in mice. J. Natl. Cancer Inst. 1967;38:771–778. [PubMed] [Google Scholar]
- 20.Turner W, Chirigos MA, Scott D. Enhancement of Friend and Rauscher leukemia virus replication in mice by Guaroa virus. Cancer Res. 1968;28:1064–1073. [PubMed] [Google Scholar]
- 21.Steeves RA, Mirand EA, Thomson S, Avila L. Enhancement of spleen focus formation and virus replication in Friend virus-infected mice. Cancer Res. 1969;29:1111–1116. [PubMed] [Google Scholar]
- 22.McDonald TL. Blocking of cell-mediated immunity to Moloney murine sarcoma virus-transformed cells by lactate dehydrogenase virus-antibody complex. J. Natl. Cancer Inst. 1983;70:493–497. [PubMed] [Google Scholar]
- 23.Robertson SJ, Ammann CG, Messer RJ, Carmody AB, Myers L, Dittmer U, Nair S, Gerlach N, Evans LH, Cafruny WA, Hasenkrug KJ. Suppression of acute anti-friend virus CD8+ T-cell responses by coinfection with lactate dehydrogenase-elevating virus. J. Virol. 2008;82:408–418. doi: 10.1128/JVI.01413-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plagemann PG, Rowland RR, Even C, Faaberg KS. Lactate dehydrogenase-elevating virus: an ideal persistent virus? Springer Semin. Immunopathol. 1995;17:167–186. doi: 10.1007/BF00196164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Onyekaba CO, Harty JT, Even C, Hu BG, Plagemann PG. Persistent infection of mice by lactate dehydrogenase-elevating virus: effects of immunosuppression on virus replication and antiviral immune responses. Virus Res. 1989;14:297–315. doi: 10.1016/0168-1702(89)90023-3. [DOI] [PubMed] [Google Scholar]
- 26.van den Broek MF, Sporri R, Even C, Plagemann PG, Hanseler E, Hengartner H, Zinkernagel RM. Lactate dehydrogenase-elevating virus (LDV): lifelong coexistence of virus and LDV-specific immunity. J. Immunol. 1997;159:1585–1588. [PubMed] [Google Scholar]
- 27.Cafruny WA, Chan SP, Harty JT, Yousefi S, Kowalchyk K, McDonald D, Foreman B, Budweg G, Plagemann PG. Antibody response of mice to lactate dehydrogenase-elevating virus during infection and immunization with inactivated virus. Virus Res. 1986;5:357–375. doi: 10.1016/0168-1702(86)90029-8. [DOI] [PubMed] [Google Scholar]
- 28.Weiland E, Weiland F, Grossmann A. Lactate Dehydrogenase-elevating Virus Induces Anti-Golgi Apparatus Antibodies. J. Gen. Virol. 1987;68:1983–1991. doi: 10.1099/0022-1317-68-7-1983. [DOI] [PubMed] [Google Scholar]
- 29.Cafruny WA, Hovinen DE. Infection of Mice with Lactate Dehydrogenase-elevating Virus Leads to Stimulation of Autoantibodies. J. Gen. Virol. 1988;69:723–729. doi: 10.1099/0022-1317-69-3-723. [DOI] [PubMed] [Google Scholar]
- 30.Bradley DS, Broen JJ, Cafruny WA. Infection of SCID mice with lactate dehydrogenase-elevating virus stimulates B-cell activation. Viral Immunol. 1991;4:59–70. doi: 10.1089/vim.1991.4.59. [DOI] [PubMed] [Google Scholar]
- 31.Rowland RRR, Even C, Anderson GW, Chen Z, Hu B, Plagemann PGW. Neonatal Infection of Mice With Lactate Dehydrogenase-elevating Virus Results in Suppression of Humoral Antiviral Immune Response but does not Alter the Course of Viraemia or the Polyclonal Activation of B Cells and Immune Complex Formation. J. Gen. Virol. 1994;75:1071–1081. doi: 10.1099/0022-1317-75-5-1071. [DOI] [PubMed] [Google Scholar]
- 32.Anderson GW, Rowland RR, Palmer GA, Even C, Plagemann PG. Lactate dehydrogenase-elevating virus replication persists in liver, spleen, lymph node, and testis tissues and results in accumulation of viral RNA in germinal centers, concomitant with polyclonal activation of B cells. J. Virol. 1995;69:5177–5185. doi: 10.1128/jvi.69.8.5177-5185.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musaji A, Meite M, Detalle L, Franquin S, Cormont F, Preat V, Izui S, Coutelier JP. Enhancement of autoantibody pathogenicity by viral infections in mouse models of anemia and thrombocytopenia. Autoimmun. Rev. 2005;4:247–252. doi: 10.1016/j.autrev.2004.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334::676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 35.Neighbors M, Hartley SB, Xu X, Castro AG, Bouley DM, O'Garra A. Breakpoints in immunoregulation required for Th1 cells to induce diabetes. Eur. J. Immunol. 2006;36:2315–2323. doi: 10.1002/eji.200636432. [DOI] [PubMed] [Google Scholar]
- 36.Bock M, Bishop KN, Towers G, Stoye JP. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J. Virol. 2000;74:7422–7430. doi: 10.1128/jvi.74.16.7422-7430.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lander MR, Chattopadhyay SK. A Mus dunni cell line that lacks sequences closely related to endogenous murine leukemia viruses and can be infected by ectropic, amphotropic, xenotropic, and mink cell focus-forming viruses. J. Virol. 1984;52:695–698. doi: 10.1128/jvi.52.2.695-698.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chesebro B, Wehrly K, Cloyd M, Britt W, Portis J, Collins J, Nishio J. Characterization of mouse monoclonal antibodies specific for Friend murine leukemia virus-induced erythroleukemia cells: friend-specific and FMR-specific antigens. Virology. 1981;112:131–144. doi: 10.1016/0042-6822(81)90619-x. [DOI] [PubMed] [Google Scholar]
- 39.Robertson MN, Spangrude GJ, Hasenkrug K, Perry L, Nishio J, Wehrly K, Chesebro B. Role and specificity of T-cell subsets in spontaneous recovery from Friend virus-induced leukemia in mice. J. Virol. 1992;66:3271–3277. doi: 10.1128/jvi.66.6.3271-3277.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chesebro B, Wehrly K, Doig D, Nishio J. Antibody-induced modulation of Friend virus cell surface antigens decreases virus production by persistent erythroleukemia cells: influence of the Rfv-3 gene. Proc. Natl. Acad. Sci. U. S. A. 1979;76:5784–5788. doi: 10.1073/pnas.76.11.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasenkrug KJ, Brooks DM, Chesebro B. Passive Immunotherapy for Retroviral Disease: Influence of Major Histocompatibility Complex Type and T-Cell Responsiveness. Proc. Natl. Acad. Sci. U. S. A. 1995;92:10492–10495. doi: 10.1073/pnas.92.23.10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasenkrug KJ. Lymphocyte Deficiencies Increase Susceptibility to Friend Virus-Induced Erythroleukemia in Fv-2 Genetically Resistant Mice. J. Virol. 1999;73:6468–6473. doi: 10.1128/jvi.73.8.6468-6473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Messer RJ, Dittmer U, Peterson KE, Hasenkrug KJ. Essential role for virus-neutralizing antibodies in sterilizing immunity against Friend retrovirus infection. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12260–12265. doi: 10.1073/pnas.0404769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawabata H, Niwa A, Tsuji-Kawahara S, Uenishi H, Iwanami N, Matsukuma H, Abe H, Tabata N, Matsumura H, Miyazawa M. Peptide-induced immune protection of CD8+ T cell-deficient mice against Friend retrovirus-induced disease. Int. Immunol. 2006;18:183–198. doi: 10.1093/intimm/dxh361. [DOI] [PubMed] [Google Scholar]
- 45.Gomez KA, Coutelier JP, Retegui LA. Changes in the specificity of antibodies in mice infected with lactate dehydrogenase-elevating virus. Scand. J. Immunol. 1997;46:168–174. doi: 10.1046/j.1365-3083.1997.d01-107.x. [DOI] [PubMed] [Google Scholar]
- 46.Hasenkrug KJ, Brooks DM, Dittmer U. Critical Role for CD4+ T Cells in Controlling Retrovirus Replication and Spread in Persistently Infected Mice. J. Virol. 1998;72:6559–6564. doi: 10.1128/jvi.72.8.6559-6564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Earl PL, Moss B, Morrison RP, Wehrly K, Nishio J, Chesebro B. T-lymphocyte priming and protection against Friend leukemia by vaccinia-retrovirus env gene recombinant. Science. 1986;234:728–731. doi: 10.1126/science.3490689. [DOI] [PubMed] [Google Scholar]
- 48.Dittmer U, Race B, Hasenkrug KJ. Kinetics of the Development of Protective Immunity in Mice Vaccinated with a Live Attenuated Retrovirus. J. Virol. 1999;73:8435–8440. doi: 10.1128/jvi.73.10.8435-8440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hessell AJ, Hangartner L, Hunter M, Havenith CEG, Beurskens FJ, Bakker JM, Lanigan CMS, Landucci G, Forthal DN, I. Parren PWH, Marx PA, Burton DR. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 50.Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, Zinkernagel RM. Control of Early Viral and Bacterial Distribution and Disease by Natural Antibodies. Science. 1999;286:2156–2159. doi: 10.1126/science.286.5447.2156. [DOI] [PubMed] [Google Scholar]
- 51.Steeves RA, Grundke-Lqbal I. Bacterial lipopolysaccharides as helper factors for Friend spleen focus-forming virus in mice. J. Natl. Cancer Inst. 1976;56:541–546. doi: 10.1093/jnci/56.3.541. [DOI] [PubMed] [Google Scholar]
- 52.Kreja L, Seidel HJ. On the role of the spleen in Friend virus (FMuLV-P) erythroleukemia. Exp. Hematol. 1985;13:623–628. [PubMed] [Google Scholar]
- 53.Olbrich ARM, Schimmer S, Dittmer U. Preinfection Treatment of Resistant Mice with CpG Oligodeoxynucleotides Renders Them Susceptible to Friend Retrovirus-Induced Leukemia. J. Virol. 2003;77:10658–10662. doi: 10.1128/JVI.77.19.10658-10662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlach N, Schimmer S, Weiss S, Kalinke U, Dittmer U. Effects of Type I Interferons on Friend Retrovirus Infection. J. Virol. 2007;81:6160. doi: 10.1128/JVI.80.7.3438-3444.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inada T, Yamazaki S. Replication of lactate dehydrogenase-elevating virus in cells infected with murine leukaemia viruses in vitro. J. Gen. Virol. 1991;72:2437–2444. doi: 10.1099/0022-1317-72-10-2437. [DOI] [PubMed] [Google Scholar]
- 56.Inada T, Kikuchi H, Yamazaki S. Comparison of the ability of lactate dehydrogenase-elevating virus and its virion RNA to infect murine leukemia virus-infected or -uninfected cell lines. J. Virol. 1993;67:5698–5703. doi: 10.1128/jvi.67.9.5698-5703.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pease LR, Murphy WH. Co-infection by lactic dehydrogenase virus and C-type retrovirus elicits neurological disease. Nature. 1980;286:398–400. doi: 10.1038/286398a0. [DOI] [PubMed] [Google Scholar]
- 58.He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, Moore JP, Cerutti A. HIV-1 Envelope Triggers Polyclonal Ig Class Switch Recombination through a CD40-Independent Mechanism Involving BAFF and C-Type Lectin Receptors. J. Immunol. 2006;176:3931–3941. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- 59.Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, Zinkernagel RM. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat. Immunol. 2003;4:343–349. doi: 10.1038/ni911. [DOI] [PubMed] [Google Scholar]